
Abstract
In recent years, the RNA molecule became one of the most promising targets for therapeutic intervention. Currently, a large number of RNA-based therapeutics are being investigated both at the basic research level and in late-stage clinical trials. Some of them are even already approved for treatment. RNA-based approaches can act at pre-mRNA level (by splicing modulation/correction using antisense oligonucleotides or U1snRNA vectors), at mRNA level (inhibiting gene expression by siRNAs and antisense oligonucleotides) or at DNA level (by editing mutated sequences through the use of CRISPR/Cas). Other RNA approaches include the delivery of in vitro transcribed (IVT) mRNA or the use of oligonucleotides aptamers. Here we review these approaches and their translation into clinics trying to give a brief overview also on the difficulties to its application as well as the research that is being done to overcome them.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Antisense oligonucleotides
- Aptamers
- CRISPR-Cas gene editing
- Modified mRNA replacement therapy
- siRNA-based drugs
- U1snRNA vectors
7.1 Introduction
The RNA molecule has traditionally been viewed as an intermediate between DNA and protein. Recently though, this reductive view has been abandoned as more classes and functions of RNA have been discovered as well as therapeutic applications involving this molecule are being developed. RNA therapeutics can either mimic or antagonize the endogenous RNA functions and have several advantages. They can act even on targets that were previously “undraggable” and, most importantly, they are easy to design, cost effective, stable and easy to combine with other drugs presenting also low immunogenicity. Despite these advantages, the use of RNAs as drugs requires the overcoming of two major obstacles: the poor pharmacological properties of RNA, which is rapidly degraded by RNases and the difficulties in its delivery to the target organs and tissues. In this chapter we present the major RNA-based therapeutics currently under research, discussing the challenges to their translation into the clinic and the recent advances in delivery strategies. RNA tools such as ribozymes, riboswitches and SINE-UP strategy are no less important but will not be discussed in this chapter.
7.2 Antisense Oligonucleotides
7.2.1 Brief Overview
Antisense oligonucleotides (AONs) are short synthetic oligonucleotides that bind to RNA through standard Watson-Crick base pairing and can modulate the function of their target RNA [1, 2]. AONs can function in various ways (Fig. 7.1). For example, AONs can mediate targeted gene knockdown through the recruitment of endogenous RNAse H to degrade mRNA at sites of DNA:RNA hybridization caused by AON binding (Fig. 7.1a). They can also be designed to bind to translation initiation sites on mRNAs in cytosol to block translation (Fig. 7.1b). Another approach uses single-stranded AONs to modulate miRNAs expression; these AONs directly bind target miRNAs to inhibit their function (anti-miRs), and thus depress their target gene (Fig. 7.1c). Moreover, AONs can also be used to modulate pre-mRNA splicing in the target gene bypassing the disease-causing mutation (Fig. 7.1d) [3, 4]. These AONs are designated Splice Switching Oligonucleotides (SSOs) and are single stranded 15–25 nucleotides long, which direct pre-mRNA splicing to a new pathway by binding sequence elements and sterically blocking access to the transcript by the spliceosome and other splicing factors [1, 5,6,7].

Antisense mechanisms of RNA-based drugs. Antisense oligonucleotides (AONs) impact in gene expression through four different mechanisms: (a) RNase H-mediated mRNA degradation ; (b) steric block of ribosome binding; (c) complementary binding to target microRNAs (miRNAs) in order to inhibit their function (antagomirs); and (d) splicing modulation
The modification of gene expression, using a synthetic single stranded DNA, resulting in inhibition of mRNA translation was demonstrated for the first time by Paterson and colleagues in 1977 in a cell-free system [8]. Almost a year later, Zamecnik and Stephenson showed that in chicken fibroblast tissue culture containing Rous Sarcoma virus, the addition of a synthetic 13-mer oligonucleotide complementary to the 3′ end of the virus, could inhibit its replication and the subsequent transformation of fibroblasts into sarcoma cells [9]. Since then, remarkable progress has been made in oligonucleotide drug development and currently, antisense technology is a powerful tool that can be used for target validation and to correct or alter RNA expression for therapeutic benefit [1, 10,11,12].
Initially, AONs were just synthetic unmodified DNA or RNA molecules, which despite delivering some promising results, would prove to be quite ineffective in biological systems due to their susceptibility to degradation by nucleases, poor affinity for their target mRNA, multiple off-target effects, inability to cross the cell membrane given their negative charge, and weak binding to plasma proteins, leading to rapid clearance by the kidney [1, 3, 13]. Therefore, a wide variety of chemically modified analogues of nucleotides have been developed since then. These chemical modifications were made in the oligonucleotides, generating three categories of AONs, commonly known as generations, with different chemical and pharmacological properties.
7.2.2 Antisense Oligonucleotides Chemistry
Several characteristics need to be fulfilled for the clinical application of antisense oligonucleotides. First, the sequence of the antisense oligonucleotide must be specific enough to avoid off-targets. Other important aspects to be taken into account are the chemistry of the AON and its resistance to degradation by nucleases in order not only to maintain the integrity of the molecule but also to ensure that it is present in an amount, which is sufficient for a true efficacy. In addition, ideal AON should have good pharmacokinetic (PK) and pharmacodynamic (PD) properties and, above all, should not be toxic. Finally, and a very fundamental thing to check, is whether the designed AONs are possible to deliver to target tissues or organs. To try to cope with these desired AON properties, several chemical modifications have been made to the backbone, ribose sugar moiety or nucleobase components, which have a profound effect on the enhanced stability, binding strength and specificity to the target RNA sequence [13] (Fig. 7.2).

Chemical modifications of antisense oligonucleotides.First generation antisense oligonucleotides (AONs) are characterized by phosphorothioate (PS) backbone; second generation AONs contain a methyl or methoxyethyl group at the 2′ position of the ribose; finally, third generation AONs are characterized by modifications of the furanose ring structure
The first generation of AONs is characterized by alterations in the backbone, the most common being the phosphorothioate (PS) backbone, accomplished by the replacement of one of the non-bridging oxygen atoms by a sulphur atom. AONs bearing PS linkages are compatible with recruitment of RNase H, which cleaves the target of AONs. This modification allows for an improved nuclease resistance, as well as strong binding to plasma proteins, reducing renal clearance, but still presents poor binding affinity, low specificity and poor cellular uptake [1, 7, 10, 13,14,15]. Despite these disadvantages, PS oligonucleotides are still the most commonly used AONs and were the first antisense-based drug approved for clinical use in 1998 with fomivirsen (Vitravene®) used for repression of cytomegalovirus mRNA translation [16]. It gained U.S. FDA (U.S. Food and Drug Administration) approval for intraocular treatment of cytomegalovirus retinitis in immunosuppressed patients in 1998 [16] and was discontinued later due to commercial considerations.
In order to surpass the downsides of the first generation oligonucleotides, a second generation was developed through modifications at the 2′ position of the ribose. The most widely studied second generation AONs are 2′-O-methyl (2′-OMe) and 2′-O-Methoxyethyl (2′-MOE), which present higher nuclease resistance and higher affinity for the target RNA, while also reducing non-specific protein binding and toxicity [7, 10, 13, 15]. These second generation AONs, however, do not support RNase H-mediated cleavage of the target mRNA, which impairs their usage for purposes of gene downregulation [1, 14, 17]. This limitation has been minimized with the development of “gapmer” structures where 2′ sugar-modified residues are present on either side of a central “gap” region comprising 8–10 PS-modified nucleotides. The external sugar modified residues thus increase affinity and nuclease resistance, while the internal “gap” region allows RNase H-mediated cleavage of the target RNA [1, 6, 18, 19].
Finally, the third generation of oligonucleotides is characterized by modifications of the furanose ring of the nucleotide, with the most common being peptide nucleic acids (PNAs), locked nucleic acids (LNAs) and phosphoroamidate morpholino oligomers (PMOs) [12, 14]. These modifications further increase nuclease and protease resistance, target affinity, specificity and in vivo stability of antisense drugs, while reduce non-specific interactions with proteins [1, 13, 14, 20, 21]. Nevertheless, PNAs and PMOs present poor cellular uptake, low water solubility and are rapidly cleared from the blood due to their uncharged nature [1, 10, 20, 22] whereas LNAs appear to generate higher toxicity than other chemical modifications, questioning their safety for therapeutic applications [1, 20].
7.2.3 Recent Successful Applications of Antisense Oligonucleotides
The therapeutic application of AONs is very promising. A huge amount of preclinical data has been produced in recent years and many studies have even undergone clinical trials (Table 7.1). Of those, four drugs with different AON chemistries and treatment targets reached, or almost reached clinical practice [12] (Fig. 7.3). One of them is Mipomersen (Kynamro®; Genzyme) that was approved by the U.S. FDA in 2013 for the treatment of familial hypercholesterolemia (FH). Mipomersen is a gapmer of 20 nucleotides and has a sequence complementary to a segment of the Apo b-100 mRNA. Its binding creates a DNA:RNA hybrid that is substrate for the enzyme RNase H thus inducing the cleavage of the human Apo b-100 mRNA. The drug has a PS backbone, with 2′-MOE-modified ends, which when compared with earlier antisense technologies, provides greater biological stability and higher affinity to the target mRNA [12, 67]. When administered subcutaneously at a dose of 200 mg per week, it was shown to reduce ApoB-100 production and low-density lipoprotein cholesterol (LDL-C) in a dose-dependent fashion [68]. In general, the results achieved with Mipomersen point to its efficacy, safety, and tolerability, demonstrating its suitability for use in the target patient population, and providing a tangible tool for use in the management of FH and severe hypercholesterolemia [11, 12, 68]. However, mild-to-moderate injection site reactions, flu-like symptoms and hepatic effects (despite transient and generally reversible) limited its utilization and therefore its commercial success [11].

Antisense drugs for clinical practice.Currently, four drugs with different AON chemistries and mechanisms of action have either obtained U.S. FDA approval (Mipomersen, Nusinersen and Eteplirsen) and reached clinical practice, or are seeking accelerated approval soon, with significant pre-clinical data supporting their rapid translation into clinic (Miraversen). (a) Mipomersen (Kynamro®; Genzyme), a 2′-MOE-modified AON approved by the U.S. FDA in 2013 for the treatment of familial hypercholesterolemia (FH); (b) Miravirsen (SPC3649, Santaris Pharma), an antagomir to miR-122, seeking approval for hepatitis C treatment; (c) Nusinersen (Spinraza®, Biogen), a fully modified 2′-MOE AON, approved by U.S. FDA in 2016 for the treatment of spinal muscular atrophy (SMA) and, (d) Eterplirsen® (EXONDYS 51TM, Sarepta), a PMO approved by the U.S. FDA in 2016 for the use in Duchenne muscular dystrophy (lightning symbol means the existence of a pathogenic alteration)
AONs complementary to mature miRNAs (antagomirs) are also being developed to counteract miRNAs implicated in disease pathogenesis. An example is Miravirsen (SPC3649, Santaris Pharma), an antagomir to miR-122, a liver-specific microRNA that the hepatitis C virus (HCV) requires for replication. Miravirsen is designed to recognize and sequester miR-122, making it unavailable to HCV. As a result, viral replication is inhibited, and the level of HCV infection is reduced [12]. Positive results were observed in a phase II study. The use of Miravirsen in patients with chronic HCV genotype 1 infection showed prolonged dose-dependent reductions in HCV RNA levels without evidence of viral resistance [50]. The updated results revealed no long-term safety issues among 27 Miravirsen-treated patients [51, 69]. Moreover, there was a prolonged decrease in miR-122 plasma levels in patients dosed with Miravirsen but the plasma levels of other miRNAs were not significantly affected by antagonizing miR-122 [69, 70].
The above examples use AONs to alter gene expression, either directly or indirectly, to change disease progression. Another precise method to alter gene expression is to manipulate pre-mRNA splicing using SSOs. This is the case of Nusinersen (Spinraza®, Biogen), a fully modified 2′-MOE AON, approved by U.S. FDA in 2016 for the treatment of spinal muscular atrophy (SMA). SMA is an autosomal recessive neuromuscular disease caused by progressive loss of alfa-motor neurons in the anterior horn of the spinal cord [12, 71]. The most severe form, infant onset or type 1, is the most common one, representing 50% of all SMA cases. Type 2 is less severe, but also very debilitating. These infants never walk, and as they grow the disease progresses and patients begin to lose the capacity of lift even their arms. In humans, a parolog gene of SMA exists, the SMN2 gene that differs of the SMN1 by 5–11 nucleotides. However, in the majority of the SMN2 transcripts the exon 7 is lacking, resulting in a truncated protein, which is rapidly degraded [72]. Nusinersen induces the inclusion of exon 7 in the SMN2 mRNA by targeting and blocking an intron 7 internal splice site. This action increases SMN protein production, thus improving its function [73]. Intrathecal injection of Nusinersen (every 4 months) allows therapeutic delivery directly into the cerebrospinal fluid (CSF) bathing the spinal cord, the site of motor neuron degeneration and, substantially prolonging survival of type 1 infants, while also resulting in improvements in all measures evaluated [32]. Similar benefit was demonstrated in patients with later onset type 2 SMA [33]. More remarkable, treatment of type 1 pre-symptomatic infants with Nusinersen has been demonstrated to result, in many cases, in achievement of motor milestones at the age expected for healthy infants. Moreover, 92% of the infants treated prior to the development of symptoms were able to sit without support, a milestone never achieved by a type 1 SMA infant before Nusinersen treatment was introduced and 50% were able to walk without support [74].
