
Abstract
Thrombosis is a major cause of morbidity and mortality in cancer patients. The pathogenesis of blood coagulation activation in oncological patients is complex and involves both clinical and biological factors. Abnormalities in one or more coagulation test are common in cancer patients, even without thrombotic manifestations, indicating an ongoing hypercoagulable condition. Moreover, venous thromboembolism (VTE) can be the first symptom of an occult malignancy in an otherwise healthy individual. The levels of laboratory markers of activation of blood coagulation parallel the development of malignancy, being the coagulant mechanisms important for both thrombogenesis and tumor progression. Besides general clinical risk factors for VTE, also disease-specific clinical factors, i.e., type and stage of the tumor, and anticancer therapies increase the thrombotic risk in these patients. Furthermore, biological factors, including the cancer cell-specific prothrombotic properties together with the host cell inflammatory response to the tumor, are relevant as well as unique players in the pathogenesis of the cancer-associated hypercoagulability. Cancer cells produce and release procoagulant and fibrinolytic proteins, inflammatory cytokines, and procoagulant microparticles. They also express adhesion molecules binding to the receptors of host vascular cells (i.e., endothelial cells, platelets, and leukocytes), thereby stimulating the prothrombotic properties of these normal cells, including the shed of cell-specific microparticles and neutrophil extracellular traps. Of interest, several genes responsible for the cellular neoplastic transformation drive the programs of hemostatic properties expressed by cancer tissues. A better understanding of such mechanisms will help the development of novel strategies to prevent and treat the Trousseau’s syndrome (i.e., cancer-associated thrombosis).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Thrombosis
- Cancer
- Procoagulant mechanisms
- Hypercoagulable state
- DIC
- Tissue factor
- Microparticles
- Platelet
- Leukocyte
- Endothelium
2.1 Introduction
The close relationship between cancer and thrombosis has been known for more than a century. Cancer patients present with many types of hemostatic abnormalities and have an increased risk of both thrombotic and hemorrhagic complications. A very intimate and reciprocal relation exists between malignant disease, the occurrence of clotting alterations and thrombosis. Indeed, malignancy most commonly induces a procoagulant shift in the host hemostatic balance [1], thus establishing a condition favorable to the development of thrombosis. Vice versa, the activation of blood coagulation favors the tumor growth and dissemination.
Cancer is associated with a four- to sevenfold increase in the risk of venous thromboembolism (VTE) [2, 3], peaking in the first 3 months following cancer diagnosis [4, 5]. The risk of thrombosis is also increased in metastatic compared with non-metastatic cancer disease. However, even without thrombosis, the majority of cancer patients present with clotting alterations detectable by laboratory tests, which reveal different degrees of coagulation activation and characterize the hypercoagulable state of these subjects [6]. Currently, it is fully recognized that cancer patients are at significant risk of developing all types of thrombotic events, spanning from venous or arterial thrombosis to systemic syndromes, such as disseminated intravascular coagulation (DIC) with severe bleeding. Preventing these complications is clinically relevant because they considerably contribute to the morbidity and mortality of these patients [7].
The pathogenesis of the cancer-associated thrombosis (CAT) is complex and multifactorial. Many clinical factors influence the thrombotic risk of these patients. Clinical factors include general risk factors (i.e., older age, cardiovascular diseases, prior VTE, infections, prolonged immobilization), as well as disease-specific factors, as the cancer site and stage and anticancer therapies. Further, biological mechanisms are involved in the pathogenesis of CAT. Indeed, tumor cells gain the capacity to activate the host hemostatic system in several ways, and this phenomenon is often driven by the same oncogenes responsible for the cellular neoplastic transformation [8]. By this process, cancer tissues become able to express different procoagulant proteins (i.e., tissue factor [TF], cancer procoagulant [CP], factor VII) and to shed procoagulant microparticles. Furthermore, they activate platelets, leukocytes, and endothelial cells, by direct cell–cell adhesion mechanisms, or through the liberation of inflammatory cytokines or proangiogenic factors. All these phenomena contribute to the pathogenesis of CAT.
In this chapter, we wish to review the most recent advances in our knowledge on the pathogenic factors underlying the development of thrombosis in patients with malignant diseases, with a particular focus on the cancer tissue-specific biological properties, by which malignant cells are capable to activate the hemostatic system.
2.2 Pathogenic Factors of Cancer-Associated Thrombosis
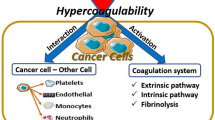
Multiple clinical factors together with biological procoagulant mechanisms expressed by cancer tissues concur to the activation of blood coagulation and importantly contribute to the overall thrombotic risk of these patients [9, 10] (Fig. 2.1). Clinical risk factors include general and biological factors, which are common to cancer and non-cancer patients, whereas in patients with malignancy there are also a number of disease-specific clinical as well biological factors, which render the pathogenesis of cancer-associated thrombosis unique.

Pathogenesis of thrombophilic state in cancer patients is multifactorial. Clinical risk factors include general and biological factors, which are common to cancer and non-cancer patients, whereas in patients with malignancy there are also a number of disease-specific clinical as well biological factors. Particularly, a unique role is played by the capacity of tumor cells to interact and activate blood coagulation
Altogether, these factors favor the shift of the hemostatic balance toward a prothrombotic condition, as shown by the appearance of subclinical coagulation changes in almost all of cancer patients, who constantly present with high levels of circulating biomarkers of hypercoagulability.
2.2.1 General Factors
General factors include both clinical and biological risk factors, which are common to all patients, with and without cancer.
As listed in Fig. 2.1, general clinical factors include older age, prior history of VTE, prolonged immobilization, obesity, infections, cardiovascular risk factors, renal and respiratory diseases, and anemia.
On the other hand, biological factors carrying a thrombotic risk in the general population comprise high leukocyte and platelet counts, the inherited thrombophilia, and the ABO blood group.
In particular, elevated numbers of platelets and neutrophils are often observed in patients with cancer, and several studies have demonstrated their association with an increased risk of thrombosis [11, 12], and with a poor cancer prognosis [13]. It is possible that granulocyte colony-stimulating growth factor (G-CSF), produced by many tumors and present in the circulation of many cancer patients, contributes to increase the number of neutrophils and induce their activation [13]. The “Khorana risk model,” an important tool that predicts the risk of cancer-associated thrombosis, includes pretreatment thrombocytosis (platelet count ≥ 350 × 109/L) and leukocytosis (leukocytes ≥ 11 × 109/L) among the five clinical risk factors associated with an increased risk of VTE, together with the site of cancer, hemoglobin < 10 g/dl, and a body mass index ≥ 35 kg/m2 [14]. These findings have now been validated by many other large studies [15,16,17].
The role of prothrombotic genotypes has been considered in cancer patients [18]. The influence of inherited thrombophilia in patients with cancer may be more difficult to demonstrate than in the general population, the risk of thrombosis due to cancer per se possibly outweighing the contribution of thrombophilia factors. However, in the presence of cancer, prothrombotic genotypes may further increase the thrombotic risk [19]. Several studies evaluating the role of factor V Leiden or G202110A prothrombin gene mutation on the risk of CAT have been published [20]. Overall, although conflicting results were obtained, it appears that patients with cancer and either of these mutations tend to exhibit a higher risk of thrombosis than patients with cancer without these mutations. Indeed, in recent large studies the risk of VTE of patients with both cancer and factor V Leiden mutation was increased from 2 to 12-fold, compared to patients without factor V Leiden [5, 21, 22]. Studies investigating on the impact of prothrombin 20210A mutation on the risk of cancer-related VTE have shown conflicting results, possibly due to the rarity of this mutation. However, a large study found that patients with cancer and prothrombin 20210A mutation had a fourfold increased risk of VTE compared to the non-carriers with cancer and an 18-fold increase compared to cancer-free non-carriers [5]. Similar results were reported for central venous catheter-related VTE [23].
Finally, an association between ABO blood groups and the risk of VTE has also been described since 1969, being non-O blood groups associated to an increased risk of VTE [24], particularly type A1 and B groups [25]. The single-nucleotide polymorphism (SNP) rs8176719 represents a site in the ABO gene essential to determine the O group and has been used to evaluate the risk of cancer-related VTE in a case–control study [22]. In cancer patients, a non-O blood type was associated with a 30% increased risk of VTE, and even higher risk (12-fold increased) if compared to cancer-free subjects with an O blood type [22].
2.2.2 Disease-Specific Factors
In this category are clinical and biological pathogenic factors that are exclusive to the malignant disease (Fig. 2.1).
Clinical factors undoubtedly include the site of cancer. Indeed, large epidemiological studies have recognized brain tumors, hematological malignancies, and pancreatic, gastric, ovarian, uterus, pulmonary and renal adenocarcinomas as having the highest risk of VTE [26]. Among hematological malignancies, multiple myeloma, Hodgkin’s disease, and non-Hodgkin lymphoma have shown the highest incidence of VTE [27].
The stage of cancer is also an important risk factor for VTE; advanced, metastatic disease is linked to a higher risk of VTE compared to localized tumors [26]. The initial period after diagnosis of cancer is at high risk of VTE as well [5].
Finally, active anticancer treatments, such as chemo-, radio-, and hormone therapies, antiangiogenetic agents, combination regimens, cancer surgery, the use of erythropoiesis-stimulating agents and blood transfusions exert a prothrombotic effect [28]. Chemotherapeutic agents and tumor-derived products can directly damage the vascular endothelium, leading to a loss of endothelial antithrombotic properties [28]. Moreover, chemotherapy can induce an overexpression of tissue factor, an increased exposure of cell membrane phosphatidylserine, and the release of procoagulant microparticles [29]. All these mechanisms, as well as the presence of central venous lines to deliver drugs and nutrients, can play a role in the pathogenesis of CAT.
2.3 Cancer Cell Prothrombotic Mechanisms
The principal procoagulant mechanisms expressed by cancer cells are schematically depicted in Fig. 2.2 and include:

Principal mechanisms of hypercoagulability in cancer. Direct blood clotting activation by the expression of hemostatic proteins with procoagulant activity by cancer cells; host cell-cancer cell interaction, through the expression of surface adhesion molecules and through the release of soluble mediators, including inflammatory cytokines and proangiogenic and growth-stimulating factors
-
1.
The activation by cancer cells of the clotting system through the expression of procoagulant properties (such as tissue factor [TF], cancer procoagulant [CP], and heparanase), TF-bearing procoagulant microparticles (TF-MP), coagulation factors, and fibrinolysis proteins;
-
2.
The activation by cancer cells of the procoagulant potential of host blood vascular cells, i.e., platelets, leukocytes, and endothelial cells. The latter mechanism can occur either by cell–cell direct contact mediated by specific surface adhesion receptors, and/or by the release of inflammatory cytokines, and proangiogenic and growth-stimulating factors (i.e., vascular endothelial growth factor [VEGF], basic fibroblast growth factor [bFGF], and G-CSF) by both cancer and host blood cells. The activation of platelets, endothelial cells, and leukocytes produces, among other procoagulant features, the release of blood cell-specific procoagulant microparticles (MP) and neutrophil extracellular traps (NETs).
2.3.1 Cancer Cell Procoagulant Properties
The procoagulant properties expressed by cancer cells include:
-
procoagulant proteins,
-
microparticles (MPs),
-
coagulation factors,
-
fibrinolysis proteins.
2.3.1.1 Cancer Cell Procoagulant Proteins
Among procoagulant proteins, tissue factor (TF) is the best characterized. TF is the primary initiator of blood coagulation in normal and pathological conditions. It forms a complex with activated factor VII to trigger blood coagulation by proteolytic activation of factors IX and X (clotting extrinsic pathway). Many solid tumors and hematologic malignancies constitutively express TF, and the levels of TF expression tend to be associated with an aggressive pattern of the tumor. Indeed, studies performed in malignant gliomas and pancreatic tumors demonstrated that the levels of TF expression correlate with the histological grade of malignancy and vascularity [30,31,32,33,34]. Moreover, TF activity can be potentiated by the expression of anionic phospholipids on the outer leaflet of glioma cells, leading to coagulation reactions through the intrinsic pathway and to explosive generation of thrombin [35]. Other studies conducted in colorectal and pancreatic cancers show that plasma levels of TF antigen correlate with the tumor size [36, 37]. TF expression by tumor cells is a consequence of cancer-causing mutations, like oncogenes activation (i.e., KRAS and MET) or tumor suppressor genes inactivation (i.e., p53, PTEN) [38]. The elevated inflammatory status of cancer patients also enhances TF production. Indeed, endothelial cells and monocyte/macrophages that do not express TF under normal conditions can be induced to express TF by proinflammatory stimuli (i.e., IL-1β, TNF-α, and bacterial lipopolysaccharides [LPS]) [39].
Tumor-cell-derived TF plays a central role in the generation of thrombin in cancer, but also contributes to tumor progression by directly influencing the expression of VEGF by both malignant cells and host vascular cells. This property regulates tumor neovascularization and provides an important link between activation of coagulation, inflammation, thrombosis, and tumor growth and metastasis [1].
Another tumor cell procoagulant is cancer procoagulant (CP) that, unlike TF, directly activates factor X independently of coagulation factor VII. CP has been detected in different malignant cells [40], from both solid and hematologic tumors, but not in normal tissues. Of interest, in patients with acute promyelocytic leukemia (APL), CP is expressed by bone marrow blast cells at the onset of disease, but disappears at remission [41]. In addition, in APL patient blasts, CP expression paralleled the degree of malignant transformation and disappeared upon cellular differentiation by therapy with all-trans-retinoic-acid (ATRA). In contrast, cells resistant to ATRA maintained their malignant phenotype and continue to express CP. Similar observations have been reported for breast cancer [42]. The relative contribution of this factor to the overall cellular procoagulant activity and/or possible interactions with TF are unknown at this time.
Among other tumor cell procoagulant activities, the role of the enzyme heparanase is gaining much relevance. Heparanase is a protease that cleaves heparan sulfate (HS) of the extracellular matrix (ECM). Its expression is restricted to platelets, activated white blood cells, and placenta. Many studies demonstrated an overexpression of this enzyme in essentially all human tumors, both solid and hematological [43], which promotes tumor dissemination and metastasis, by remodeling ECM barrier [44, 45], releasing VEGF-A and bFGF bound to HS [46, 47], and by facilitating endothelial cells migration and proliferation [48, 49]. Additionally, it upregulates the expression of the blood coagulation initiator TF and interacts with the tissue factor pathway inhibitor (TFPI) on the cell surface of endothelial and tumor cells, leading to dissociation of TFPI with resulting increased cell surface TF activity [50]. Finally, heparanase directly activates the extrinsic coagulation pathway, increasing the level of factor Xa in the presence of TF/VIIa, acting as a cofactor of TF [50].
2.3.1.2 Tumor Microparticles
Tumor-cell-shed microparticles (MP) represent an emerging mechanism of tumor-promoted clotting activation. MP are plasma membrane vesicles of 0.1–1 μm in diameter, composed of lipids, proteins, and nucleic acids, released from virtually all types of blood cells upon activation, apoptosis, malignant transformation, and stress [51]. MP display typical surface cell proteins derived from the cell of origin, but they can also carry proteins acquired from other cell types by a fusion process. Platelet-derived MP (PMP) constitute the majority (>80%) of circulating MP, whereas less than 10% originate from granulocytes and less than 5% from endothelial cells, red blood cells, and monocytes. However, in pathological conditions, an overall increment of MP occurs also from other sources, including tumor cells. Under normal conditions, MP express anionic phosphatidylserine (PS) on their outer leaflet, though several reports show that a subset of circulating PMP may also express TF [52]. In healthy individuals, the majority (>95%) of circulating PMP express PS, whereas only a very low number express TF, and circulate at low levels. MP undergo phenotypic and quantitative changes in several clinical conditions, most of which associated with an increased risk of both arterial and venous thrombosis (i.e., diabetes, acute coronary syndrome, disseminated intravascular coagulation, antiphospholipid syndrome) [53,54,55,56]. The increased number and thrombogenic activity of MP in prothrombotic disorders indicate their important role in the pathogenesis of thrombosis. In the cancer setting, TF-bearing MP are of particular interest, since, due to the abundance of negatively charged phospholipids on their surface, they display TF in its “active” form. Elevated levels of circulating MP have been described in cancer patients, with both solid and hematologic malignancies [57, 58], and different reports suggest an additional role for these elements in the establishment of a thrombotic state in cancer [59,60,61]. Several studies demonstrated that TF-positive MP can be derived from tumor cells. The increased production of MP by cancer cells seems to be controlled by definite genetic events occurring in tumorigenesis, including activating and inactivating mutations in oncogenes and tumor repressor genes [62]. Studies in animal and human models showed that tumor-derived TF-positive MP contribute to cancer-associated thrombosis [63,64,65,66]. The intravenous injection into mice of MP derived from human tumor cells and expressing high levels of TF induced a TF-dependent activation of coagulation, which resulted in a DIC-like syndrome [37]. Elevated TF-positive MP have been reported in patients with solid tumors and VTE gastric and pancreatic cancers being the most studied [59, 61, 67]. Fewer reports have been published in the setting of hematological malignancies, where high levels of blast cell-derived MP have been confirmed in acute promyelocytic leukemia [68] and acute myeloid leukemia [69]. In patients with multiple myeloma, TF-positive MP activity was higher in those developing VTE [70]. In one study of essential thrombocythemia (ET), MP numbers were significantly higher in ET patients than controls, and the thrombin generation potential of MP-rich plasma from these patients was significantly increased [71].