Another SSO, already in the market is Eterplirsen® (EXONDYS 51™, Sarepta) that was approved by the U.S. FDA in 2016 for use in Duchenne muscular dystrophy (DMD) patients, a severe, childhood-onset disease that results mostly from deletions within the dystrophin gene. DMD is a progressive, neuromuscular disease, occurring mainly in males (1 in 3500–5000 males born worldwide) [75, 76]. It is caused by an absence of the protein dystrophy, a membrane-associated protein that forms a network with sarcolemmal glycoproteins by linking the cytoskeleton actin in muscle fibers within the first few extracellular matrix [77], which results in altered myocyte integrity, muscle wasting and relentlessly progressive weakness. Becker muscular dystrophy (BMD) is a milder disease caused by dystrophin truncations (due to “in frame” deletions) rather than its absence. A viable strategy for generating truncated, but functional, dystrophin protein involves the skipping of exons to correct DMD-linked mutations (which includes 83% of mutations in DMD) [78]. This can reduce the severity of the disease and produce a milder phenotype, similar to that of BMD. Eteplirsen, the first PMO drug ever approved, binds to the exon/intron splice site at the beginning of exon 51, resulting in its skipping, giving origin to an in frame transcript, which prevents the unwanted degradation of the mutant transcripts by the nonsense-mediated mRNA decay (NMD) pathway, and allowing the production of an internally deleted but functional dystrophin protein [79]. Eterplirsen is applicable for approximately 14% of patients with DMD mutations. It is administered via intravenous infusion and was found to be well tolerated, with no adverse effects, in several clinical trials [79]. In addition, over 3 years of follow-up, Eterplirsen-treated patients showed a slower rate of decline in ambulation assessed by the 6-min walk test compared to untreated matched historical controls from two DMD natural history cohorts: the Leuven Neuromuscular Reference Center (LNMRC) and the Italian Telethon registry [80]. Previously, the ability of Eteplirsen to induce expression of dystrophin had been demonstrated by an observed increase of dystrophin-positive fibers in skeletal muscle of DMD patients [35]. Recently, Kinane and coworkers compared the pulmonary function data from DMD patients, who received Eteplirsen in studies 201/202 (included 12 patients treated with Eteplirsen over 5 years) with the natural history data published. This study verified that the deterioration of respiratory muscle function with Eteplirsen treatment as measured by forced vital capacity was half of that seen in natural history. Maximum expiratory pressure and maximum inspiratory pressure also declined more slowly in Eteplirsen treated patients compared to natural history, thus demonstrating its potential to preserve respiratory function in patients with DMD [81].
7.2.4 Antisense Oligonucleotides Delivery
One of the major issues for the use of AONs for therapeutic purposes is the efficient delivery to their target site. AONs need to reach the target tissue and, once there, they must reach the appropriate intracellular compartment [2, 7]. Parenteral injection, such as intravenous infusion or subcutaneous injection is the main method at the moment of delivery of PS modified single-stranded AONs formulated in a simple saline solution [15, 21]. However, even though AON activity has been observed in many tissues such as lung, stomach, bladder, and heart, AONs predominantly accumulate in liver, kidney, bone marrow, adipocytes, and lymph nodes [21]. Therefore, delivery problems must be considered in terms of sets of barriers to movement of AON within the body. Tissue barriers to delivery include the vascular endothelial barrier, first-pass renal excretion (which strongly affects PK and bio-distribution of AONs), and the blood brain barrier (BBB) that AONs cannot cross due to their size and charge, limiting their access to the central nervous system (CNS), in the case of CNS diseases. The one exception to this is intrathecal injection of single-stranded AONs with specific chemical modifications into the CSF, which allows AONs into the CNS [21].
Two main strategies are being developed to improve AON delivery: viral and non-viral delivery. Despite viral vectors are efficient systems for the delivery of genetic material and for the capability to infect a large number of cell types, they also showed some constraints, such as immunogenicity, tumorogenicity risks, limited loading capacity and scaling-up problems [7]. However, adeno-associated viruses (AAVs), which are non-integrative vectors and therefore present a low risk of genomic insertions, have been used in in vitro cells and in animal models to efficiently deliver AONs sequences embedded into modified snRNA systems (modified U1 snRNAs and modified U7 snRNAs). Indeed, promising therapeutic results were obtained with this strategy to induce exon-skipping in diseases like Leber Congenital Amaurosis [82] and DMD [83,84,85,86,87] and exon-inclusion in SMA [88, 89] (for a more extended review on this subject see [90]). Non-viral delivery also represents a good alternative and the conjugation of free AONs with non-viral delivery carriers can be achieved by different strategies. One option is to conjugate AONs (or their carrier) to a ligand that interacts selectively with a cell surface receptor. Ideally, one such receptor should be expressed only in the tissue to be targeted. Additionally, it should also be abundantly expressed, rapidly and extensively internalized, and have a high affinity to its ligands, so that they become readily available. Some receptors used to target AONs include integrins, G protein-coupled receptors (GPCRs), human receptor tyrosine kinase (RTK), scavenger receptors, asialoglycoprotein receptor (ASGR), Toll-like receptors (TLRs) and folate receptor [91,92,93,94,95,96,97], as reviewed in [98]. However, receptors fulfilling all the above-referred criteria are not available for the majority of tissues and, once the AONs reach the cell surface of the target cell, they must be internalized by endocytosis and packed into a vesicle, termed endosome. Then, the endosomes fuse with lysosomes, organelles rich in hydrolases, which ultimately degrade a high portion of the internalized AONs. In fact, one strategy to improve delivery of AONs is the use of short cell-penetrating peptides (CPPs), sequences of short cationic and/or amphipathic peptides (fewer than 30 amino acids) that translocate small drugs/cargo across cell membranes. CPPs are attached to their cargo through covalent linkages or through the formation of noncovalent nanoparticle complexes [99] that can promote uptake of macromoleclues via endocytosis. CPPs covalently conjugated to AONs were already used for therapeutic purposes in DMD [100,101,102,103,104,105] Myotonic Dystrophy type I [106] and SMA [107, 108]. However, CPPs have a limited endosomal escape and overcoming the rate-limiting step of endosomal escape into the cytoplasm remains a major challenge to their successful use. Several studies tried to overcome this and some relevant results have been obtained. For instance, specific synthetic endosomal escape domains (EEDs) significantly enhanced cytoplasmic delivery in the absence of cytotoxicity [109] and a CPP-adaptor system capable of efficient intracellular delivery was also recently developed [110]. Another possibility is to incorporate AONs into nanoparticles (NPs) that based on their size and materials, will determine the AON biodistribution and interaction. In fact, the progress of nanotechnology has provided several nanosystems with the aim to increase the drug targeting efficacy. The most common types used for drug delivery are solid lipid nanoparticles (SLNs), polymer nanoparticles, lipid-based nanoparticles (LNPs) and carbon-based nanomaterials [110]. For example, cationic core-shell NPs named T1 and ZM2 (a type of polymer nanoparticles) were used to conjugate AONs for exon skipping application in preclinical studies in DMD mice [110, 111]. As these obstacles of delivery are overcome, the advantages of antisense technology will warrant that antisense oligonucleotide therapeutics will be one of the most promising clinical approaches to genetic diseases in the future.
7.3 U1 snRNA-Mediated Therapy
7.3.1 Brief Overview
Since its discovery in the early days of splicing research, U1 snRNA has been recognized as a crucial player in the first stages of the splicing process [112,113,114]. U1 snRNA is a 164 nucleotides long molecule with a well-defined structure consisting of four stem-loops, which primarily exerts its function in the form of a ribonucleoprotein (RNP) complex (termed U1 snRNP) containing seven Smith antigen (Sm) proteins and three U1-specific proteins U1A, U1C and U1-70K [115] (Fig. 7.4a). It is now well-established that U1 snRNP initiates spliceosome assembly by binding to the 5′ splice donor site (ss) through base pairing between the single stranded 5′ tail of the U1 snRNA molecule and the moderately conserved stretch of nucleotides at the 5′ss (CAG/GURAGU; R-purine) marking the exon-intron boundary [116]. However, not all base pairs at different 5′ss positions are equally important, and their contribution to splicing roughly correlates with their conservation (Fig. 7.4b). In the 9 nucleotides consensus sequence the most conserved 5′ss positions lie at the first two intronic nucleotides (+1 and +2), and the sequence GU at these positions accounts for ~99% of all 5′ss. The next most conserved 5′ss positions are −1G and +5G, which form strong G-C base pairing with U1 [117]. Once the donor site does not always conform to the consensus sequence, but can instead have a degenerate pattern feature, it is understandable that many other additional elements such as splicing silencer and enhancer motifs, the presence of alternative splice sites, secondary structures and regulatory proteins can influence the splice site selection (Fig. 7.4a) [117, 118].

Role of U1 small nuclear ribonucleoproteins (snRNPs) in splicing . (a) The 5′ end of U1 snRNA base pairs to the 5′ splice site (ss) in order to define a functional splice donor site. This process is positively and negatively modulated by different splicing factors, which bind to exonic and intronic splicing enhancer and silencer motifs (ESE, ISE, ESS and ISS, respectively). (b) The 5′ss motif. The height of each nucleotide corresponds to its conservation at the corresponding positon (−3 to −1 are exonic positions, while +1 to +8 correspond to intronic positions). The most conserved 5′ss positions are +1 and +2, which determine the 5′ss subtype: the GU subtype accounts for ~99% of 5’ss. Minoritary subtypes have a mismatch to U1 at either +1 or +2 and include the GC and the very rare AU 5′ss. The next most conserved 5′ ss positions are -1G and +5G, which form strong G-C base pairs with U1 through three hydrogen bonds. Consensus nucleotides −2A, +3A, +4A, and + 6U are also conserved but have a lesser although important contribution to 5’ss strength because their base pairing to U1 involves only the formation of two hydrogen bonds. The 5’ss positions +7 and +8 do not exhibit substantial conservation in humans, yet several lines of evidence indicate that these positions can base-pair to U1 and contribute to splicing
U1 snRNA is classically known for its role in pre-mRNA splicing events. However, the finding that U1 snRNA levels far exceed other spliceosomal associated snRNA levels led to the notion that it may have additional roles in the cell apart from splicing regulation [115, 119]. Indeed, emerging evidence suggests that U1 snRNA plays a key role in transcription initiation and in the protection of pre-mRNAs from degradation, as also has a regulatory function in the 3′-end formation, protecting pre-mRNA transcripts against premature polyadenylation and contributing to the regulation of alternative polyadenylation [115, 119,120,121].
Splicing mutations at the 5′ss, which are frequent among defects that cause human disease , compromise U1 snRNA binding and can prevent spliceosome assembly and subsequent splicing, which results in exon skipping, intron retention or activation of cryptic splice sites [117, 122]. The most deleterious mutations at a 5′ss are those affecting the nearly invariant GU dinucleotide at the positions +1 and +2. For the remaining nine positions the effects on splicing are less understood. Indeed, nucleotide substitutions in the less conserved positions can cause splicing defects in several but not all 5′ss, suggesting that this 5′ss positions and/or the general context define at what level splicing is changed [117, 123].
7.3.2 Two Generations of Engineered U1 snRNAs to Correct Splicing Defects
As donor splice site mutations disrupt the complementarity of the donor site with the endogenous U1 snRNA, restoring the complementarity through engineered modification of the U1 snRNA represents a valuable approach (Fig. 7.5a1, a2). In fact, in the mid-80s, Zhuang and Weiner [124] demonstrated for the first time that modified U1 snRNAs were able to suppress 5′ss mutations. Since then, the physiological role of the U1 snRNA to promote exon inclusion in the presence of 5′ss mutations affecting different positions of the donor site, has been extensively exploited as a possible therapy for numerous diseases. Significant correction levels have been achieved for mutations located in less conserved 5′ss positions in diseases like Neurofibromatosis type 1 [125], Coagulation factor VII deficiency [126, 127], Retinitis pigmentosa [128, 129], Propionic acidemia [130], Phenylketonuria [131] and Bardet-Biedl syndrome [132, 133]. Furthermore, recent studies have also demonstrated the feasibility of this approach in the more conserved GU region. In fact, partial correction of splicing defects caused by mutations in the +1 position of 5′ss was observed, not only in a Fanconi anemia case [134], but also in Sanfilippo C disease patient cells [135]. Also, for a mutation in the +2 position causing Hemophilia B, the treatment with a modified U1 snRNA led to an increase in the proportion of correct transcripts (~20%) [136]. In general, though, results of U1 snRNA therapeutic approaches can vary depending on the nature of the mutation and on the overall genomic context.

U1 snRNA-mediated therapy for mutations affecting 5′ splice site (ss).(a1) A wild-type endogenous U1 small nuclear ribonucleoprotein (snRNP) does not bind to 5′ss due to the presence of a 5′ss mutation. (a2) An exogenous U1 snRNP (first generation particle) is modified in 5′ tail with a compensatory alteration (semi-circle) that allows for base-pairing with the mutated 5′ss and the restoration of exon recognition and inclusion. (b) The presence of a 5′ss mutation does not allow the correct 5′ss recognition by U1 snRNP but an exon-specific U1 snRNP (second generation particle) with an engineered 5′ tail which binds a downstream non-conserved intronic region can activate the mutated 5′ss, through a mechanism not yet fully understood, allowing the correct exon recognition and inclusion
Until now, modified U1s effects in vivo were only addressed in two studies. In the first one, Balestra and co-workers [137] showed the rescue of the expression of a splicing-defective human factor VII (hFVII) mutant by a mutation-adapted U1 snRNA which improved hFVII circulating levels in mice, highlighting the potential of this strategy as a therapy for FVII coagulation deficiency. In the second study, Lee et al. [138] demonstrated the therapeutic effect of a mutation-adapted U1 snRNA in a knock-in mouse model of Aromatic L-amino acid decarboxylase (AADC) deficiency.
In common with other rescue strategies based on targeting RNA by complementarity, modified U1 snRNAs have to deal with potential off-target effects. This is particularly dangerous for modified U1 snRNAs with only one base change from the natural U1 snRNA, which might activate normally silent cryptic donor splice sites and induce aberrant splicing in other genes [139]. The consequences of such unwanted side reactions are hard to predict and depend on the function of the spliced transcript. However, the binding site sequence screening and mapping against the human genome to rule out sequence homologies should extensively decrease nonspecific events although their total exclusion cannot be guaranteed [140]. Therefore, experimental analysis should be performed whenever possible to test the effect of the U1 treatment on non-target transcripts. In mutation-adapted U1 snRNA in vitro approaches to correct 5′ splicing defects in Retinitis pigmentosa [129] and Bardet-Biedl syndrome [133], this type of test was performed and no missplicing events were found in the non-target transcripts. Also, in the in vivo U1 snRNA therapeutic strategy for AADC deficiency, the treatment was well tolerated and no toxic effects were seen within the study period [138]. However, in the in vivo study for hFVII deficiency [137], the authors observed hepatotoxicity, most probably caused by the binding of the engineered U1 to similar consensus 5′ss in other genes.
It was previously shown that U1 snRNAs do not necessarily have to bind at the 5′ss to promote exon definition. Some atypical 5′ss are recognized by U1 snRNA shifted by one nucleotide [141] and U1 snRNAs complementary to intronic sequences downstream of the 5′ss were originally reported to enhance the recognition of 5′ss in model gene systems [142, 143]. Given this, to reduce the possible interaction of modified U1 snRNAs with non-target 5′ss, a second generation of engineered U1s called Exon-Specific U1 snRNAs (ExSpeU1s) was developed. The ExSpeU1s have engineered 5′ tails that direct their loading into non-conserved intronic regions downstream of the 5′ss of a specific exon, and are expected to improve specificity and reduce potential off-target events [139, 144] (Fig. 7.5b). In different cellular models (i.e. minigene assays, patient’s cells or iPSC’s), a number of ExSpeU1s has been successfully applied, allowing an efficient rescue of exon skipping caused by various types of splicing mutations in Hemophilia B [144, 145], Cystic Fibrosis [144], SMA [144, 146, 147], Fanconi anemia [148] and Netherton syndrome [149]. The ExSpeU1 strategy has also been investigated in mouse models. For SMA, Dal Mas et al. [146], reported that AAV-mediated delivery of ExSpeU1 corrects splicing, increasing the inclusion of SMN2 exon 7 in different tissues/organs. In another study, Rogalska and colleagues [150] created a mouse expressing a particular ExSpeU1 and, after crossing it with a severely affected SMA mouse, observed increased inclusion of the missing exon, followed by SMN protein production and increased mice lifespan. Possible gene expression side effects were also addressed and, from a panel of 12,414 analysed genes, only 12 had altered expression after treatment.
ExSpeU1 molecules have also successfully rescued splicing in a transgenic mouse model of Familial Dysautonomia, a rare genetic disease with no treatment [151]. For Hemophilia B, another ExSpeU1 was explored in mice expressing two natural F9 splicing defective variants at 5′ss or 3′ss, and efficiently rescued human F9 splicing in liver resulting in an increase of the target protein and coagulation activity [152]. This study, as the pivotal one developed by Fernandez Alanis et al. with the ExSpeU1 strategy [144], interestingly showed that a single ExSpeU1 can be used to correct exon-skipping mutations at the consensus 5′ss (apart from canonical GU dinucleotide), the polypyrimidine tract, and even at exonic regulatory elements, thus extending the applicability of ExSpeU1s to panels of mutations and cohorts of patients with the same genetic disorder.
Despite the promising results obtained with ExSpeU1s in different studies, its precise mechanism of action for splicing correction is not totally clear. Pagani and co-workers demonstrated that ExSpeU1s are assembled as U1-like particles and that their splicing rescue activity is dependent on the U1-70K protein and on the loop IV structure of the U1 snRNA; not on the recruitment of endogenous U1 snRNP to the upstream 5′ss [150]. This may indicate that ExSpeU1s promote correct exon recognition through the recruitment of splicing factors that subsequently activate the mutated 5′ss [144, 146, 149,150,151,152]. However, it is important to stress that the splicing stimulator activity of ExSpeU1s was also responsible for the activation of a cryptic 5′ss in an approach attempted to correct a splicing defect causing Intrahepatic Cholestasis, which resulted in the production of an additional splice transcript with intron retention [153].
Globally, both mutation-adapted U1 snRNA and exon-specific U1 snRNA constitute a novel therapeutic strategy to correct splicing defects associated to defective exon definition in several human disorders. Once the U1 snRNA-mediated approaches act at pre-mRNA level, they have the main advantage of maintaining the regulated expression of the targeted gene in the normal chromosomal context [139, 154]. Also, given that the U1 snRNA gene used for splicing rescue includes promoter and regulatory sequences, it has the capability of guaranteeing long term correction of the genetic defect [139]. Despite these advantages, U1 snRNA-mediated therapies may also face some problems such as the presence of off-target effects and low efficacies. Therefore, in a near future, it will be imperative not only to develop a specific method or tool to search for off-target effects, but also to adjust the expression levels of U1 snRNA therapeutic particles in pre-clinical in vivo studies [154].
7.3.3 Engineered U1 snRNAs Delivery
U1 snRNA-mediated therapies also have to deal with the challenge of an efficient delivery to a target tissue. In the in vivo studies already developed, one of the most successful gene therapy systems available nowadays – AAV vectors – has been chosen as the method for U1 snRNA-engineered particles delivery into mice [137, 138, 146, 150, 151]. AAV vectors allow a highly efficient delivery to various tissues following systemic injection, even though dependent on the viral serotype used [90, 155]. Also, the low packaging capacity of AAV vectors is quite adequate for U1 snRNA-based approaches given the small cassette size to package [90]. However, despite the modifications that have been introduced in viruses, the potential for antiviral immunity and phenotoxicity of the transgene are still major limitations to the use of viral vectors for therapy. Possible alternatives to viruses are liposomes and nanoparticle delivery [155].
Among the several RNA tools enabling the rescue of splicing , both mutation-adapted U1 snRNA and ExSpeU1 snRNA therapeutic strategies have already shown their efficacy to repair different types of splicing defects at least in animal models of disease. Still, further developments will be necessary for this therapeutic approach to be translated to human trials.
7.4 siRNA-Based Drugs
7.4.1 Brief Overview
The last decade of the twentieth century has also witnessed the discovery of a new mechanism of gene regulation whose therapeutic potential is still being unveiled: RNA interference (RNAi). Interestingly, the first experimental observation of this mechanism came up from a failed genetic experiment aimed at developing more attractive petunia flowers. In fact, in 1990, Jorgensen and co-workers attempted to genetically engineer flower pigmentation genes, to be inserted into the target plant genome. To their surprise, however, instead of generating more colorful flowers, they ended up producing a generation of plants that had virtually lost all pigmentation, thus becoming white. This observation prompted additional studies to check the expression levels of endogenous genes involved in the natural pigmentation biosynthetic pathway and most of them were strongly reduced. Thus, a concept of co-suppression, whereby sequence-related genes could negatively regulate each other, was born [156, 157]. Still, little was known on its underlying mechanism. The first major breakthrough came from the pivotal studies by Andrew Fire and Craig Mello. By introducing various forms of long RNA molecules into C. elegans, their team observed that those with a double-stranded presentation (double stranded RNAs, dsRNAs) were the actual inducers of the silencing phenomenon, which was then coined RNAi [158]. Thus, the work by Fire and Mello, which earned them the 2006 Nobel Prize of Medicine, has not only represented a major advance in the understanding of RNAi basic mechanism, but also provided a simple and reproducible method by which long dsRNAs could be used to induce specific gene silencing in lower organisms commonly used in genetic research, such as C. elegans [159] and D. melanogaster [160]. In the meantime, other teams kept their focus in plant systems, aiming at a better understanding of the role that RNAi and additional silencing processes assume in plant homeostasis. Soon it became clear that gene silencing operating at the RNA level has roles in adaptative protection against viruses [161], genome defense against mobile DNA elements [162, 163] and developmental regulation of gene expression (reviewed in [164]). A second component of RNA silencing, in addition to dsRNAs, was then identified and coined short interfering RNAs, which resulted from the processing of dsRNAs into 21–26 nt counterparts [165]. Interestingly, those short interfering RNA molecules could be sorted into two classes depending on their size, and soon it became clear that each of those classes assumed different functions. The long ones (24–26 nt) were dispensable for sequence specific mRNA degradation , but essential for systemic silencing and methylation of homologous DNA [164]. Another interesting contribution to the deeper understanding of the overall RNA silencing process came from a work of Cogoni and co-workers, who described a new biological function for RNA silencing in Neurospora called quelling, which can be activated upon the introduction of transgenic DNA. These authors observed that quelling targets preferentially transgenes arranged in large tandem arrays and its effectors are also short interfering RNAs [166], reviewed in [167]. Altogether, these works unveiled an unexpected complexity in the RNA silencing process in plants, prompting additional studies to check whether the same would also apply in animals. By this time, however, no one foresaw that the RNAi mechanism would also work in mammalian systems because long dsRNAs were already known to induce a strong interferon response. The first demonstrations that RNAi also works in humans came from the work of two independent groups in Germany, one operating at the Max Planck Institute, and the other at the University of Bayreuth, and a third one in the United States, operating at the NIH, Bethesda. The team at the Max Plank Institute showed that synthetic versions of short dsRNA molecules were able to trigger a strong gene silencing effect in mammalian cells without inducing the interferon response. Moreover, they tested a series of design features for those short dsRNAs including length, blunt or sticky ends and chemical modifications, finding that structurally defined 21–23 base-pair small RNAs, with 2 nucleotide unpaired overhangs at the 3′ ends, were the most efficient mediators of RNAi [168, 169]. This fundamental work was published in Nature in 2001, and became the scientific content for a key patent in the field called “Tuschl II”. In parallel, the NIH team came up with another demonstration that synthetic siRNAs can induce gene-specific inhibition of expression in C. elegans and in cell lines from humans and mice. They did it by systematically comparing the level of gene expression decrease caused by siRNAs versus that caused by single stranded AONs [170]. Their work, published in PNAS, was another step to open a path toward the use of siRNAs as a reverse genetic and therapeutic tool in mammalian cells, as the authors themselves have stated. Around the same time, at the University of Bayreuth, Kreutzer and Limmer had also reasoned that short fragments of dsRNA would putatively mediate a RNAi response similar to the one originally described by Fire and Mello and, even though their findings have never been published, they did file key patents around the discovery. Additional studies on the subject ended up unveiling the endogenous RNA silencing pathway that was being fed by small dsRNAs, from now on called small interfering RNAs (siRNAs). It also became evident that the same pathway is also able to process microRNAs (miRNAs), as previously seen (Fig. 7.1c). Here, we will focus solely on the RNAi process triggered by siRNAs.
The siRNA pathway starts with the cytoplasmic cleavage of long dsRNAs by an enzyme called Dicer. As a result, short dsRNA duplexes are formed. Then, those dsRNAs are incorporated into the RNA-induced silencing complex (RISC), where the strands are separated, and one strand guides RISC to the complementary region of target mRNA (Fig. 7.6). The heart of RISC is the Argonaute (AGO) proteins. In humans there are 8 AGO proteins, 4 from AGO clade (AGO1-4) and 4 from P-element induced wimpy testis (PIWI) clade (PIWI1-4; [171]). Still, not all AGO proteins are cleavage competent. In fact, AGO2 is the sole executer that accomplishes siRNA-induced silencing. Thus, whenever the siRNA strand loaded into RISC has complete sequence complementarity with its target mRNA sequence, it triggers site-specific mRNA cleavage, which ultimately results in a reduced expression of that mRNA and of the target protein (Fig. 7.4; reviewed in [172]). This exact same process can also be induced by direct exogenous supply of synthetic siRNAs. Over the years, a series of empirical and rational guidelines started accumulating from the analysis of hundreds of functional siRNAs. There are now a number of guidelines one should follow in order to design an effective siRNA, which have been well reviewed elsewhere [173]. There are also many websites and companies that either offer reliable methods for the design of effective siRNAs or even design them on demand. Because of their small size, the chemical synthesis of siRNAs is relatively easy and nowadays, several companies offer them delivered in ready-to-transfect format. This is, therefore, a simple, easy-to-handle RNAi-effector for virtually every lab need.

The RNA interference (RNAi) mechanism. Entry of double-stranded RNA (dsRNA) into eukaryotic cells results in targeted RNA-induced silencing complex (RISC)-mediated cleavage of messenger RNA (mRNA) through activation of the endogenous RNAi mechanism: dsRNAs are recognized and cleaved into shorter fragments by Dicer, and subsequently loaded into a multiprotein conglomerate called RISC, which facilitates the separation of the two RNA strands. Once the double-stranded RNA is separated, one strand gets degraded while the other associated with RISC acts as a template for RISC-mediated cleavage of complementary RNA, thus reducing protein translation
Since the half-life of siRNA is short, an alternative RNAi-effector molecule has also been developed: short hairpin RNAs (shRNAs), which are not directly transfected into their target cells. Instead, shRNAs are transcribed in the nucleus from an exogenous DNA expression vector bearing a palindromic sequence with a spacer in between, whose transcript folds into a short dsRNA with a terminal loop. The shRNA transcript is processed by Drosha, an RNase III endonuclease. The resulting pre-shRNA is exported to the cytoplasm, where it can then be processed by another RNase III, called Dicer, and incorporated into RISC, thus triggering the same RNAi process previously described (reviewed in [172]). In general, shRNAs are harder to complex/internalize. Still, by delivering DNA instead of the effector RNA molecule, they take advantage of the cell’s transcription machinery to produce specific shRNA transcripts, they allow for high potency sustainable effects using low-copy numbers. One such approach results in less off-target effects, putatively ensuring greater safety. Additionally, a shRNA expression vector does also cost less than the bulk manufacturing of siRNAs (reviewed in [174]).