MP can also play a role in cancer progression, especially due to their capacity to influence angiogenesis [72]. In one study, it has been shown that PMP isolated from healthy donors can promote proliferation, survival, and capillary tube formation of human endothelial cells [73]. In addition, PMP can stimulate the expression of proangiogenic factors by tumor cells [74]. Finally, the expression of TF by circulating MP represents per se an important mechanism of MP-promoted tumor progression, by means of TF role in tumor growth, angiogenesis, and metastasis [75,76,77].
The clinical significance of MP as a predictive biomarker of VTE risk in cancer patients has not been fully elucidated. For this reason, some trials are evaluating the utility of measuring TF-MP to predict VTE in cancer [78]. Since MP are clearly involved in thrombosis and cancer, potential modulation of their release and activity may have important therapeutic implications.
2.3.1.3 Coagulation Factors
The plasma protein factor VII (FVII), under normal conditions, is constitutively expressed in the liver, mainly by hepatocytes [79]. However, FVII can be expressed also by monocytes and macrophages in inflammatory conditions [80, 81], and in cancer, where the expression of ectopic FVII has been described in hepatocellular carcinoma cells [82], bladder cancer [83], ovarian cancer [84], and laryngeal carcinoma [85]. More recent studies on FVII mRNA expression in different cancer cell lines have demonstrated a frequent expression of endogenous FVII in various cancer cells [86], especially in colon cancer cell lines [87]. In these experiments, ectopic FVII was functionally active due to cancer cell expression of γ-glutamyl carboxylase, which facilitates the post-translational edits required for proper positioning of FVII on the cell membrane [86]. Coagulant-active ectopically expressed TF:FVIIa was also found on TF-positive ovarian cancer cells, making this complex a plausible trigger of VTE at distant sites, which is a frequent complication in patients with ovarian cancer [88].
Factor VIII (FVIII) plays a key role in the coagulation cascade. Several studies have shown that high factor VIII activity indicates an increased risk for primary and recurrent venous thromboembolism [89, 90]. High FVIII levels have been observed in patients with multiple myeloma, breast cancer, and colorectal cancer [91,92,93]. A small retrospective study reported higher FVIII levels in patients with various types of cancer and a history of thrombosis in comparison to a matched control group without thrombosis [94]. A subsequent prospective cohort study confirmed that high FVIII plasma level is a significant risk factor for symptomatic VTE in cancer patients [95]. In this study, the risk conferred by FVIII correlated with the FVIII levels. A significant difference in FVIII according to the tumor site was described, being FVIII levels highest in cancers of the stomach or pancreas, in which an association with disease stage was also seen. Similar findings were reported in patients with multiple myeloma [91]. However, to what extent the malignant disease contributes to FVIII plasma levels needs to be further elucidated.
2.3.1.4 Fibrinolysis Proteins
In this context, it is important to consider that tumor cells also generate anticoagulant forces and interact with the host fibrinolytic system. Indeed, cancer cells can express fibrinolytic proteins such as the plasminogen activators [i.e., urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA)], their inhibitors [i.e., plasminogen activator inhibitor-1 (PAI-1) and plasminogen activator inhibitor-2 (PAI-2)], and receptors (i.e., urokinase-type plasminogen activator receptor, and annexin II, a co-receptor for plasminogen and tPA). Elevated levels of PAI-1 antigen and activity have been found in patients with pancreatic cancer and malignant glioma [96, 97], predisposing to VTE. In a mouse model xenografted with a human lung adenocarcinoma cell line, the anti-VEGF drug, bevacizumab, induced an increase in PAI-1 levels and enhanced thrombosis, which was reduced by a PAI-1 inhibitor [98]. In APL, the increased annexin II expression by leukemic cells favors the assembly of the fibrinolytic cascade proteins on the cell surface and has been linked to excessive activation of fibrinolysis and bleeding complications [99].
Likely, depending on which side, pro- or antifibrinolytic, prevails, the clinical manifestations may be quite different, from bleeding symptoms, as observed in leukemia, to VTE, as evidenced in solid tumors.
2.3.2 Host Cell Procoagulant Properties Elicited by Cancer Cells
A strong interaction occurs between cancer cells and the host normal vascular cells, particularly platelets, leukocytes, and endothelial cells, which generally results in the expression of a procoagulant phenotype by normal cells. As schematically shown in Fig. 2.2, cancer cells activate the procoagulant potential of host normal vascular cells by two principal mechanisms, i.e., (1) the expression by cancer cells of surface adhesion molecules and counter-receptors by which they anchor other blood cells and attach to the vessel wall and (2) the release of soluble mediators, including inflammatory cytokines (i.e., TNF-α, IL-1β), proangiogenic and growth-stimulating factors (i.e., VEGF, bFGF, G-CSF), and platelet aggregation agonists.
2.3.2.1 Platelets’ Activation by Tumor Cells
There is growing evidence that platelets are very important in promoting the hypercoagulable state of patients with cancer [100]. However, a fundamental step occurring in malignancy is platelet activation by direct cancer cell–platelet adhesion [17, 100, 101], and/or by tumor secretion of platelet-activating molecules (i.e., ADP, thrombin, matrix metalloproteinases, IL-6) [102], which lead to platelet adhesion/aggregation (Fig. 2.3). Among adhesion mechanisms, selectins expressed on platelets, leukocytes, and endothelium can bind tumor cells to form aggregates [103]. Specifically, P-selectin expressed on the surface of activated platelets binds to many types of human cancer cells [104], and this interaction can also promote tumor growth and metastasis [104]. In general, platelet activation, aggregation, coagulation, and thrombus formation are crucial events in limiting blood loss after tissue damage but are also major determinants of hematogenous tumor metastasis [105]. Increased platelet activation and aggregation correlate with the metastatic potential of cancer cells in both in vitro and in vivo models of experimental metastasis. Indeed, platelet aggregation protects the tumor cell surface from immunological recognition in the circulation. Tumor-cell-induced platelet aggregation can result in a “platelet coating” of cancer cells shielding them from natural killer (NK) cells [106]. Some tumor cells may use podoplanin, a transmembrane sialoglycoprotein, to activate platelets [107]. Podoplanin is a ligand of the platelet receptor C-type lectin receptor type-2 (CLEC-2) and induces platelet aggregation in normal conditions. Podoplanin is present on the surface of certain tumor cells, including melanoma, squamous cell carcinoma, seminoma, and brain tumor cells [107]. Increased levels of podoplanin are associated with tumor metastasis or malignant progression, however, recent data clearly show that podoplanin-positive MP from brain tumors activate circulating platelets, and are associated with platelet aggregation and increased thrombotic risk in these patients [108]. Strategies are now being developed to inhibit podoplanin–CLEC2 interactions in preclinical models of solid tumors [109].

Platelet activation by tumor cells. Tumor cells activate platelets through direct cancer cell–platelet adhesion, tumor secretion of platelet-activating molecules or by the expression of podoplanin on cancer cells’ surface. Activated platelets can mediate the onset of hypercoagulability in cancer patients by direct clotting activation and thrombus formation or by interacting with other blood cells
Activated platelets can mediate the onset of hypercoagulability in cancer patients by interacting with other blood cells. First, the interaction of platelets with leukocytes is likely involved in inducing a procoagulant state in cancer patients. In an animal model of CAT, platelet–leukocyte interaction, as mediated by P-selectin on platelets and P-selectin glycoprotein ligand (PSLGI) on leukocytes, was necessary for the formation of mucin-induced lung microthrombi [110]. Moreover, platelets stimulate the release of neutrophil extracellular DNA traps (NETs) by leukocytes, which promote venous thrombosis (see Sect. 2.3.2.2). Second, the interaction of platelets with endothelial cells is relevant for platelet-mediated CAT. A study in a mouse model of deep vein thrombosis suggests that platelets have a critical role in thrombus formation in a condition of flow restriction, through the interaction with von Willebrand factor (vWF) bound to the endothelium [111]. Finally, platelets promote thrombosis in cancer patients by the activation of the coagulation cascade leading to thrombin generation. Indeed, activated platelet exposes phosphatidylserine (PS) on their outer membrane, which provides a negatively charged surface for initiation of fibrin clot formation [112]. Furthermore, adherent activated platelets release procoagulant PMPs, which further contribute to the fibrin deposition and microthrombi formation, as previously described (see Sect. 2.3.1.2).