Once all cells have the RNAi machinery and, in principle, any gene can be knocked down, soon siRNAs became invaluable tools in the lab, enabling the easy genetic knockdown of any sequence. RNAi was rapidly exploited as a tool to promote unbiased genome-wide screening to search for relevant genes involved in specific biological processes, first in invertebrate cells [170, 175,176,177] and latter in mammalian cells [178,179,180,181,182]. In fact, this knockdown technique provides a valuable tool for the functional annotation of mammalian genes [183, 184], for the creation of knockout animals [185] and for the identification of new drug targets (reviewed in [186]), but these are far from being the major application of this technology. In fact, RNAi has been regarded as one of the major breakthroughs in the field of molecular medicine, and its potential as a therapeutic effector has been largely tested over the last decades.
The need to optimize the technique and take it from bench to clinic is also prompting extra research efforts to gain a deeper understanding of the overall RNAi mechanism. For example, in order to function, siRNAs need to escape to the cytosol, where the RISC works. Thus, release from the endosome is an important barrier. Understanding the mechanism(s) that promotes and limits endosomal release may help to optimize this limiting step. This remains, though, a major area of investigation for all nucleic acid therapeutics [11].
7.4.2 Recent Successful Applications of siRNA-Based Drugs
Being a naturally occurring post-transcriptional gene silencing process, this mechanism has several advantages when compared to other AON technologies, and that recognition triggered major investments in RNAi-based drug development by large pharmaceutical and biotechnological companies [187]. The potential of siRNA therapeutics was first demonstrated by Song and co-workers 15 years ago, when injection of Fas siRNAs protected mice from autoimmune hepatitis. Fas-mediated apoptosis is implicated in a broad spectrum of liver diseases, where inhibiting hepatocyte death can be life-saving. These authors investigated the silencing effect of siRNA duplexes targeting the gene encoding the Fas receptor (Fas), to protect mice from liver failure and fibrosis in two models of autoimmune hepatitis. Intravenous injection of Fas siRNA specifically reduced both Fas mRNA and Fas protein expression levels in mouse hepatocytes, and the effects persisted without diminution for 10 days [188]. This pioneer work has not only shown that siRNA-directed Fas silencing could work in vivo and be of therapeutic effect for preventing and/or treating acute and chronic liver injury [188], but also provided the proof-of-principle on the potential of the overall RNAi technology to treat or prevent disease (reviewed in [189]). Since then, drug development has been rapid, with siRNAs facing virtually the same obstacles as AONs . Fortunately, some of the AON strategies could be adapted to siRNA therapeutics, thus accelerating siRNA preclinical drug development and clinical evaluation. In general, RNAi clinical trials are progressing well. Clinical Phase I and II studies of siRNA therapeutics have demonstrated potent (as high as 98%) and persistent (lasting for weeks) gene knockdown effects, especially in liver, with some signs of clinical improvement and without unacceptable toxicity (reviewed in [189]). There are also several trials in Phase III development (Table 7.2; reviewed in [11]).
Early this year Alnylam has announced U.S. FDA acceptance of New Drug Application (NDA) and Priority Review Status for Patisiran, an investigational RNAi therapeutic for the treatment of hereditary transthyretin amyloidosis (hATTR) [225]. Almost at the same time, the company presented new clinical results from the APOLLO Phase III study of this drug at the 16th International Symposium on Amyloidosis. The APOLLO Phase III trial was a randomized, double-blind, placebo-controlled, global study designed to evaluate the efficacy and safety of Patisiran in hATTR amyloidosis patients with polyneuropathy. The primary endpoint of the study was the change from baseline in modified Neurologic Impairment Score +7 (mNIS+7) relative to placebo at 18 months. According to the general manager of the transthyretin (TTR) program at Alnylam, “the clinical results presented further highlight the robust profile of Patisiran and provide evidence supporting Patisiran as a potentially transformative treatment approach for patients with hATTR amyloidosis”. Also the results obtained in the cardiac subpopulation, which corresponded to approximately 50% of the patients enrolled in the APOLLO study, revealed significant improvements in measures of cardiomyopathy, the leading cause of death in patients with hATTR amyloidosis, relative to placebo [201]. Finally, in August 2018, the drug got its U.S. FDA approval, and is now commercialized under the designation Onpattro™ [226].
Hopefully, the approval of the first RNAi therapeutic will pave the way for approval of other targets (reviewed in [227]), especially if we take into account that there are several other siRNA drugs under evaluation, which have recently advanced for phase III development (Table 7.2; reviewed in [11]). The most relevant examples include Revusiran, which is a second siRNA drug under evaluation as a treatment option for patients with familial amyloid cardiomyopathy by reducing plasma TTR levels [220]; QPI-1002, which is being developed for the treatment of delayed graft function for kidney transplants [192]; Fitusiran, a potent siRNA drug under study for patients with hemophilia, which aims at ameliorating the disorder by reducing the plasma levels of anti-thrombin [198], and Inclisiran, which targets proprotein convertase subtilisin/kexin type 9 (PCK9) to reduce the risk of cardiovascular disease [200], as reviewed in [11].
7.4.3 Delivery of siRNA-Based Drugs
Intracellular delivery of double-stranded siRNAs is more challenging than delivery of single stranded AONs [186]. Still, it is also worth mentioning that, as suggested by the best estimates, only a few hundred cytosolic siRNAs per cell are needed for efficient and sustained gene knockdown [228, 229]. This happens because the guide strand of the siRNA remains stable within the RISC for weeks, even though it gets diluted with every cell division [230]. Thus, the same siRNA molecule can target multiple transcripts, knocking down gene expression in slowly dividing or non-dividing cells over the same period. Overall, as noticed by several authors, this actually contributes to turn the delivery obstacle into a less formidable one than that faced by other antisense mechanisms, which act on a one-to-one basis [230] (reviewed in [229]).
Still, knowing that the translation of siRNAs from the bench to the clinic would be hindered by their limited cellular uptake, low biological stability and unfavorable pharmacokinetics, the development of appropriate delivery methods became mandatory to proceed with preclinical studies. Therefore, different approaches have been (and are being) attempted to ensure safer and long-lasting delivery methods for siRNA-based drugs, for both systemic and targeted delivery. Most of these developments were made in parallel with siRNA drug development and only through the combined efforts of several independent teams were these drugs modified in ways that allowed their clinical evaluation, with the promising results highlighted in the previous section. Whatever the case, an effective delivery system must fulfill a series of criteria, which have already been listed by Tatiparti et al. amongst other author: be stable at the body temperature and pH variations, have an endocytosis promoting shape, cannot be toxic, must exhibit high siRNA loading abilities and have a size that avoids rapid renal and hepatic clearance [231]. In general, all the delivery systems developed for gene therapy may also be adapted for siRNA delivery [232].
7.4.4 Non-targeted Delivery
Early strategies for solving the dual problems of intracellular delivery and rapid excretion involved incorporating siRNAs into LNPs – smaller, more homogeneous analogues of lipoplexes used for laboratory transfection [233,234,235], (reviewed in [189]). LPNs were first shown to be effective in targeting the hepatitis B virus (HBV) in mice, where the LPN-formulated siRNA was given in 3 daily injections of 3 mg/Kg/day. This treatment regimen resulted in a decrease of HBV levels by 1–2 orders of magnitude [236], as reviewed in [237]. Nevertheless, these complexes (and other nanoparticle strategies for siRNA delivery) accumulate in the liver and other filtering organs, which limits their effectiveness in penetrating other tissues [235, 238] (reviewed in [189]). Furthermore, the administration of siRNAs with LNP delivery vehicles is quite pro-inflammatory. In fact, lipid-based vehicles can become entrapped in endosomes [237], where the Toll-like receptors (TLR) will recognize various moieties in dsRNAs, modified siRNAs or even from their degradation products [239], eliciting an undesirable innate inflammatory response. So, in most circumstances the siRNAs require pretreatment regimens including antihistamines, non-steroidal anti-inflammatories and even relatively high doses of glucocorticoids [190, 191, 240] (reviewed in [11]).
Still, recent developments by several independent teams have demonstrated the feasibility of systemic administration of either chemically modified or complexed siRNAs. In fact, even though unmodified siRNAs do not distribute broadly to tissues after systemic administration (reviewed in [11]), simple chemical modifications of the 2′-position of the ribose and substitution of phosphorothioate linkages, such as the ones described for AONs in the first section of this chapter (2′-OMe and 2′-MOE), and 2′-fluoro (2′-F), protect siRNAs from nuclease digestion, thus prolonging their half-lives, both in serum and other body fluids [236, 241] (reviewed in [189]). 2′-modifications can also prevent recognition by innate immune receptors by blocking the binding to TLR [242,243,244] and reduce off-target effects that could arise from the suppression of partially complementary sequences [245] (reviewed in [189]). As already referred, these modifications had been previously designed for use in AONs and not siRNAs and, even though they did show effective in improving stability, specificity and immunogenic properties, they do not improve potency. Recently, however, two novel, siRNA-optimized 2′-O modifications, were shown to increase in vivo activity of siRNAs, not only by increasing their potency but also their in vivo duration compared to their unmodified counterparts when delivered using LNPs: 2′-O-benzyl and 2′-O-methyl-4-pyrimidine (2′-O-CH2Py(4); [246]). Several teams have also been assessing different complexation methods with functional peptides [247] and/or different vectors: exosomes [248] including lipid nanocarriers such as pegylated immunoliposomes (PILs; [249]), stable-nucleic-acid-lipid-particles (SNALPs; [250]), or polyhydroxyalkanoate-based nanovehicles [251], reviewed in [252]. Also under consideration are siRNA delivery strategies that use viral particles. The viral delivery of siRNAs is composed of two main strategies: siRNAs are either chemically synthesized and loaded into a viral capsule, or they can be expressed from the DNA of a recombinant virus (reviewed in [253]).
Meanwhile, second generation LPNs were also developed. Constructed with the anionic lipid dilinol eylmethyl-4-dimethylaminoburyate (DLin-MC3-DMA), they mediate potent gene knockdown at reduced doses compared with first generation LNPs, while improving delivery [190, 217]. Partisiran (previously termed ALN-TTR02; ChemIDplus-Partisiran), for example, is exemplary of a minimally chemically modified siRNA delivered primarily to the liver in a second generation liposome formulation.
7.4.5 Targeted Delivery
Overall, there has been a huge progress over the last decade concerning not only non-targeted but also targeted delivery of siRNA drugs. In fact, siRNAs can also be targeted for uptake in selected tissues or cell types by taking advantage of high-affinity antibody or antibody fragments [254,255,256], aptamers (nucleic acids selected for high-affinity binding; [257,258,259] or receptor ligands [260,261,262,263,264], which bind to specific cell surface receptors and mediate cell-specific uptake. The targeting moieties can be either directly conjugated to siRNAs (bound non-covalently) or incorporated into LPNs or other nanoparticles (reviewed in [189]). In general, targeted uptake has the advantages of being effective at a lower dose while exhibiting lower toxicity, which may potentially occur from knockdown effects in unintended tissues. It is also a tool of great advantage for the treatment of non-systemic diseases. The easiest organ to target is the liver, which is a filtering organ that traps nanoparticles. It is also the primary site of synthesis of many circulating proteins. That is why it has been the target organ in most early clinical attempts at translating RNAi (reviewed in [186]). Furthermore, several diseases, which directly affect this organ may benefit from a straightforward liver targeting method. The most successful possibility under study includes a series of more drastic chemical modifications, where siRNAs have a trivalent N-acetylgalactosamine (GalNAc) moiety conjugated to the 3′ terminus of one of the strands [220, 264] (reviewed in [11]). GalNAc mediates hepatocyte uptake through the hepatocyte-restricted asialoglycoprotein receptor (ASGPR), thus being a suitable mediator for whole-liver delivery [265] (reviewed in [189]). Uptake by this receptor is primarily through clathrin-dependent endocytosis [97]. Examples of GalNAc-modified siRNAs include Revusiran and Fitusiran (ChemIDplus-Revusiran; ChemIDplus-Fitusiran (reviewed in [11]). Also, local delivery to the CNS, a region that is difficult to deliver drugs to due to the BBB, is being addressed, with promising results. First preliminary evidence that in vivo downregulation of specific genes by RNAi could work at the CNS level came from studies in rats and mice using invasive local delivery methods (reviewed in [266]). Lately, however, evidence is accumulating on the successful brain delivery of si/shRNAs using specifically designed vectors and/or modifications that include the use of enzyme-sensitive LPNs [267], carbosilane dendrimers [268], cholesterol modifications [269], and recombinant fusion proteins [270]. Also, the pharmaceutical industry is investing in developing BBB-directed vectors. One of those examples is a family of vectors that take advantage on the existence of specific receptors and transport systems, which are highly expressed at the BBB to provide essential substances to brain cells. These vectors comprise a full-length protein (Melanotransferrin) and may be used to facilitate receptor mediated drug delivery into the brain to treat CNS disorders [271]. Recently, the application of this new peptide vector to siRNA and ongoing studies addressing the brain delivery of Iduronate 2-sulfatase (I2S) for the treatment of Hunter Syndrome, a rare X-linked lysosomal storage disorder, was discussed and its results in knockout mice were quite promising [272].
7.5 CRISPR-Cas Gene Editing
In addition to the most well known RNA-based therapeutics (antisense drugs and siRNA-based drugs ) several other mechanisms of action are also potential strategies. Recently, a new gene editing technology, Clustered Regulatory Interspaced Short Palindromic Repeats (CRISPR) and the CRISPR-associated protein 9 (Cas9) (CRISPR-Cas 9) system, has received unprecedented acceptance in the scientific community for a variety of genetic applications (reviewed in [273]) (Fig. 7.7a). Even though this technology lies beyond the scope of this chapter, it does deserve some attention, as it may become a leading method for gene editing and even RNA-based therapeutics , in the long term.


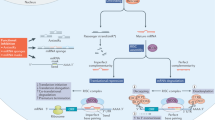
Additional RNA-based drug mechanisms. (a) CRISPR/Cas9: CRISPR/Cas9 induces double strand breaks (DBS) when targeted to a specific genomic site by an appropriate guide RNA (sgRNA). This property may be used for mutation correction, by adding a donor DNA sequence that has homologous overlaps to the DBS (typically 100–1000 bp of overlap is used), thus promoting homologous repair of the cleaved genomic DNA; (b) Modified mRNAs: This approach consists in introducing chemically modified, stabilized mRNAs into cells to be translated to protein. Once internalized, sense-RNA drugs can be used for transient in vivo transcription (IVT) of mRNAs to replace mutated proteins or for vaccination without the risk of genomic alteration; (c) Aptamers : Aptamers take advantage of their selection for high-affinity binding to molecular ligands, often in the nanomolar or subnanomolar range. They can be compared to nucleic acid antibodies, having many of the advantages of conventional protein antibodies. They can be either agonists or antagonists, linked for bifunctional targeting and conjugated to other RNAs, small-molecule drugs, toxins or peptides. However, unless modified, they are rapidly excreted and do not activate immune functions, as other antibodies do
Similarly to what had already happened with RNAi, the CRISPR-Cas system was not specifically developed as a method for gene editing. Instead, it is a naturally occurring prokaryotic immune defense strategy against non-self DNA based invasions (e.g., viruses, plasmids), which was recently discovered in bacteria and archaea [273,274,275,276], and latter adapted for bench applications [277, 278]. Also like RNAi, the specificity of CRISPR-Cas relies on the antisense pairing of RNAs (here termed single guide RNA, sgRNA) to specific genes but instead of binding directly to RNA, sgRNAs bind to chromosomal DNA. Another relevant difference between the RNAi and CRISPR technologies has to do with the transiency of their effect. In fact, unlike siRNAs, sgRNAs induce stable changes in gene expression, which are invaluable for in vivo gene screening. Thus, genomic targeting through CRISPR-Cas creates indels that can be adapted for stable eukaryotic genome engineering, namely Cas-mediated gene knockdown (reviewed in [186, 273]). In general, the application of CRISPR/Cas9 for DNA editing as well as for mammalian gene editing was established in the 2012–2013 period and, in just 3 years, this technique has revolutionized the entire gene editing field. Currently, CRISPR-Cas gene knockdown in zygotes provides a fast method for the development of different animal models, when compared to homologous recombination. Nevertheless, it does hold a series of drawbacks and raises a number of concerns, particularly when its therapeutic potential is considered. In fact, since this technique has the ability to modify the genome, its ethical and safe concerns are enormous. Furthermore (and like every other antisense technology), CRISPR-Cas holds potential for both on- and off-target effects. Moreover, by creating double-stranded DNA breaks, the Cas endonuclease can also lead to oncogenic gene translocations and trigger a DNA damage response, ultimately causing cell-cycle arrest or even cell death. Finally, depending on the repair pathway which is activated, gene editing may be imprecise [186]. Still, it must be noticed that in the 5 years following the publication of the method, several improvements to reduce off-target effects and provide a better control of the whole mechanism, while enhancing its efficiency have been developed and reported (reviewed in [186, 279]). It should also be mentioned that CRISPR/Cas9 is certainly a more versatile technique than RNAi, has it may not only induce indels but also repress or activate gene expression and cause both heritable and non-heritable genomic changes [280,281,282]. In fact, CRISPR/Cas9 can be adapted to upregulate gene expression in different ways: the first, most obvious approach consists in using this technology to stably introduce constitutive active promoter elements to a gene, thus stably enhancing its expression. Alternatively, a modified Cas9 (fused to a transcriptor activator protein) may be targeted to any gene of interest, driving a transient enhancement in gene expression known as CRISPR activation (CRISPRa; [273]). In addition, the emergence of newer gene editing tools such as the Cpf1 enzyme, which is a single RNA-guided endonuclease, will eventually strengthen the portfolio of applications that may be achieved by CRISPR mediated genome engineering [283]. Therefore, it is becoming clear that clinical CRISPR-Cas studies will also be a trend in research over the next years, starting with ex vivo editing of differentiated cells, which may then be infused into patients. Furthermore, as other authors have already stated, it will also greatly benefit from the accumulated knowledge on other non-RNA-based gene editing tools, such as zinc-finger nucleases and on the delivery methods previously developed for AON - and siRNA-based drugs .
7.6 Messenger RNA as a Novel Therapeutic Approach
Another RNA-based approach is to introduce chemically modified stabilized mRNAs into cells, where those exogenous mRNAs will eventually be translated to protein (Fig. 7.7b). In fact, in vitro transcribed (IVT) mRNA has recently come into focus as a potential new drug class to deliver genetic information. Such synthetic mRNAs can be engineered to transiently express proteins by structurally resembling natural mRNAs [186, 284]. One advantage of mRNA-based therapy over viral gene delivery is that mRNA does not transit to the nucleus, thereby mitigating insertional mutagenesis risks. Moreover, mRNA provides transient, half-life-dependent protein expression, while avoiding constitutive gene activation and maintaining dose responsiveness. Because of these advantages, IVT mRNA treatment is an emerging class of therapy, with multiple mRNA-based cancer immunotherapies and vaccines currently in clinical trials [284,285,286]. However, the fact that IVT mRNA, despite its strong resemblance to naturally occurring mRNA, can be recognized by the innate immune system may play an important part in its applicability. For vaccination approaches, the inflammatory cytokine production resulting from mRNA-induced immune stimulation might add to the effectiveness of the evoked immune response. For non-immunotherapy approaches, however, the story is different and so far, cancer immunotherapy is the only field in which mRNA based therapeutics have reached clinical trials [287,288,289,290,291,292,293,294]. Nevertheless, the potential of IVT mRNA is currently being explored for a variety of applications, ranging from inherited or acquired disorders to regenerative medicine, all of which remain at the preclinical stage [295, 296]. In fact, an increasing number of preclinical studies has evaluated mRNA-based therapy for a wide range of diseases such as surfactant B deficiency, myocardial infarction [297] sensory nerve disorders [298], fulminant hepatitis [299] hemophilia B [300, 301], congenital lung disease [302], cancer [303], liver and lung fibrosis [304], and methylmalonic acidemia [305]. However, the main hurdle in implementation of mRNA for therapeutics, the systemic delivery of mRNA molecules to target cells, remains a challenge. Better understanding of the factors that determine translational efficiency as well as RNA recognition by innate immune receptors, has improved the intracellular stability and functionality of mRNA transfected to cells. Still, when aiming to harness mRNA molecules for gene therapy purposes, this progress was insufficient. The need for mRNA protection from degradation in extracellular compartments, as well as for enabling its entry to the cell, has raised the demand for suitable delivery platforms [285, 295]. A possible solution for this challenge relies in the rapidly evolving field of nucleic acid-loaded NPs. In fact, the progress in the field of NPs-mediated RNAi-based therapy, has led to similar development of nanocarriers for mRNA. Particularly, the widely investigated family of LNPs was proposed to be such appropriate mRNA nanocarriers [296, 305, 306]. Moreover, the use of polyplex nanomicelles has also been explored [298, 299]. In order to achieve high efficacy in vivo some IVT mRNA specific formulation adjustments should be done in a near future. These adjustments are more important when systemic administration is required. Moreover, in order to expand the variety of mRNA-based therapies, cell specific targeted delivery systems are also needed especially in diseases involving a certain organ, which is inaccessible by standard LNPs, as well as in many types of solid tumors [296]. In conclusion, innovative design of nanocarriers for IVT mRNAs delivery will help to increase their potential and turn them into a valid therapeutic approach.
7.7 Aptamer-Based Drugs
Another potential class of RNA therapeutics are oligonucleotide aptamers (see Fig. 7.7c). The term aptamer comes from the Latin word “aptus”, which means “to fix”, as a clear reference to the lock and key relationship of aptamers and their targets [307, 308].
Aptamers are short (20–70 bases) single stranded oligonucleotides (ssRNA/ssDNA), which bind to their targets through 3D conformational complementarities with high affinity and specificity. Unlike the previously referred strategies, aptamers can be tailored selectively against a variety of targets, from nucleotides to amino acids, proteins, small molecules or even live cells [309]. Still, proteins are the major targets in aptamer research (reviewed in [310]). Oligonucleotide aptamers have affinity and specificity capacities, which are comparable to those of monoclonal antibodies, whilst having minimal immunogenicity, high production, low cost and high stability. These oligonucleotides can be selected trough an in vivo process called Systematic Evolution of Ligands by Exponential enrichment (SELEX), which dates back to 1990. This method was originally described and performed by Szostak and Gold [307, 308]. The whole process starts with the synthesis of a screening library formed by a large number of randomly combinatorial ssDNA and/or ssRNAs. Each one of those random ssDNA/ssRNAs has one conserved sequence at each end. That sequence allows primer binding and amplification. The random library is then incubated with the target proteins, under proper conditions. Then, through a partition step, the sequences that had bind to target proteins are separated from those that did not bind. In the third step, the binding sequences are eluted and amplified with primers complementary to their conserved sequences, either by PCR (for ssDNA) or RT-PCR (for ssRNA). All these steps form a single SELEX cycle. This selection process is then repeated for about 7–20 rounds of incubation, partitioning and amplification. Ultimately, this results in the identification of a small number of binding sequences with high affinity and specificity for further processing and optimization. Generally, the binding sequences are then transformed into bacteria (E. coli) for further sequencing and characterization (reviewed in [310]). Naturally, in the post-SELEX process, the synthesized aptamers (as every other AON ) can be chemically modified for therapeutic purposes, to stabilize and protect them against nucleases in vivo. Recent advances in SELEX technology, with the introduction of chemically modified bases and the use of deep sequencing to analyze enriched RNAs in early rounds of selection, have greatly reduced the time needed and the likelihood of identifying high-affinity aptamers (reviewed in [186]).
Over approximately 10 years, starting in 2005, when the first aptamer Pegaptanib (PubChem, Pegaptanib) was approved for wet age-related macular degeneration (AMD) therapy by U.S. FDA, oligonucleotide aptamers were growing more and more popular. Until 2016, when the last estimates were published online, there had been over 900 aptamers developed against various targets for diagnostic and therapeutic purposes [311]. Nevertheless, drug development of aptamers is currently not very active, with big pharmacological companies being much more focused on the technologies reviewed in the previous sections of this chapter. Still, it is worth mentioning that these oligonucleotides could substitute for some applications of therapeutic antibodies, with lower risk of developing immunological responses. They could also be used for targeted intracellular delivery of other molecules, including RNA-based drugs.
7.8 Conclusion
Over the last decades, an exceptional increase on the understanding of the versatile roles of RNAs has sparked the development of new classes of RNA-based drugs. Therapeutic RNA-based applications are emerging, in different fields, from inherited genetic diseases , oncology, viral infections and diabetes to neurological, cardiovascular, bone-related and ocular diseases. Over the last years in particular, much effort has been focused on the development of RNA-based therapeutics . Currently, even though there are a number of RNA-based therapeutic strategies, which may be attempted in order to either correct or modulate gene expression, there has been a clear prevalence of studies focused on splicing modification and gene expression inhibition using different types of AONs . Actually, the first AON-based drugs were recently approved, closely followed by the first siRNA-based therapeutic drug, which was approved last year. Still, there is a strong need to optimize the delivery steps of RNA-based technologies and to improve the drug-like properties of therapeutic nucleic acids. Expanding the range of targeted cells and tissues will require the development of robust strategies for cytosolic delivery, thus overcoming the two major hurdles of getting across the plasma membrane and out of the endosome.
In conclusion, as the first generation of nucleic acid therapeutics become drugs, the barrier for investing in RNA-based therapeutics will be lowered, and more resources will become available for exploring other mechanisms of action for RNA-based drugs apart from splicing modulation and single-gene knockdown. As already pointed out by other authors, the flexibility of RNA design should allow for the facile construction of potent multifunctional drugs that have more than one mode of action and disrupt multiple targets. One such multifunctional drug may hold the promise of substituting for drug cocktails in a future not so distant. There is also the largely unexplored potential of targeting other RNA species and disrupting their functions. Therefore, in the near future, RNA-based drugs may become an increasing component of the pharmacopoeia, greatly expanding the universe of druggable targets and providing affordable treatment options for previously untreatable diseases. Ultimately, this kind of drugs may hold potential to actually cure genetic diseases [186].