2.3.2.2 Leukocytes Activation by Tumor Cells
Leukocyte numbers are frequently elevated in cancer patients, but also they circulate in an activated status as they are challenged by tumor cells to exhibit a procoagulant phenotype. The most important subpopulations of leukocytes involved in the clotting activation are neutrophils and monocytes. Tumor cells can activate leukocytes by direct cell–cell adhesion or by the release of cytokines and growth factors in the bloodstream (Fig. 2.4). In particular, G-CSF is produced by many tumors and is found elevated in the circulation of many cancer patients [13]. G-CSF increases the number of neutrophils and induces their activation.

Leukocytes’ procoagulant activities elicited by tumor cells. The subpopulations of leukocytes involved in the clotting activation in cancer patients are neutrophils and monocytes. Tumor cells can activate leukocytes by direct cell–cell adhesion or by the release of cytokines and growth factors in the bloodstream. Neutrophils activated by tumor cells release procoagulant enzymes and expose on their surface high levels of TF, adhesion molecules for platelets and endothelial cells, and neutrophil extracellular traps (NETs), thus promoting the activation of blood clotting cascade. Moreover, activated platelets can provide signals that promote formation of NETs, which provide a scaffold and a stimulus for platelet adhesion and thrombus formation. Activated monocytes/macrophages express highly procoagulant TF and coagulation factors on their surface upon activation by cytokines, leading to fibrin formation and deposition
Tumor-cell-activated neutrophils release procoagulant enzymes, including elastase and myeloperoxidase (MPO). They also expose on their surface high levels of TF and adhesion counter-receptors for platelet and endothelial cell adhesion molecules, as documented in myeloproliferative neoplasms [113, 114].
There is emerging evidence that cancer cells also predispose neutrophils to the release of DNA extracellular traps (“neutrophil extracellular traps” or NETs) [115]. NETs were first identified as a host defense mechanism against pathogens. They are the result of externalized DNA (nuclear or mitochondrial) released from the nucleus of neutrophils, decorated by histones and granular proteases following activation by bacterial LPS or cytokines. During sepsis, this mechanism named NETosis creates a high local concentration of proteases and provides a method of entrapment and killing of pathogens. However, NETs are also known to promote coagulation, providing a scaffold and a stimulus for platelet adhesion and thrombus formation [116]. The prothrombotic effect of NETs can be explained by their high content of negatively charged nucleic acids and histones, providing a strong activation signal for platelets, which translates into platelet aggregation and thrombosis [116]. At the same time, activated platelets can provide signals that promote formation of NETs [117]. In addition to its implication in thrombosis, the formation of NETs in cancer may affect the tumor biology. Tumor infiltrating-neutrophils can exert a role in promoting different steps of tumor progression. Of interest, the procoagulant activity of NETs leads to the generation of thrombin, which can affect all aspects of cancer progression [118].
The role of activated monocytes/macrophages in CAT is well known. Since the 80s, different studies have demonstrated that macrophages infiltrating the tumor are locally activated toward a procoagulant activity that contributes to fibrin deposition within malignant tissues [119, 120]. A study in ovarian cancer showed that in advanced disease, blood monocytes were activated to a procoagulant phenotype, adding to the activation of intravascular coagulation and thrombo-embolic complications [121]. Notably, monocytes are the only circulating blood cells that are able to synthetize and express highly procoagulant TF on their surface upon activation by cytokines (i.e., IL-lβ, TNF-α) and LPS [122]. Cancer cells can secrete these mediators, thus triggering the monocyte-induced mechanism of thrombosis. Moreover, macrophages infiltrating the tumor have been found to express coagulation factors II, V, VII, and X on their surface [122]. More recent studies have shown that blood monocytes are also capable to release extracellular traps (ETs) in response to several inflammatory stimuli. Monocyte ETs display a morphology similar to NETs, being associated to MPO, lactoferrin, citrullinated histones, and elastase, and a procoagulant activity [123]; however, their role in CAT needs to be further elucidated. Finally, a recent study in lung cancer patients with VTE found a relation between a high absolute monocyte count with a refractoriness to anticoagulant therapy and poor prognosis [124].
2.3.2.3 Endothelium Activation by Tumor Cells
Physiologically, the endothelium facilitates the blood flow by providing an antithrombotic surface that inhibits platelets’ adhesion and coagulation activation. Several factors can perturb the resting state of endothelium in cancer patients (Fig. 2.5). Tumor cells can activate endothelial cells directly by cell–cell adhesion, as demonstrated in studies in non-small cell lung and colorectal carcinomas [125], or by the release of proinflammatory cytokines and acute phase proteins, which trigger the activation of endothelial cells as well as of monocytes [126]. In addition, in malignancy, reactive oxygen species and intracellular proteases released by activated neutrophils can induce detachment or lysis of endothelial cells, affecting functions involved in thrombomodulation.

Endothelium prothrombotic activation by cancer cells. Tumor cells can activate endothelial cells directly by cell–cell adhesion or by the release of proinflammatory cytokines. Cytokines regulate the expression of endothelial cell products active in hemostasis, including thrombomodulin (TM), TF, vWF, selectins, and fibrinolysis proteins (PAI-1)
Among cytokines, interleukin-lβ (IL-lβ) and tumor necrosis factor (TNF-α) can regulate the expression of endothelial cell products active in hemostasis, including thrombomodulin (TM), TF, vWF, adhesive receptors (i.e., selectins), and fibrinolysis proteins (i.e., fibrinolysis inhibitor PAI-1) [127]. TM is a membrane receptor of vascular endothelial cells with a potent anticoagulant function [128], since it binds and forms a complex with thrombin to activate the natural anticoagulant protein C. In cancer patients, increased levels of soluble TM and reduced expression of surface TM have been observed [129], leading to a loss of anticoagulant membrane TM at the endothelial outward. Furthermore, soluble TF released from endothelial cells in response to TNF-α has been demonstrated and is a marker of increased expression of TF on endothelial cells surface, a potent mechanism of prothrombotic response to inflammation [130]. Upregulation of the procoagulant TF with downregulation of the anticoagulant TM/protein C system converts the normal anticoagulant endothelium into a prothrombotic endothelium. Increased levels of vWF released from the endothelium are also described in cancer patients, and are of particular relevance in the pathogenesis of thrombosis in myeloproliferative neoplasms: Once platelets bind to vWF, they become activated and are able to aggregate and strengthen a clot [131]. Activated endothelium can also shed soluble adhesion molecules like selectins, which are commonly expressed by endothelial cells (P-selectin, E-selectin), platelets (P-selectin), and leukocytes (L-selectin). Increased levels of circulating E-selectin and P-selectin have been described in patients with myeloproliferative neoplasms and thrombosis [132], as well as in patients with lung and breast cancers [133, 134]. Moreover, reduced plasma levels of nitric oxide (NO) produced by endothelial cells, which inhibits platelet adhesion, activation, and aggregation, represent another mechanism of thrombus formation in myeloproliferative neoplasms [135]. Finally, circulating endothelial cells (CECs) have been established as markers of endothelial damage or dysfunction [136]. CEC levels increase in many types of solid tumors and hematological malignancies, and correlate with angiogenetic activity and tumor progression [137, 138], although their role in CAT has yet to be established.
2.4 The Oncogene Perspectives
In the last decade, molecular studies have demonstrated that oncogenes and repressor genes responsible for neoplastic transformation (i.e., mutation/induction of KRAS, EGFR, or MET, loss of PTEN, or TP53) also drive the programs for the expression of hemostatic proteins in cancer tissues (Fig. 2.6). Coagulopathy and thrombosis have been regarded for a long time as unspecific consequences of cancer-related disruption in tissue anatomy and vascular continuity, or driven by vascular hyperpermeability, inflammation, stasis, and toxic side effect [8]. Recent studies, however, suggest that activated coagulation may possess cancer-specific properties [8]. Rak and colleagues proposed that the type of cancer cell influenced the state of the coagulation system, as different cancers differ greatly in terms of risk of VTE [139]. Indeed, pancreatic, brain, gastrointestinal, ovarian, and hematological malignancies are all associated with a higher risk of VTE compared to skin, breast, and prostate cancer [26].