Abbreviations
- 2′-F:
-
2′-fluoro
- 2′-MOE:
-
2′-O-methoxyethyl
- 2′-O-CH2Py(4):
-
2′-O-methyl-4-pyrimidine
- 2′-OMe:
-
2′-O-methyl
- AADC:
-
Aromatic L-amino Acid Decarboxylase
- AAVs:
-
Adeno-associated Viruses;
- AGO:
-
Argonaute
- AMD:
-
Age-related Macular Degeneration
- AONs:
-
Antisense Oligonucleotides
- ASGPR:
-
Asialoglycoprotein receptor
- ASGR:
-
Asialoglycoprotein Receptor
- BBB:
-
Blood Brain Barrier
- BMD:
-
Becker Muscular Dystrophy
- CNS:
-
Central Nervous System
- CPPs:
-
Cell-penetrating Peptides
- CRISPR:
-
Clustered Regulatory Interspaced Short Palindromic Repeats
- CRISPR-Cas 9:
-
CRISPR-associated protein 9 (Cas9)
- CSF:
-
Cerebrospinal Fluid
- DLin-MC3-DMA:
-
Anionic lipid dilinol eylmethyl-4-dimethyl-aminoburyate
- DMD:
-
Duchenne Muscular Dystrophy
- dsRNAs:
-
double-stranded RNAs
- ExSpeU1s:
-
Exon-Specific U1 snRNAs
- FH:
-
Familial Hypercholesterolemia
- GalNAc:
-
N-acetylgalactosamine
- GPCRs:
-
G Protein-coupled Receptors
- HCV:
-
Hepatitis C Virus
- hFVII:
-
Human Factor VII
- hTTRA:
-
Hereditary Transthyretin Amyloidosis
- I2S:
-
Iduronate 2-sulfatase
- IVT:
-
in vitro transcribed
- LDL-C:
-
Low-density Lipoprotein Cholesterol
- LNAs:
-
Locked Nucleic Acids
- LNPs:
-
Lipid-based Nanoparticles
- miRNAs:
-
microRNAs
- mNIS+7:
-
Modified Neurologic Impairment Score +7
- mRNA:
-
messenger RNA
- NDA:
-
New Drug Application
- PCK9:
-
Proprotein Convertase Subtilisin/Kexin Type 9
- PD:
-
Pharmacodynamics
- PILs:
-
Pegylated immunoliposomes
- PIWI:
-
P-element induced wimpy testis
- PK:
-
Pharmacokinetics
- PMOs:
-
Phosphoroamidate Morpholino Oligomers
- PNAs:
-
Peptide Nucleic Acids
- PS:
-
Phosphorothioate
- RIS:
-
RNA-induced silencing complex
- RNAi:
-
RNA interference
- RNP:
-
Ribonucleoprotein
- RTK:
-
Human Receptor Tyrosine Kinase
- SELEX:
-
Systematic Evolution of Ligands by Exponential enrichment
- sgRNA:
-
single guide RNA
- shRNA:
-
short hairpin RNA
- siRNAs:
-
small interference RNAs
- SLNs:
-
Solid Lipid Nanoparticles
- Sm:
-
Smith antigen
- SMA:
-
Spinal Muscular Atrophy
- SNALPs:
-
Nucleic-acid-lipid-particles
- SSOs:
-
Splice Switching Oligonucleotides
- TLRs:
-
Toll-like Receptors
- TTR:
-
Transthyretin
- U.S. FDA:
-
U.S. Food and Drug Administration
References
Bennett CF, Swayze EE (2010) RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol 50:259–293
Godfrey C et al (2017) Delivery is key: lessons learnt from developing splice-switching antisense therapies. EMBO Mol Med 9(5):545–557
McClorey G, Wood MJ (2015) An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr Opin Pharmacol 24:52–58
Rossor AM, Reilly MM, Sleigh JN (2018) Antisense oligonucleotides and other genetic therapies made simple. Pract Neurol 18(2):126–131
Bauman JA et al (2010) Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res 38(22):8348–8356
Kole R, Krainer AR, Altman S (2012) RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 11(2):125–140
Sardone V et al (2017) Antisense oligonucleotide-based therapy for neuromuscular disease. Molecules 22(4):E563
Paterson BM, Roberts BE, Kuff EL (1977) Structural gene identification and mapping by DNA-mRNA hybrid-arrested cell-free translation. Proc Natl Acad Sci USA 74(10):4370–4374
Zamecnik PC, Stephenson ML (1978) Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci USA 75(1):280–284
Schoch KM, Miller TM (2017) Antisense oligonucleotides: translation from mouse models to human neurodegenerative diseases. Neuron 94(6):1056–1070
Crooke ST et al (2018) RNA-targeted therapeutics. Cell Metab 27(4):714–739
Oberemok VV et al (2018) A half-century history of applications of antisense oligonucleotides in medicine, agriculture and forestry: we should continue the journey. Molecules 23(6):1302
Khvorova A, Watts JK (2017) The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 35(3):238–248
Chan JH, Lim S, Wong WS (2006) Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol 33(5–6):533–540
Chery J (2016) RNA therapeutics: RNAi and antisense mechanisms and clinical applications. Postdoc J 4(7):35–50
Roehr B (1998) Fomivirsen approved for CMV retinitis. J Int Assoc Phys AIDS Care 4(10):14–16
Kurreck J (2003) Antisense technologies. Improvement through novel chemical modifications. Eur J Biochem 270(8):1628–1644
Rigo F, Seth P, Bennett C (2014) Antisense oligonucleotide-based therapies for diseases caused by pre-mRNA processing defects. In: Yeo G (ed) Systems biology of RNA binding proteins, advances in experimental medicine and biology. Springer, New York, pp 303–352
Monia BP et al (1993) Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J Biol Chem 268(19):14514–14522
Evers MM, Toonen LJ, van Roon-Mom WM (2015) Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv Drug Deliv Rev 87:90–103
Geary RS et al (2015) Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev 87:46–51
Aartsma-Rus A (2012) Overview on AON design. Methods Mol Biol 867:117–129
Vitravene Study G (2002) A randomized controlled clinical trial of intravitreous fomivirsen for treatment of newly diagnosed peripheral cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol 133(4):467–474
Vitravene Study G (2002) Randomized dose-comparison studies of intravitreous fomivirsen for treatment of cytomegalovirus retinitis that has reactivated or is persistently active despite other therapies in patients with AIDS. Am J Ophthalmol 133(4):475–483
Vitravene Study G (2002) Safety of intravitreous fomivirsen for treatment of cytomegalovirus retinitis in patients with AIDS. Am J Ophthalmol 133(4):484–498
Duell PB et al (2016) Long-term mipomersen treatment is associated with a reduction in cardiovascular events in patients with familial hypercholesterolemia. J Clin Lipidol 10(4):1011–1021
Raal FJ et al (2010) Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 375(9719):998–1006
Raal FJ et al (2016) Pediatric experience with mipomersen as adjunctive therapy for homozygous familial hypercholesterolemia. J Clin Lipidol 10(4):860–869
Santos RD et al (2015) Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arterioscler Thromb Vasc Biol 35(3):689–699
Chiriboga CA et al (2016) Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 86(10):890–897
Finkel RS et al (2016) Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388(10063):3017–3026
Finkel RS et al (2017) Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med 377(18):1723–1732
Mercuri E et al (2018) Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 378(7):625–635
Cirak S et al (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378(9791):595–605
Mendell JR et al (2013) Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 74(5):637–647
Miner P et al (2004) An enema formulation of alicaforsen, an antisense inhibitor of intercellular adhesion molecule-1, in the treatment of chronic, unremitting pouchitis. Aliment Pharmacol Ther 19(3):281–286
Monteleone G et al (2015) Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N Engl J Med 372(12):1104–1113
Ackermann EJ et al (2016) Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid 23(3):148–157
Digenio A et al (2016) Antisense-mediated lowering of plasma apolipoprotein C-III by Volanesorsen improves dyslipidemia and insulin sensitivity in type 2 diabetes. Diabetes Care 39(8):1408–1415
Gaudet D et al (2017) The APPROACH study: a randomized, double-blind, placebo-controlled, phase 3 study of volanesorsen administered subcutaneously to patients with familial chylomicronemia syndrome (FCS). J Clin Lipidol 11:814–815
Gaudet D et al (2014) Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 371(23):2200–2206
Gaudet D et al (2015) Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N Engl J Med 373(5):438–447
Buller HR et al (2015) Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med 372(3):232–240
Bhanot S et al (2013) ISIS-GCCRRX, a novel glucocorticoid (GC) receptor antisense drug reduces cholesterol and triglycerides and attenuates dexamethasone induced hepatic insulin resistance without systemic GC antagonism in normal subjects. Diabetologia 56:S282
Morgan E, et al (2014) ISIS-GCGRRx, an antisense glucagon receptor antagonist, caused rapid, robust, and sustained improvements in glycemic control without changes in BW, BP, lipids, or hypoglycemia in T2DM patients on stable metformin therapy. Diabetes 56(11):2183–2195
Chi KN et al (2016) A phase I dose-escalation study of apatorsen (OGX-427), an antisense inhibitor targeting heat shock protein 27 (Hsp27), in patients with castration-resistant prostate cancer and other advanced cancers. Ann Oncol 27(6):1116–1122
Limmroth V et al (2014) CD49d antisense drug ATL1102 reduces disease activity in patients with relapsing-remitting MS. Neurology 83(20):1780–1788
Hong D et al (2015) AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci Transl Med 7(314):314ra185
Chowdhury S et al (2016) A phase I dose escalation, safety and pharmacokinetic (PK) study of AZD5312 (IONISARRx), a first-in-class Generation 2.5 antisense oligonucleotide targeting the androgen receptor (AR). Eur J Cancer 69:S145
Janssen HL et al (2013) Treatment of HCV infection by targeting microRNA. N Engl J Med 368(18):1685–1694
van der Ree MH et al (2014) Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antivir Res 111:53–59
Viney NJ et al (2016) Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 388(10057):2239–2253
Graham MJ et al (2017) Cardiovascular and metabolic effects of ANGPTL3 antisense oligonucleotides. N Engl J Med 377(3):222–232
Sermet-Gaudelus I et al (2018) Antisense oligonucleotide eluforsen improves CFTR function in F508del cystic fibrosis. J Cyst Fibros S1569–1993(18)30914–7
Querfeld C et al (2017) Ph 1 trial evaluating MRG-106, a microrna-155 inhibitor, administered by intratumoral, subcutaneous, or intravenous delivery in cutaneous T-cell lymphoma (CTCL) patients. Hematol Oncol 35:275
Pfeiffer N et al (2017) First-in-human phase I study of ISTH0036, an antisense oligonucleotide selectively targeting transforming growth factor beta 2 (TGF-β2), in subjects with open-angle glaucoma undergoing glaucoma filtration surgery. PLoS One 12(11):e0188899
McCaleb M et al (2017) Systemic pharmacodynamic efficacy of a complement factor B antisense oligonucleotide in preclinical and phase 1 clinical studies. Invest Ophthalmol Vis Sci 58:1952
Goemans NM et al (2016) Long-term efficacy, safety, and pharmacokinetics of drisapersen in Duchenne muscular dystrophy: results from an open-label extension study. PLoS One 11(9):e0161955
Voit T et al (2014) Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol 13(10):987–996
Saad F et al (2011) Randomized phase II trial of Custirsen (OGX-011) in combination with docetaxel or mitoxantrone as second-line therapy in patients with metastatic castrate-resistant prostate cancer progressing after first-line docetaxel: CUOG trial P-06c. Clin Cancer Res 17(17):5765–5773
Tsimikas S et al (2015) Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet 386(10002):1472–1483
van Meer L et al (2016) Renal effects of antisense-mediated inhibition of SGLT2. J Pharmacol Exp Ther 359(2):280–289
Miller TM et al (2013) An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol 12(5):435–442
Sewell KL et al (2002) Phase I trial of ISIS 104838, a 2′-methoxyethyl modified antisense oligonucleotide targeting tumor necrosis factor-alpha. J Pharmacol Exp Ther 303(3):1334–1343
Mignon L et al (2016) ISIS-DMPKRx in healthy volunteers: a placebo-controlled, randomized, single ascending-dose phase 1 Study. Neurology 86(16 Supplement):P3.166
Bianchini D et al (2013) First-in-human Phase I study of EZN-4176, a locked nucleic acid antisense oligonucleotide to exon 4 of the androgen receptor mRNA in patients with castration-resistant prostate cancer. Br J Cancer 109(10):2579–2586
Toth PP (2011) Antisense therapy and emerging applications for the management of dyslipidemia. J Clin Lipidol 5(6):441–449
Visser ME et al (2012) Mipomersen, an apolipoprotein B synthesis inhibitor, lowers low-density lipoprotein cholesterol in high-risk statin-intolerant patients: a randomized, double-blind, placebo-controlled trial. Eur Heart J 33(9):1142–1149
Nguyen DD, Chang S (2017) Development of novel therapeutic agents by inhibition of oncogenic microRNAs. Int J Mol Sci 19(1):E65
van der Ree MH et al (2016) Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment Pharmacol Ther 43(1):102–113
Hua Y et al (2010) Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev 24(15):1634–1644
Lefebvre S et al (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16(3):265–269
Hua Y et al (2008) Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet 82(4):834–848
De Vivo DC et al (2017) Interim efficacy and safety results from the phase 2 NURTURE study evaluating nusinersen in presymptomatic infants with spinal muscular atrophy. Neurology 88:S46.003.75
Mendell JR et al (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 71(3):304–313
Emery AE (1991) Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul Disord 1(1):19–29
Ervasti JM (2007) Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta 1772(2):108–117
Kole R, Krieg AM (2015) Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev 87:104–107
Lim KR, Maruyama R, Yokota T (2017) Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther 11:533–545
Mendell JR et al (2016) Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 79(2):257–271
Kinane TB et al (2018) Long-term pulmonary function in Duchenne muscular dystrophy: comparison of Eteplirsen-treated patients to natural history. J Neuromuscul Dis 5(1):47–58
Garanto A et al (2016) In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet 25(12):2552–2563
Denti MA et al (2006) Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc Natl Acad Sci USA 103(10):3758–3763
Denti MA et al (2008) Long-term benefit of adeno-associated virus/antisense-mediated exon skipping in dystrophic mice. Hum Gene Ther 19(6):601–608
Goyenvalle A et al (2009) Enhanced exon-skipping induced by U7 snRNA carrying a splicing silencer sequence: promising tool for DMD therapy. Mol Ther 17(7):1234–1240
Goyenvalle A et al (2012) Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol Ther 20(6):1212–1221
Vulin A et al (2012) Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol Ther 20(11):2120–2133
Geib T, Hertel KJ (2009) Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS One 4(12):e8204
Odermatt P et al (2016) Somatic therapy of a mouse SMA model with a U7 snRNA gene correcting SMN2 splicing. Mol Ther 24(10):1797–1805
Imbert M, Dias-Florencio G, Goyenvalle A (2017) Viral vector-mediated antisense therapy for genetic diseases. Genes (Basel) 8(2):E51
Krause DS, Van Etten RA (2005) Tyrosine kinases as targets for cancer therapy. N Engl J Med 353(2):172–187
Cornelio DB, Roesler R, Schwartsmann G (2007) Gastrin-releasing peptide receptor as a molecular target in experimental anticancer therapy. Ann Oncol 18(9):1457–1466
Low PS, Kularatne SA (2009) Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol 13(3):256–262
McGettrick AF, O’Neill LA (2010) Localisation and trafficking of Toll-like receptors: an important mode of regulation. Curr Opin Immunol 22(1):20–27
Millard M, Odde S, Neamati N (2011) Integrin targeted therapeutics. Theranostics 1:154–188
Canton J, Neculai D, Grinstein S (2013) Scavenger receptors in homeostasis and immunity. Nat Rev Immunol 13(9):621–634
D’Souza AA, Devarajan PV (2015) Asialoglycoprotein receptor mediated hepatocyte targeting – strategies and applications. J Control Release 203:126–139
Juliano RL (2016) The delivery of therapeutic oligonucleotides. Nucleic Acids Res 44(14):6518–6548
McClorey G, Banerjee S (2018) Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics. Biomedicine 6(2):E51
Yin H et al (2008) Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function. Hum Mol Genet 17(24):3909–3918
Yin H et al (2009) A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum Mol Genet 28(4):699
Wu B et al (2008) Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc Natl Acad Sci USA 105(39):14814–14819
Betts C et al (2012) Pip6-PMO, a new generation of peptide-oligonucleotide conjugates with improved cardiac exon skipping activity for DMD treatment. Mol Ther Nucleic Acids 1:e38
Gao X et al (2014) Effective dystrophin restoration by a novel muscle-homing peptide-morpholino conjugate in dystrophin-deficient mdx mice. Mol Ther 22(7):1333–1341
Jirka SMG et al (2014) Peptide conjugation of 20-O-methyl phosphorothioate antisense oligonucleotides enhances cardiac uptake and exon skipping in mdx mice. Nucleic Acid Ther 24:25–36
Leger AJ et al (2013) Systemic delivery of a Peptide-linked morpholino oligonucleotide neutralizes mutant RNA toxicity in a mouse model of myotonic dystrophy. Nucleic Acid Ther 23(2):109–117
Hammond SM et al (2016) Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci USA 113(39):10962–10967
Shabanpoor F et al (2017) Identification of a peptide for systemic brain delivery of a morpholino oligonucleotide in mouse models of spinal muscular atrophy. Nucleic Acid Ther 27(3):130–143
Lönn P et al (2016) Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci Rep 6:32301
Salerno JC et al (2016) Novel cell-penetrating peptide-adaptors effect intracellular delivery and endosomal escape of protein cargos. J Cell Sci 129(12):2473–2474
Falzarano MS, Passarelli C, Ferlini A (2014) Nanoparticle delivery of antisense oligonucleotides and their application in the exon skipping strategy for Duchenne muscular dystrophy. Nucleic Acid Ther 24(1):87–100
Lerner MR et al (1980) Are snRNPs involved in splicing? Nature 283(5743):220–224
Rogers J, Wall R (1980) A mechanism for RNA splicing. Proc Natl Acad Sci USA 77(4):1877–1879
Mount SM et al (1983) The U1 small nuclear RNA-protein complex selectively binds a 5′ splice site in vitro. Cell 33(2):509–518
West S (2012) The increasing functional repertoire of U1 snRNA. Biochem Soc Trans 40(4):846–849
Buratti E, Baralle D (2010) Novel roles of U1 snRNP in alternative splicing regulation. RNA Biol 7(4):412–419
Roca X, Krainer AR, Eperon IC (2013) Pick one, but be quick: 5′ splice sites and the problems of too many choices. Genes Dev 27(2):129–144
Raponi M, Baralle D (2008) Can donor splice site recognition occur without the involvement of U1 snRNP? Biochem Soc Trans 36.(Pt 3:548–550
Guiro J, O’Reilly D (2015) Insights into the U1 small nuclear ribonucleoprotein complex superfamily. Wiley Interdiscip Rev RNA 6(1):79–92
Spraggon L, Cartegni L (2013) U1 snRNP-dependent suppression of polyadenylation: physiological role and therapeutic opportunities in Cancer. Int J Cell Biol 2013:846510
Valadkhan S, Gunawardane LS (2013) Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem 54:79–90
Buratti E et al (2007) Aberrant 5′ splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res 35(13):4250–4263
Roca X et al (2008) Features of 5′-splice-site efficiency derived from disease-causing mutations and comparative genomics. Genome Res 18(1):77–87
Zhuang Y, Weiner AM (1986) A compensatory base change in U1 snRNA suppresses a 5′ splice site mutation. Cell 46(6):827–835
Baralle M et al (2003) Identification of a mutation that perturbs NF1 agene splicing using genomic DNA samples and a minigene assay. J Med Genet 40(3):220–222
Pinotti M et al (2008) U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency. Blood 111(5):2681–2684
Pinotti M et al (2009) Rescue of coagulation factor VII function by the U1+5A snRNA. Blood 113(25):6461–6464
Tanner G et al (2009) Therapeutic strategy to rescue mutation-induced exon skipping in rhodopsin by adaptation of U1 snRNA. Hum Mutat 30(2):255–263
Glaus E et al (2011) Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol Ther 19(5):936–941
Sánchez-Alcudia R et al (2011) Overexpression of adapted U1snRNA in patients’ cells to correct a 5′ splice site mutation in propionic acidemia. Mol Genet Metab 102(2):134–138
Martínez-Pizarro A et al (2018) Intronic PAH gene mutations cause a splicing defect by a novel mechanism involving U1snRNP binding downstream of the 5′ splice site. Plos Genet 14:e1007360
Schmid F et al (2011) U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum Mutat 32(7):815–824
Schmid F et al (2013) A gene therapeutic approach to correct splice defects with modified U1 and U6 snRNPs. Hum Gene Ther 24(1):97–104
Hartmann L et al (2010) Correct mRNA processing at a mutant TT splice donor in FANCC ameliorates the clinical phenotype in patients and is enhanced by delivery of suppressor U1 snRNAs. Am J Hum Genet 87(4):480–493
Matos L et al (2014) Therapeutic strategies based on modified U1 snRNAs and chaperones for Sanfilippo C splicing mutations. Orphanet J Rare Dis 9:180
Scalet D et al (2019) Disease-causing variants of the conserved +2T of 5′ splice sites can be rescued by engineered U1snRNAs. Hum Mutat 40(1):48–52
Balestra D et al (2014) An engineered U1 small nuclear RNA rescues splicing defective coagulation F7 gene expression in mice. J Thromb Haemost 12(2):177–185
Lee NC et al (2016) Mutation-adapted U1 snRNA corrects a splicing error of the dopa decarboxylase gene. Hum Mol Genet 25(23):5142–5147
Pinotti M et al (2011) RNA-based therapeutic approaches for coagulation factor deficiencies. J Thromb Haemost 9(11):2143–2152
Wally V, Murauer EM, Bauer JW (2012) Spliceosome-mediated trans-splicing: the therapeutic cut and paste. J Invest Dermatol 132(8):1959–1966
Roca X, Krainer AR (2009) Recognition of atypical 5′ splice sites by shifted base-pairing to U1 snRNA. Nat Struct Mol Biol 16(2):176–182
Cohen JB et al (1994) Suppression of mammalian 5′ splice-site defects by U1 small nuclear RNAs from a distance. Proc Natl Acad Sci USA 91(22):10470–10474
Hwang DY, Cohen JB (1997) U1 small nuclear RNA-promoted exon selection requires a minimal distance between the position of U1 binding and the 3′ splice site across the exon. Mol Cell Biol 17(12):7099–7107
Fernandez Alanis E et al (2012) An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet 21(11):2389–2398
Tajnik M et al (2016) Molecular basis and therapeutic strategies to rescue factor IX variants that affect splicing and protein function. PLoS Genet 12(5):e1006082
Dal Mas A et al (2015) Exon-specific U1s correct SPINK5 exon 11 skipping caused by a synonymous substitution that affects a bifunctional splicing regulatory element. Hum Mutat 36(5):504–512
Nizzardo M et al (2015) Spinal muscular atrophy phenotype is ameliorated in human motor neurons by SMN increase via different novel RNA therapeutic approaches. Sci Rep 5:p. 11746
Mattioli C et al (2014) Unusual splice site mutations disrupt FANCA exon 8 definition. Biochim Biophys Acta 1842(7):1052–1058
Dal Mas A et al (2015) Improvement of SMN2 pre-mRNA processing mediated by exon-specific U1 small nuclear RNA. Am J Hum Genet 96(1):93–103
Rogalska ME et al (2016) Therapeutic activity of modified U1 core spliceosomal particles. Nat Commun 7:11168
Donadon I et al (2018) Exon-specific U1 snRNAs improve ELP1 exon 20 definition and rescue ELP1 protein expression in a familial dysautonomia mouse model. Hum Mol Genet 27(14):2466–2476
Balestra D et al (2016) An exon-specific U1snRNA induces a robust factor IX activity in mice expressing multiple human FIX splicing mutants. Mol Ther Nucleic Acids 5(10):e370
van der Woerd WL et al (2015) Analysis of aberrant pre-messenger RNA splicing resulting from mutations in ATP8B1 and efficient in vitro rescue by adapted U1 small nuclear RNA. Hepatology 61(4):1382–1391
Hwu WL, Lee YM, Lee NC (2017) Gene therapy with modified U1 small nuclear RNA. Expert Rev Endocrinol Metab 12(3):171–175
Suñé-Pou M et al (2017) Targeting splicing in the treatment of human disease. Genes (Basel) 8(3):E87
Napoli C, Lemieux C, Jorgensen R (1990) Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell 2(4):279–289
van der Krol AR et al (1990) Flavonoid genes in petunia: addition of a limited number of gene copies may lead to a suppression of gene expression. Plant Cell 2(4):291–299
Fire A et al (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391(6669):806–811
Montgomery MK, Xu S, Fire A (1998) RNA as a target of double-stranded RNA-mediated genetic interference in Caenorhabditis elegans. Proc Natl Acad Sci USA 95(26):15502–15507
Kennerdell JR, Carthew RW (1998) Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell 95(7):1017–1026
Voinnet O (2001) RNA silencing as a plant immune system against viruses. Trends Genet 17(8):449–459
Ketting RF et al (1999) Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell 99(2):133–141
Wu-Scharf D et al (2000) Transgene and transposon silencing in Chlamydomonas reinhardtii by a DEAH-box RNA helicase. Science 290(5494):1159–1162
Hamilton A et al (2002) Two classes of short interfering RNA in RNA silencing. EMBO J 21(17):4671–4679
Hamilton AJ, Baulcombe DC (1999) A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 286(5441):950–952
Cogoni C et al (1996) Transgene silencing of the al-1 gene in vegetative cells of Neurospora is mediated by a cytoplasmic effector and does not depend on DNA-DNA interactions or DNA methylation. EMBO J 15(12):3153–3163
Cecere G, Cogoni C (2009) Quelling targets the rDNA locus and functions in rDNA copy number control. BMC Microbiol 9:44
Elbashir SM et al (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411(6836):494–498
Elbashir SM, Lendeckel W, Tuschl T (2001) RNA interference is mediated by 21- and 22-nucleotide RNAs. Genes Dev 15(2):188–200
Caplen NJ et al (2000) dsRNA-mediated gene silencing in cultured Drosophila cells: a tissue culture model for the analysis of RNA interference. Gene 252(1–2):95–105
Hutvagner G, Simard MJ (2008) Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol 9(1):22–32
Deng Y et al (2014) Therapeutic potentials of gene silencing by RNA interference: principles, challenges, and new strategies. Gene 538(2):217–227
Sandy P, Ventura A, Jacks T (2005) Mammalian RNAi: a practical guide. BioTechniques 39(2):215–224
Aagaard L, Rossi JJ (2007) RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev 59(2–3):75–86
Fraser AG et al (2000) Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408(6810):325–330
Lum L et al (2003) Identification of Hedgehog pathway components by RNAi in Drosophila cultured cells. Science 299(5615):2039–2045
Boutros M et al (2004) Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303(5659):832–835
Paddison PJ et al (2004) A resource for large-scale RNA-interference-based screens in mammals. Nature 428(6981):427–431
Silva JM et al (2008) Profiling essential genes in human mammary cells by multiplex RNAi screening. Science 319(5863):617–620
Schlabach MR et al (2008) Cancer proliferation gene discovery through functional genomics. Science 319(5863):620–624
Brass AL et al (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319(5865):921–926
Luo J et al (2009) A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 137(5):835–848
Mittal V (2004) Improving the efficiency of RNA interference in mammals. Nat Rev Genet 5(5):355–365
Hannon GJ, Rossi JJ (2004) Unlocking the potential of the human genome with RNA interference. Nature 431(7006):371–378
Rytlewski JA, Beronja S (2015) RNAi in the mouse: rapid and affordable gene function studies in a vertebrate system. Wiley Interdiscip Rev Dev Biol 4(1):45–57