Oncogenes, tumor suppressor genes, and microRNA implicated in hypercoagulability in cancer. Genes for neoplastic transformation also drive the programs for the expression of hemostatic proteins in cancer tissues
Specific genotypes of cancer cells may affect the coagulation system either directly or through changes in tumor environment [140]. For instance, it is documented that several dominant oncogenes, such as RAS, EGFRvIII, MET, and many other genetic lesions frequently upregulate VEGF, enhancing neo-angiogenesis [141]. Oncogenes’ pathways also influence the recruitment of inflammatory cells, which themselves may exhibit proangiogenic and procoagulant phenotypes [142, 143]. However, oncogenic pathways also deregulate coagulation effectors more directly, through several different types of effects, i.e., by the abnormally high/constitutive expression of TF [144], by triggering the ectopic expression of coagulation genes [140, 145, 146], or by the emission of TF-bearing large and small microparticles that can enter biofluids and the general circulation [147]. In addition, mutations in different oncogenes and the loss of some repressor genes in different types of tumors may activate the coagulation system using one or another of these mechanisms. For example, in colorectal cancer, mutant KRAS is able to upregulate the expression of TF-bearing MPs; nonetheless, in the same type of cells the deletion of TP53 has been associated to enhanced TF exposure and shedding [36]. Overexpression of EGFR and HER-2, both upstream activators of the RAS signaling cascade, resulted in increased TF production in glioma and carcinoma cells, respectively [77, 146]. TF may also be upregulated by the mutation of the oncogene MET in hepatoma [148] and by the loss of PTEN tumor suppressor, especially under hypoxia, in glioblastoma [149]. Finally, several types of microRNA (miRNA) have been implicated in alterations of coagulant properties of cancer cells (i.e., expression and regulation of TF, heparanase, PAR-1, PAI-1, and COX-2) [150,151,152,153,154].
Genetic regulation of coagulation factors in cancer cells implies that molecularly different subtypes of cancer should exhibit different coagulation patterns (or “coagulomes”). Indeed, molecular profiling of human glioblastoma (GBM) has recently revolutionized the classification of this malignancy in four molecular subgroups, which effectively constitute different disease entities but also display a different pattern of activation of the coagulation system [108, 140]. These data suggest that activation of oncogenic pathways contributes to both quantitative and qualitative rearrangements of the cellular “coagulome,” which seems to be specific for each tumor type. It may also be possible that in different patients, even if affected by the same disease, thrombosis could be triggered by somewhat different mechanisms and could, hypothetically, be opposed using approaches based on the “coagulome” of the underlying disease. Thus, the coagulant phenotype of cancer cells could be viewed as one of several important effector mechanisms that link genetic progression of the disease and its biological and clinical behavior [8]. This does not necessarily imply a direct proportionality between procoagulant properties and clinical aggressiveness, but suggests that deregulation of hemostatic proteins may influence the tumor microenvironment in pathogenically significant ways. Some authors have also postulated that the coagulation system could play a role at preclinical, or otherwise occult stage of malignancy, and in particular that thrombin might trigger the growth of dormant cancer cells [155]. Dormant cells could be awakened by tissue injury, cardiovascular disease, or other conditions that may activate the clotting system. In this regard, interestingly, a higher frequency of colorectal cancer was recently described in certain forms of thrombophilia, especially in association with the homozygous mutation of the factor V Leiden [156]. Importantly, a recent study has demonstrated that in glioblastoma, exogenous expression of TF disrupts the dormant state of transformed but indolent tumor cells, both by recruitment of inflammatory cells and blood vessels and by facilitating gene mutations and silencing [38].
All these evidences contribute to postulate that procoagulant events are probably not an accompanying phenomenon in cancer, but an effector in tumor growth and progression, and possibly an initiator of malignant transformation. For instance, it is arguable that a better control of hemostatic perturbations may offer new means of therapy, control, and prevention in cancer.
2.5 Conclusions
Cancer-associated thrombosis is a major clinical issue, since thrombotic events increase morbidity and mortality of cancer patients. There is now growing knowledge about the mechanisms of hypercoagulability which predisposes to thrombotic complications, and this does not only translate into a better understanding of the pathogenesis but also offers new potential therapeutic targets of cancer-associated thrombosis. A very important advance in our knowledge has been the discovery that these cellular events can be genetically driven, involving the same genes driving tumorigenesis, as a mechanism of tumor growing and survival. Thus, targeting the mechanisms of coagulation activation could be beneficial for the treatment of the tumor itself. Finally, the biological markers of activation of the clotting system can be a clinical tool, which will help to identify the subgroups of patients at higher risk of VTE and to establish more accurate and targeted anticoagulation strategies to prevent thrombosis in cancer patients.
References
Falanga A, Marchetti M, Vignoli A (2013) Coagulation and cancer: biological and clinical aspects. J Thromb Haemost 11:223–233
Lee AYY, Levine MN (2003) Venous thromboembolism and cancer: risks and outcomes. Circulation 107(Suppl 23)
Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC (2013) Epidemiology of venous thrombosis. Blood 122(10):1712–1723
Blom JW, Vanderschoot JPM, Oostindiër MJ, Osanto S, van der Meer FJM, Rosendaal FR (2006) Incidence of venous thrombosis in a large cohort of 66,329 cancer patients: results of a record linkage study. J Thromb Haemost 4(3):529–535
Blom J, Doggen C, Osanto S, Rosendaal F (2005) Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA 9(293):715–722
Falanga A, Russo L (2012) Epidemiology, risk and outcomes of venous thromboembolism in cancer. Hamostaseologie 32:115–125
Falanga A, Russo L, Milesi V (2014) The coagulopathy of cancer. Curr Opin Hematol 21:423–429
Magnus N, D’Asti E, Meehan B, Garnier D, Rak J (2014) Oncogenes and the coagulation system—forces that modulate dormant and aggressive states in cancer. Thromb Res 133(Suppl 2):S1–S9
Khorana AA, McCrae KR (2014) Risk stratification strategies for cancer-associated thrombosis: an update. Thromb Res 133(Suppl 2):35–38
Falanga A, Marchetti M, Russo L (2015) The mechanisms of cancer-associated thrombosis. Thromb Res 135:S8–S11
Khorana AA, Francis CW, Culakova E, Lyman GH (2005) Risk factors for chemotherapy-associated venous thromboembolism in a prospective observational study. Cancer 104(12):2822–2829
Simanek R, Vormittag R, Ay C, Alguel G, Dunkler D, Schwarzinger I et al (2010) High platelet count associated with venous thromboembolism in cancer patients: results from the Vienna Cancer and Thrombosis Study (CATS). J Thromb Haemost 8(1):114–120
Demers M, Wagner DD (2003) Neutrophil extracellular traps: a new link to cancer-associated thrombosis and potential implications for tumor progression. Oncoimmunology 2(2)
Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW (2008) Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood 111(10):4902–4907
Ay C, Dunkler D, Marosi C, Chiriac A-L, Vormittag R, Simanek R et al (2010) Prediction of venous thromboembolism in cancer patients. Blood 116(24):5377–5382
Moore RA, Adel N, Riedel E, Bhutani M, Feldman DR, Tabbara NE et al (2011) High incidence of thromboembolic events in patients treated with cisplatin-based chemotherapy: A large retrospective analysis. J Clin Oncol 29(25):3466–3473
Menter DG, Tucker SC, Kopetz S, Sood AK, Crissman JD, Honn KV (2014) Platelets and cancer: a casual or causal relationship: Revisited. Cancer Metastasis Rev 33(1):231–269
Gran OV, Braekkan SK, Hansen JB (2018) Prothrombotic genotypes and risk of venous thromboembolism in cancer. Thromb Res
Rosendaal F (1999) Venous thrombosis: a multicausal disease. Lancet 353(9159):1167–1173
Decousus H, Moulin N, Quenet S, Bost V, Rivron-Guillot K, Laporte S et al (2007) Thrombophilia and risk of venous thrombosis in patients with cancer. Thromb Res 120(Suppl 2):51–61
Pabinger I, Ay C, Dunkler D, Thaler J, Reitter EM, Marosi C et al (2015) Factor V Leiden mutation increases the risk for venous thromboembolism in cancer patients—results from the Vienna Cancer And Thrombosis Study (CATS). J Thromb Haemost 13(1):17–22
Gran OV, Smith EN, Brækkan SK, Jensvoll H, Solomon T, Hindberg K et al (2016) Joint effects of cancer and variants in the factor 5 gene on the risk of venous thromboembolism. Haematologica 101(9):1046–1053
Dentali F, Gianni M, Agnelli G, Ageno W (2007) Association between inherited thrombophilic abnormalities and central venous catheter thrombosis in patients with cancer: results of the CAVECCAS study. J Thromb Haemost 6:70–75
Jick H, Slone D, Westerholm B, Inman W, Vessey M, Shapiro S et al (1969) Venous thromboembolic disease and ABO blood type. A cooperative study. Lancet 15(1):539–542
Sode BF, Allin KH, Dahl M, Gyntelberg F, Nordestgaard BG (2013) Risk of venous thromboembolism and myocardial infarction associated with factor V Leiden and prothrombin mutations and blood type. CMAJ 185(5):E229–E237
Wun T, White RH (2009) Epidemiology of cancer-related venous thromboembolism. Best Pr Res Clin Haematol 22(1):9–23
Khorana AA, Francis CW, Culakova E, Kuderer NM, Lyman GH (2007) Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer 110(10):2339–2346
Falanga A, Marchetti M (2012) Anticancer treatment and thrombosis. Thromb Res 129(3):353–359
Lechner D, Weltermann A (2008) Chemotherapy-induced thrombosis: a role for microparticles and tissue factor? Semin Thromb Hemost 34(2):199–203
Kakkar A, Lemoine N, Scully M, Tebbutt S, Williamson R (1995) Tissue factor expression correlates with histological grade in human pancreatic cancer. Br J Surg 82(8):1101–1104
Hamada K, Kuratsu J, Saitoh Y, Takeshima H, Nishi T, Ushio Y (1996) Expression of tissue factor correlates with grade of malignancy in human glioma. Cancer 77:1877–1883
Takano S, Tsuboi K, Tomono Y, Mitsui Y, Nose T (2000) Tissue factor, osteopontin, alphavbeta3 integrin expression in microvasculature of gliomas associated with vascular endothelial growth factor expression. Br J Cancer 82(12):1967–1973
Guan M, Jin J, Su B, Liu W, Lu Y (2002) Tissue factor expression and angiogenesis in human glioma. Clin Biochem 35(4):321–325
Ishimaru K, Hirano H, Yamahata H, Takeshima H, Niiro M, Kuratsu J (2003) The expression of tissue factor correlates with proliferative ability in meningioma. Oncol Rep 10:1133–1137
Fernandes RS, Kirszbeg C, Rumjanek M, Monteiro RQ (2006) On the molecular mechanisms for the highly procoagulant pattern of C6 glioma cells. J Thromb Haemost 4:1546–1552
Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI et al (2005) Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood 105(4):1734–1742
Davila M, Amirkhosravi A, Coll E, Desai H, Robles L, Colon J et al (2008) Tissue factor-bearing microparticles derived from tumor cells: impact on coagulation activation. J Thromb Haemost 6:1517–1524
Magnus N, Garnier D, Meehan B, Mcgraw S, Hoon T, Caron M (2014) Tissue factor expression provokes escape from tumor dormancy and leads to genomic alterations. PNAS 111(9):3544–3549
Falanga A, Russo L, Milesi V, Vignoli A (2017) Mechanisms and risk factors of thrombosis in cancer. Crit Rev Oncol Hematol 118:79–83
Falanga A, Consonni R, Marchetti M, Locatelli G, Garattini E, Gambacorti Passerini C et al (1998) Cancer procoagulant and tissue factor are differently modulated by all-trans-retinoic acid in acute promyelocytic leukemia cells. Blood 92(1):143–151
Falanga A, Iacoviello L, Evangelista V, Belotti D, Consonni R, D’Orazio A et al (1995) Loss of blast cell procoagulant activity and improvement of hemostatic variables in patients with acute promyelocytic leukemia administered all-trans-retinoic acid. Blood 86(3):1072–1081
Falanga A, Toma S, Marchetti M, Palumbo R, Raffo P, Consonni R et al (2002) Effect of all-trans-retinoic acid on the hypercoagulable state of patients with breast cancer. Am J Hematol 70:9–15
Nadir Y, Brenner B (2014) Heparanase multiple effects in cancer. Thromb Res. Elsevier Masson SAS 133:S90–S94
Xu X, Quiros RM, Maxhimer JB, Jiang P, Marcinek R, Ain KB et al (2003) Inverse correlation between heparan sulfate composition and heparanase-1 gene expression in thyroid papillary carcinomas: a potential role in tumor metastasis. Clin Cancer Res 9:5968–5979
El-assal ON, Yamanoi A, Ono T, Kohno H, Nagasue N (2001) The clinicopathological significance of heparanase and basic fibroblast growth factor expressions in hepatocellular carcinoma. Clin Cancer Res 7(May):1299–1305
Folkman J, Klagsbrun M, Sasse J, Wadzinski M, Ingber D, Vlodavsky I (1988) A heparin-binding angiogenic protein-basic fibroblast growth factor is stored within basement membrane. Am J Pathol 130(2):393–400
Vlodavsky I, Folkmantt J, Sullivant R, Fridman R, Ishai-michaeli R, Sasset J et al (1987) Endothelial cell-derived basic fibroblast growth factor: synthesis and deposition into subendothelial extracellular matrix. Proc Nati Acad Sci USA 84(April):2292–2296
Friedmann Y, Vlodavsky I, Aingorn H, Aviv A, Peretz T, Pecker I, et al (2000) Expression of heparanase in normal, dysplastic, and neoplastic human colonic mucosa and stroma. evidence for its role in colonic tumorigenesis. Am J Pathol 157(4):1167–1175
Elkin M, Ilan N, Ishai-Michaeli R, Friedmann Y, Papo O, Pecker I et al (2001) Heparanase as mediator of angiogenesis: mode of action. FASEB J 15(9):1661–1663
Nadir Y, Brenner B (2016) Heparanase procoagulant activity in cancer progression. Thromb Res 140:S44–S48
Ahn YS (2005) Cell-derived microparticles: “Miniature envoys with many faces”. J Thromb Haemost 3:884–887
Horstman LL, Jy W, Jimenez JJ, Bidot C, Ahn YS (2004) New horizons in the analysis of circulating cell-derived microparticles. Keio J Med 53(4):210–230
Nomura S, Suzuki M, Katsura K, Xie GL, Miyazaki Y, Miyake T et al (1995) Platelet-derived microparticles may influence the development of atherosclerosis in diabetes mellitus. Atherosclerosis 116:235–240
Mallat Z, Benamer H (2000) Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood. Circulation 101:841–843
Nieuwland R, Berckmans J, Mcgregor S, Bo AN, Romijn FPHTM, Westendorp RGJ, et al (2000) Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood 95(3):930–936
Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, et al (2005) Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol. Elsevier Masson SAS 45(9):1467–1471
Piccioli A, Falanga A, Baccaglini U, Marchetti M, Prandoni P (2006) Cancer and venous thromboembolism. Semin Thromb Hemost 32(7):694–699
Falanga A, Marchetti M (2009) Venous thromboembolism in the hematologic malignancies. J Clin Oncol 27(29):4848–4857
Kim H, Song K, Park Y, Kang Y, Lee Y, Lee K et al (2003) Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: possible role of a metastasis predictor. Eur J Cancer 39(2):184–191
Del Conde I, Bharwani LD, Dietzen DJ, Pendurthi U, Thiagarajan P, LóPez JA (2007) Microvesicle-associated tissue factor and Trousseau’s syndrome. J Thromb Haemost 5(1):70–74
Tilley RE, Holscher T, Belani R, Nieva J, Mackman J (2008) Tissue factor activity is increased in a combined platelet and microparticle sample from cancer patients. Thromb Res 122(5):604–609
Falanga A, Barbui T, Rickles FR (2008) Hypercoagulability and tissue factor gene upregulation in hematologic malignancies. Semin Thromb Hemost 34(2):204–210
Dvorak HF, Quay SC, Orenstein NS, Dvorak AM, Hahn P, Bitzer AM, et al (1981) Tumor shedding and coagulation. Science 212(4497):923–924
Bastida E, Ordinas A, Escolar G, Jamieson GA (1984) Tissue factor in microvescicles shed from U87MG human glioblastoma cells induces coagulation, platelet aggregation, and thrombogenesis. Blood 64(1):177–184
Yu JL, Rak JW (2004) Shedding of tissue factor (TF)-containing microparticles rather than alternatively spliced TF is the main source of TF activity released from human cancer cells. J Thromb Haemost 2(11):2065–2067
Tesselaar MET, Romijn FPHTM, Van Der Linden IK, Prins FA, Bertina RM, Osanto S (2006) Microparticle-associated tissue factor activity: a link between cancer and thrombosis? J Thromb Haemost 5:520–527
Manly DA, Wang J, Glover SL, Kasthuri R, Liebman HA, Key S et al (2010) Increased microparticle tissue factor activity in cancer patients with venous thromboembolism. Thromb Res 125(6):511–512
Kwaan HC, Magalha E (2010) Role of microparticles in the hemostatic dysfunction in acute promyelocytic leukemia. Semin Thromb Hemost 36(8):917–924
van Aalderen MC, Trappenburg MC, van Schilfgaarde M, Molenaar PJ (2011) Procoagulant myeloblast-derived microparticles in AML patients: changes in numbers and thrombin generation potential during chemotherapy. J Thromb Haemost 9:223–234
Auwerda JJ, Yuana Y, Osanto S, de Maat MP, Sonneveld P, Bertina RM et al (2011) Microparticle-associated tissue factor activity and venous thrombosis in multiple myeloma. Thromb Haemost 105(1):14–20
Trappenburg MC, Van Schilfgaarde M, Marchetti M, Spronk HM, Cate H, Leyte A et al (2009) Elevated procoagulant microparticles expressing endothelial and platelet markers in essential thrombocythemia. Haematologica 94(7):911–998
Martinez MC, Andriantsitohaina R (2011) Microparticles in angiogenesis: therapeutic potential. Circ Res 109:110–119
Kim HK, Song KS, Chung J, Lee KR, Lee S (2004) Platelet microparticles induce angiogenesis in vitro. Br J Haematol 124:376–384
Janowska-wieczorek A, Wysoczynski M, Kijowski J, Marquez-curtis L, Machalinski B, Ratajczak J et al (2004) Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int J Cancer 113:752–760
Falanga A, Marchetti M, Vignoli A, Balducci D (2003) Clotting mechanisms and cancer: implications in thrombus formation and tumor progression. Clin Adv Hematol Oncol 1(11):673–678
Ruf W, Yokota N, Schaffner F (2010) Tissue factor in cancer progression and angiogenesis. Thromb Res 2018(125):S36–S38
Milsom CC, Yu JL, Mackman N, Micallef J, Anderson GM, Guha A et al (2008) Tissue factor regulation by epidermal growth factor receptor and epithelial-to-mesenchymal transitions: effect on tumor initiation and angiogenesis. Cancer Res 68(24):10068–10077
Zwicker JI (2010) Predictive value of tissue factor bearing microparticles in cancer associated thrombosis. Thromb Res. Elsevier Ltd 125:S89–S91
Erdmann D, Heim J (1995) Orphan nuclear receptor HNF-4 binds to the human coagulation factor VII promoter. J Biol Chem 270(39):22988–22996
Tsao BYBP, Fair DS, Curtiss LK, Edgington TS (1984) Monocytes can be induced by lipopolysaccharide-triggered T lymphocytes to express functional factor VII/VIIa protease activity. J Exp Med 159:1042–1057
Chapman HA Jr, Allen CL, Stone OL, Fair DS. Human alveolar macrophages synthesize factor VII in vitro. Possible role in interstitial lung disease. J Clin Invest 75:2030–2037
Neaud V, Hisaka T, Monvoisin A, Bedin C, Balabaud C, Foster DC et al (2000) Paradoxical pro-invasive effect of the serine proteinase inhibitor tissue factor pathway inhibitor-2 on human hepatocellular carcinoma cells. J Biol Chem 275(45):35565–35569
Fischer EG, Riewald M, Huang HY, Miyagi Y, Kubota Y, Mueller BM et al (1999) Tumor cell adhesion and migration supported by interaction of a receptor-protease complex with its inhibitor. J Clin Invest 104(9):1213–1221
Zacharski LR, Memoli VA, Ornstein DL, Rousseau SM, Kisiel W, Kudryk BJ (1993) Tumor cell procoagulant and urokinase expression in carcinoma of the ovary. J Natl Cancer Inst 85(15):1225–1230
Wojtukiewicz MZ, Zacharski LR, Ruciñska M, Zimnoch L, Jaromin J, Rózañska-Kudelska M, et al (1999) Expression of tissue factor and tissue factor pathway inhibitor in situ in laryngeal carcinoma. Thromb Haemost 82:1659–1662
Koizume S, Jin M-S, Miyagi E, Hirahara F, Nakamura Y, Piao J-H et al (2006) Activation of cancer cell migration and invasion by ectopic synthesis of coagulation factor VII. Cancer Res 66(19):9453–9460
Tang J, Fan Q, Wu W, Jia Z, Li H, Yang Y et al (2010) Extrahepatic synthesis of coagulation factor VII by colorectal cancer cells promotes tumor invasion and metastasis. Chin Med J 123(24):3559–3565
Yokota N, Koizume S, Miyagi E, Hirahara F, Nakamura Y, Kikuchi K et al (2009) Self-production of tissue factor-coagulation factor VII complex by ovarian cancer cells. Br J Cancer 101(12):2023–2029
Kyrle PA, Minar E, Hirschl M, Bialonczyk C, Stain M, Schneider B et al (2000) High plasma levels of factor VIII and the Risk of recurrent venous thromboembolism. N Engl J Med 343(7):457–462
Legnani C, Cosmi B, Cini M, Frascaro M, Giuliana G, Palareti G (2004) High plasma levels of factor VIII and risk of recurrence of venous thromboembolism. Br J Haematol 124(4):504–510
Auwerda JJA, Sonneveld P, De Maat MPM, Leebeek FWG (2007) Prothrombotic coagulation abnormalities in patients with newly diagnosed multiple myeloma. Haematologica 92(2):279–280
Yigit E, Gönüllü G, Yücel I, Turgut M, Erdem D, Çakar B (2008) Relation between hemostatic parameters and prognostic/predictive factors in breast cancer. Eur J Intern Med 19(8):602–607
Battistelli S, Stefanoni M, Lorenzi B, Dell’Avanzato R, Varrone F, Pascucci A et al (2008) Coagulation factor levels in non-metastatic colorectal cancer patients. Int J Biol Markers 23(1):36–41
Dogan M, Demirkazik A, Konuk N, Yalcin, Buyukcelik A, Utkan G, et al (2006) The effect of venous thromboembolism on survival of cancer patients and its relationship with serum level of factor VIII and vascular endothelial growth factor: a prospective matched-paired study. Int J Biol Markers 21(4):206–210
Vormittag R, Simanek R, Ay C, Dunkler D, Quehenberger P, Marosi C et al (2009) High factor VIII levels independently predict venous thromboembolism in cancer patients: The cancer and thrombosis study. Arterioscler Thromb Vasc Biol 29(12):2176–2181
Andrén-Sandberg A, Lecander I, Martinsson G, Astedt B (1992) Peaks in plasma plasminogen activator inhibitor-1 concentration may explain thrombotic events in cases of pancreatic carcinoma. Cancer 69(12):2884–2887
Sciacca FL, Ciusani E, Silvani A, Corsini E, Frigerio S, Pogliani S et al (2004) Genetic and plasma markers of venous thromboembolism in patients with high grade glioma. Clin Cancer Res 10(4):1312–1317
Chen N, Ren M, Li R, Deng X, Li Y, Yan K et al (2015) Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol Cancer 14(1):1–7
Liu Y, Wang Z, Jiang M, Dai L, Zhang W, Wu D et al (2011) The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leuk Res 35(7):879–884
Connolly GC, Phipps RP, Francis CW (2014) Platelets and cancer-associated thrombosis. Semin Oncol. Elsevier 41(3):302–310
Falanga A, Russo L, Verzeroli C (2013) Mechanisms of thrombosis in cancer. Thromb Res. Elsevier Ltd 131:S59–S62
Lee EC, Cameron SJ (2017) Cancer and thrombotic risk: the platelet paradigm. Front Cardiovasc Med. 4(November):1–6
Läubli H, Borsig L (2010) Selectins promote tumor metastasis. Semin Cancer Biol 20(3):169–177
Chen M, Geng JG (2006) P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arch Immunol Ther Exp (Warsz) 54(2):75–84
Stegner D, Dütting S, Nieswandt B (2014) Mechanistic explanation for platelet contribution to cancer metastasis. Thromb Res 133(Suppl 2):149–157
Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW (2005) Platelets and fibrin (ogen) increase metastatic potential by impeding natural killer cell—mediated elimination of tumor cells. Blood J 105(1):178–185
Raica M, Cimpean AM, Ribatti D (2008) The role of podoplanin in tumor progression and metastasis. Anticancer Res 28(5B):2997–3006
Riedl J, Preusser M, Nazari PMS, Posch F, Panzer S, Marosi C et al (2017) Podoplanin expression in primary brain tumors induces platelet aggregation and increases risk of venous thromboembolism. Blood 129(13):1831–1839
Chang Y-W, Hsieh P, Chang Y, Lu M, Huang T-F, Chong K-Y et al (2015) Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget 6(40):42733–42748
Shao B, Wahrenbrock MG, Yao L, David T, Coughlin SR, Xia L et al (2011) Carcinoma mucins trigger reciprocal activation of platelets and neutrophils in a murine model of Trousseau syndrome. Blood 118(15):4015–4023
Brill A, Fuchs T a, Chauhan AK, Yang JJ, Meyer SF De, Ko M, et al (2011) von Willebrand factor—mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 117(4):1400–1407
Vanschoonbeek K, Feijge MAH, Van Kampen RJW, Kenis H, Hemker HC, Giesen PLA et al (2004) Initiating and potentiating role of platelets in tissue factor-induced thrombin generation in the presence of plasma: Subject-dependent variation in thrombogram characteristics. J Thromb Haemost 2(3):476–484
Falanga A, Marchetti M, Evangelista V, Vignoli A, Licini M, Balicco M et al (2000) Polymorphonuclear leukocyte activation and hemostasis in patients with essential thrombocythemia and polycythemia vera. Blood 96(13):4261–4266
Barbui T, Finazzi G, Falanga A (2013) Myeloproliferative neoplasms and thrombosis. Blood 122(13):2176–2184
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al (2012) Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA 109(32):13076–13081
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD et al (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA 107(36):15880–15885
Clark S, Ma A, Tavener S, McDonald B, Goodarzi Z, Kelly M et al (2007) Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 13(4):463–469
Snyder KM, Kessler CM (2008) The pivotal role of thrombin in cancer biology and tumorigenesis. Semin Thromb Hemost 34(8):734–741
Edwards R, Rickles F (1984) Macrophage procoagulants. Prog Hemost Thromb 7:183–209
Semeraro N (1988) Different expression of procoagulant activity in macrophages associated with experimental and human tumors. Haemostasis 18:47–54
Semeraro N, Montemurro P, Conese M, Giordano D, Stella M, Restaino A et al (1990) Procoagulant activity of mononuclear phagocytes from different anatomical sites in patients with gynecological malignancies. Int J Cancer 45(2):251–254
Pabinger I, Posch F (2014) Flamethrowers: blood cells and cancer thrombosis risk. Hematol Am Soc Hematol Educ Program 2014 2014(1):410–417
Granger V, Faille D, Marani V, Noël B, Gallais Y, Szely N et al (2017) Human blood monocytes are able to form extracellular traps. J Leukoc Biol 102(3):775–781
Go S, Kim R, Song H, Kang M, Lee U, Choi H et al (2015) Prognostic significance of the absolute monocyte counts in lung cancer patients with venous thromboembolism. Tumour Biol 36(10):7631–7639
Tas F, Duranyildiz D, Argon A, Og H (2005) Serum levels of leptin and proinflammatory cytokines in advanced-stage non-small cell lung cancer. Med Oncol 22(4):353–354
Zwicker JI, Furie BC, Furie B (2007) Cancer-associated thrombosis. Crit Rev Oncol Hematol 62:126–136
Falanga A, Marchetti M, Giovanelli S, Barbui T (1996) All-trans-retinoic acid counteracts endothelial cell procoagulant activity induced by a human promyelocytic leukemia-derived cell line (NB4). Blood 87(2):613–618
Van de Wouwer M, Collen D, Conway EM (2004) Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol 24(8):1374–1383
Lindahl AK, Boffa MC, Abildgaard U (1993) Increased plasma thrombomodulin in cancer patients. Thromb Haemost 69(2):112–114
Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U (2005) Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res 96(12):1233–1239
Friedenberg WR, Roberts RC, Alonso-Escolano D (1992) Relationship of thrombohemorrhagic complications to endothelial cell function in patients with chronic myeloproliferative. Am J Hematol 40(4):283–289
Karakantza M, Giannakoulas NC, Zikos P, Sakellaropoulos G, Kouraklis A, Aktypi A et al (2004) Markers of endothelial and in vivo platelet activation in patients with essential thrombocythemia and polycythemia vera. Int J Hematol 79(3):253–259
Gogali A, Charalabopoulos K, Zampira I, Konstantinidis AK, Tachmazoglou F, Daskalopoulos G et al (2010) Soluble adhesion molecules E-cadherin, intercellular adhesion molecule-1, and E-selectin as lung cancer biomarkers. Chest 138(5):1173–1179
Silva HC, Garcao F, Coutinho EG, De Oliveira CF, Regateiro FJ (2006) Soluble VCAM-1 and E-selectin in breast cancer: relationship with staging and with the detection of circulating cancer cells. Neoplasma. 53(6):538–543
Cella G, Marchetti M, Vianello F, Panova-Noeva M, Vignoli A, Russo L et al (2010) Nitric oxide derivatives and soluble plasma selectins in patients with myeloproliferative neoplasms. Thromb Haemost 104(1):151–156
Goon PKY, Lip GYH, Boos CJ, Stonelake PS, Blann AD (2006) Circulating endothelial cells, endothelial progenitor cells, and endothelial microparticles in cancer. Neoplasia 8(2):79–88
Mancuso P, Burlini A, Pruneri G, Goldhirsch A, Martinelli G, Bertolini F (2001) Resting and activated endothelial cells are increased in the peripheral blood of cancer patients. Blood 97(11):3658–3662
Fleitas T, Martínez-Sales V, Vila V, Reganon E, Mesado D, Martín M et al (2012) Circulating endothelial cells and microparticles as prognostic markers in advanced non-small cell lung cancer. PLoS ONE 7(10):1–6
Rak J, Klement G (2000) Impact of oncogenes and tumor suppressor genes on deregulation of hemostasis and angiogenesis in cancer. Cancer Metastasis Rev 19(1–2):93–96
Magnus N, Gerges N, Jabado N, Rak J (2013) Coagulation-related gene expression profile in glioblastoma is defined by molecular disease subtype. J Thromb Haemost 11(6):1197–1200
Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T et al (1995) Advances in brief mutant ras oncogenes upregulate VEGFIVPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res 55:4575–4580
Sparmann A, Bar-Sagi D (2004) Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 6(5):447–458
Phan VT, Wu X, Cheng JH, Sheng RX, Chung AS, Zhuang G et al (2013) Oncogenic RAS pathway activation promotes resistance to anti-VEGF therapy through G-CSF-induced neutrophil recruitment. Proc Natl Acad Sci USA 110(15):6079–6084
Rickles FR (2009) Cancer and thrombosis in women—molecular mechanisms. Thromb Res 123(Suppl 2):16–20
Koizume S, Yokota N, Miyagi E, Hirahara F, Nakamura Y, Sakuma Y et al (2009) Hepatocyte nuclear factor-4-independent synthesis of coagulation factor VII in breast cancer cells and its inhibition by targeting selective histone acetyltransferases. Mol Cancer Res 7(12):1928–1936
Magnus N, Garnier D, Rak J (2010) Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells sure to FVIIa and PAR1- or PAR2-activat-. Blood 116(5):815–819
Garnier D, Magnus N, Lee TH, Bentley V, Meehan B, Milsom C et al (2012) Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J Biol Chem 287(52):43565–43572
Provençal M, Berger-Thibaul N, Labbé D, Veitch R, Boivin D, Rivard G et al (2010) Tissue factor mediates the HGF/Met-induced anti-apoptotic pathway in DAOY medulloblastoma cells. J Neurooncol 97(3):365–372
Rong Y, Belozerov VE, Tucker-Burden C, Chen G, Durden DL, Olson JJ et al (2009) Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/activator protein-1 transcriptional activity. Cancer Res 69(6):2540–2549
Zhang L, Sullivan PS, Goodman JC, Gunaratne PH, Marchetti D (2011) MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res 71(3):645–654
Chu HW, Cheng CW, Chou WC, Hu LY, Wang HW, Hsiung CN et al (2014) A novel estrogen receptor-microRNA 190a-PAR-1-pathway regulates breast cancer progression, a finding initially suggested by genome-wide analysis of loci associated with lymph-node metastasis. Hum Mol Genet 23(2):355–367
Botla SK, Savant S, Jandaghi P, Bauer AS, Mucke O, Moskalev EA et al (2016) Early epigenetic downregulation of microRNA-192 expression promotes pancreatic cancer progression. Cancer Res 76(14):4149–4159
Wu X-L (2013) MicroRNA-143 suppresses gastric cancer cell growth and induces apoptosis by targeting COX-2. World J Gastroenterol 19(43):7758
D’Asti E, Rak J (2016) Biological basis of personalized anticoagulation in cancer: oncogene and oncomir networks as putative regulators of coagulopathy. Thromb Res 140(Suppl 1):37–43
Karpatkin S (2004) Does hypercoagulability awaken dormant tumor cells in the host? J Thromb Haemost 2(12):2103–2106
Vossen CY, Hoffmeister M, Chang-Claude JC, Rosendaal FR, Brenner H (2011) Clotting factor gene polymorphisms and colorectal cancer risk. J Clin Oncol 29(13):1722–1727
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Falanga, A., Schieppati, F., Russo, L. (2019). Pathophysiology 1. Mechanisms of Thrombosis in Cancer Patients. In: Soff, G. (eds) Thrombosis and Hemostasis in Cancer. Cancer Treatment and Research, vol 179. Springer, Cham. https://doi.org/10.1007/978-3-030-20315-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-20315-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-20314-6
Online ISBN: 978-3-030-20315-3
eBook Packages: MedicineMedicine (R0)