Lieberman J (2018) Tapping the RNA world for therapeutics. Nat Struct Mol Biol 25(5):357–364
Hayden EC (2014) RNA interference rebooted. Nature 508(7497):443
Song E et al (2003) RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med 9(3):347–351
Wittrup A, Lieberman J (2015) Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet 16(9):543–552
Coelho T et al (2013) Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med 369(9):819–829
Suhr OB et al (2015) Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis 10:109
Demirjian S et al (2017) Safety and tolerability Study of an intravenously administered small interfering ribonucleic acid (siRNA) post on-pump cardiothoracic surgery in patients at risk of acute kidney injury. Kidney Int Rep 2(5):836–843
Peddi V, Ratner L, Cooper M, Gaber O, Feng S, Tso P, Bowers V, Naraghi R, Budde K, Polinsky M et al (2014) Treatment with QPI-1002, a short interfering (SI) RNA for the prophylaxis of delayed graft function. Transplantation 98:153
Benitez-Del-Castillo JM et al (2016) Safety and efficacy clinical trials for SYL1001, a novel short interfering RNA for the treatment of dry eye disease. Invest Ophthalmol Vis Sci 57(14):6447–6454
Antoszyk A, Katz B, Singh R, Gurses-Ozden R, Erlich S, Rothenstein D, Sharon N, Hodge J, Levin L, Miller N et al (2013) A phase I open label, dose escalation trial of QPI-1007 delivered by a single intravitreal (IVT) injection to subjects with low visual acuity and acute non-arteritic anterior ischemic optic neuropathy (NAION). Invest Ophthalmol Vis Sci 54(4575):4575
Solano EC et al (2014) Toxicological and pharmacokinetic properties of QPI-1007, a chemically modified synthetic siRNA targeting caspase 2 mRNA, following intravitreal injection. Nucleic Acid Ther 24(4):258–266
Pasi KJ et al (2016) Fitusiran, an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia: updated results from phase 1 and phase 2 extension studies in patients with inhibitors. Blood 128:1397
Ragni, et al (2016) Fitusiran, an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia: updated results from a phase 1 and phase 1/2 extension study in patients without inhibitors. Blood 128:2572
Fitzgerald K et al (2017) A highly durable RNAi therapeutic inhibitor of PCSK9. N Engl J Med 376(1):41–51
Ray KK et al (2017) Inclisiran in patients at high cardiovascular risk with elevated LDL cholesterol. N Engl J Med 376(15):1430–1440
Drugs.com. Alnylam announces FDA acceptance of New Drug Application (NDA) and priority review status for patisiran, an investigational RNAi therapeutic for the treatment of hereditary ATTR (hATTR) amyloidosis. 2018 10 Aug 2018. Available from: https://www.drugs.com/nda/patisiran_180201.html
Golan T et al (2015) RNAi therapy targeting KRAS in combination with chemotherapy for locally advanced pancreatic cancer patients. Oncotarget 6(27):24560–24570
Zorde Khvalevsky E et al (2013) Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci USA 110(51):20723–20728
Gonzalez, et al (2014) Phase 2 of bamosiran (SYL040012), a novel RNAi based compound for the treatment of increased intraocular pressure associated to glaucoma. Invest Ophthalmol Vis Sci 55(13):564
Moreno-Montanes J et al (2014) Phase I clinical trial of SYL040012, a small interfering RNA targeting beta-adrenergic receptor 2, for lowering intraocular pressure. Mol Ther 22(1):226–232
Demuere et al (2016) A phase I/II study of TKM-080301, a PLK1-targeted RNAi in patients with adrenocortical cancer (ACC). J Clin Oncol 34:2547
Libertine, et al. (2015) Update on phase 2 clinical trial results of RXI-109 treatment to reduce the formation of hypertrophic dermal scars. J Am Acad Dermatol 72:AB273
Nguyen QD et al (2012) Dose-ranging evaluation of intravitreal siRNA PF-04523655 for diabetic macular edema (the DEGAS study). Invest Ophthalmol Vis Sci 53(12):7666–7674
Hill, et al (2016) A subcutaneously administered investigational RNAi Therapeutic (ALN-CC5) targeting complement C5 for treatment of PNH and complement-mediated diseases: preliminary phase 1/2 study results in patients with PNH. Blood 128:3891
Hulton, et al. (2016) A phase 1/2 trial of ALN-GO1, an investigational RNAi therapeutic for primary hyperoxaluria type 1 (PH1). Pediatr Nephrol 31:1763
Sterinu-Cercel et al (2017) A phase 2a study evaluating the multi-dose activity of ARB-1467 in HBeAg-Positive and –negative virally suppressed subjects with hepatitis B. J Hepatol p 66:S688–S689
Lawitz E et al (2015) Safety, pharmacokinetics, and biologic activity of ND-L02-s0201, a novel targeted lipid-nanoparticle to deliver HSP47 siRNA for the treatment of patients with advanced liver fibrosis: interim results from clinical phase 1b/2 studies. S Hepatology 62(909A):S688–S689
Salzberg et al (2016) Adoptive cellular immunotherapy with APN401, autologous Cbl-b-silenced peripheral blood mononuclear cells, in patients with solid tumors. J Clin Oncol 34:e14541
Triozzi, et al. (2015) Phase I clinical trial of adoptive cellular immunotherapy with APN401 in patients with solid tumors. Immun Cancer 3(Suppl 2):175
Alsina M et al (2012) Open-label extension study of the RNAi therapeutic ALN-VSP02 in cancer patients responding to therapy. J Clin Oncol 30(4):3062
Cervantes et al (2011) Phase I dose escalation study of ALN-VSP02, a novel RNAi therapeutic for solid tumors with liver involvement. J Clin Oncol 29:3025
Fitzgerald K et al (2014) Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet 383(9911):60–68
Leachman SA et al (2010) First-in-human mutation-targeted siRNA phase Ib trial of an inherited skin disorder. Mol Ther 18(2):442–446
Gillmore, et al (2016) Phase 2 open-label extension study of revusiran, an investigational RNAi therapeutic for the treatment of patients with transthyretin amyloidosis with cardiomyopathy: updated interim results. 2016: Abstract book, international symposium of amyloidosis, p PA84
Zimmermann TS et al (2017) Clinical proof of concept for a novel hepatocyte-targeting GalNAc-siRNA conjugate. Mol Ther 25(1):71–78
Xu CF et al (2015) Targeting glucose uptake of glioma cells by siRNA delivery with polymer nanoparticle. J Control Release 213:e23–e24
Zuckerman JE et al (2014) Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc Natl Acad Sci USA 111(31):11449–11454
Yuen et al (2017) Prolonged RNA interference therapy with ARC-520 Injection in treatment naı¨ve, HBeAg positive and negative patients with chronic HBV results in significant reductions of HBs antigen. J Hepatol 66:S27
Turner, et al. (2017) Hepatic targeted RNA interference provides deep and prolonged knockdown of alpha-1 antitrypsin levels in ZZ patients. J. Hepatol 66:S92
Drugs.com. Alnylam presents new clinical results from the APOLLO phase 3 study of patisiran at the 16th international symposium on amyloidosis. 2018 10 Aug 2018. Available from: https://www.drugs.com/clinical_trials/alnylam-presents-new-clinical-results-apollo-phase-3-study-patisiran-16th-international-symposium-17805.html
Drugs.com (2018) Available from: https://www.drugs.com/history/onpattro.html
Bobbin ML, Rossi JJ (2016) RNA interference (RNAi)-based therapeutics: delivering on the promise? Annu Rev Pharmacol Toxicol 56:103–122
Gilleron J et al (2013) Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol 31(7):638–646
Wittrup A et al (2015) Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat Biotechnol 33(8):870–876
Bartlett DW, Davis ME (2006) Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res 34(1):322–333
Tatiparti K et al (2017) siRNA delivery strategies: a comprehensive review of recent developments. Nanomaterials (Basel) 7(4):322–33
Han L, Tang C, Yin C (2014) Oral delivery of shRNA and siRNA via multifunctional polymeric nanoparticles for synergistic cancer therapy. Biomaterials 35(15):4589–4600
Akinc A et al (2008) A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat Biotechnol 26(5):561–569
Semple SC et al (2010) Rational design of cationic lipids for siRNA delivery. Nat Biotechnol 28(2):172–176
Shi B et al (2011) Biodistribution of small interfering RNA at the organ and cellular levels after lipid nanoparticle-mediated delivery. J Histochem Cytochem 59(8):727–740
Morrissey DV et al (2005) Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology 41(6):1349–1356
Gooding M et al (2012) siRNA delivery: from lipids to cell-penetrating peptides and their mimics. Chem Biol Drug Des 80(6):787–809
Akinc A et al (2010) Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther 18(7):1357–1364
Czech MP, Aouadi M, Tesz GJ (2011) RNAi-based therapeutic strategies for metabolic disease. Nat Rev Endocrinol 7(8):473–484
Robbins M, Judge A, MacLachlan I (2009) siRNA and innate immunity. Oligonucleotides 19(2):89–102
Chiu YL, Rana TM (2003) siRNA function in RNAi: a chemical modification analysis. RNA 9(9):1034–1048
Morrissey DV et al (2005) Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat Biotechnol 23(8):1002–1007
Judge AD et al (2006) Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol Ther 13(3):494–505
Judge AD et al (2009) Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J Clin Invest 119(3):661–673
Jackson AL et al (2006) Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 12(7):1197–1205
Kenski DM et al (2012) siRNA-optimized modifications for enhanced in vivo activity. Mol Ther Nucleic Acids 1:e5
Tai W, Gao X (2017) Functional peptides for siRNA delivery. Adv Drug Deliv Rev 110-111:157–168
Ha D, Yang N, Nadithe V (2016) Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B 6(4):287–296
Boado RJ (2005) RNA interference and nonviral targeted gene therapy of experimental brain cancer. NeuroRx 2(1):139–150
Rossi JJ (2006) RNAi therapeutics: SNALPing siRNAs in vivo. Gene Ther 13(7):583–584
Li Z, Loh XJ (2016) Recent advances of using polyhydroxyalkanoate-based nanovehicles as therapeutic delivery carriers. Nanomed Nanobiotechnol 9(3):e1429
Coutinho MF et al (2016) Genetic substrate reduction therapy: a promising approach for lysosomal storage disorders. Diseases 4(4):1429
Buduru S et al (2018) RNA interference: new mechanistic and biochemical insights with application in oral cancer therapy. Int J Nanomedicine 13:3397–3409
Song E et al (2005) Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol 23(6):709–717
Peer D et al (2007) Selective gene silencing in activated leukocytes by targeting siRNAs to the integrin lymphocyte function-associated antigen-1. Proc Natl Acad Sci USA 104(10):4095–4100
Peer D et al (2008) Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science 319(5863):627–630
McNamara JO 2nd et al (2006) Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat Biotechnol 24(8):1005–1015
Berezhnoy A et al (2014) Aptamer-targeted inhibition of mTOR in T cells enhances antitumor immunity. J Clin Invest 124(1):188–197
Wheeler LA et al (2011) Inhibition of HIV transmission in human cervicovaginal explants and humanized mice using CD4 aptamer-siRNA chimeras. J Clin Invest 121(6):2401–2412
Soutschek J et al (2004) Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432(7014):173–178
Kortylewski M et al (2009) In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol 27(10):925–932
Davis ME et al (2010) Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 464(7291):1067–1070
Wong SC et al (2012) Co-injection of a targeted, reversibly masked endosomolytic polymer dramatically improves the efficacy of cholesterol-conjugated small interfering RNAs in vivo. Nucleic Acid Ther 22(6):380–390
Nair JK et al (2014) Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc 136(49):16958–16961
Manoharan M(2014) GalNAc-siRNA with enhanced stabilization chemistry: ESC-GalNAc-siRNA. Available from: http://www.alnylam.com/web/assets/ALNY-ESC-GalNAc-siRNA-TIDES-May2014-Capella.pdf
Roberts TC et al (2016) Synthetic SiRNA delivery: progress and prospects. Methods Mol Biol 1364:291–310
Bruun J et al (2015) Investigation of enzyme-sensitive lipid nanoparticles for delivery of siRNA to blood-brain barrier and glioma cells. Int J Nanomedicine 10:5995–6008
Serramia MJ et al (2015) In vivo delivery of siRNA to the brain by carbosilane dendrimer. J Control Release 200:60–70
Bruck J et al (2015) Cholesterol modification of p40-specific small interfering RNA enables therapeutic targeting of dendritic cells. J Immunol 195(5):2216–2223
Haroon MM et al (2016) A designed recombinant fusion protein for targeted delivery of siRNA to the mouse brain. J Control Release 228:120–131
Gabathuler R (2012) New protein vectors for physiological transfer of therapeutic agents to the central nervous system. Biol Aujourdhui 206(3):191–203
Gabathuler R (2015) Using a peptide derived from transcend (mtf, p97) to deliver biologics to the cns using a physiologic pathway. Available from: http://www.brains4brain.eu/wp-content/uploads/2015/01/9th-B4B-Workshop-Scientific-Programme.pdf
Unniyampurath U, Pilankatta R, Krishnan MN (2016) RNA interference in the age of CRISPR: will CRISPR interfere with RNAi? Int J Mol Sci 17(3):291
Barrangou R et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315(5819):1709–1712
Sorek R, Kunin V, Hugenholtz P (2008) CRISPR–a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat Rev Microbiol 6(3):181–186
Grissa I, Vergnaud G, Pourcel C (2007) The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8:172
Mali P et al (2013) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826
Cong L et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Cornu TI, Mussolino C, Cathomen T (2017) Refining strategies to translate genome editing to the clinic. Nat Med 23(4):415–423
Yin H et al (2014) Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 32(6):551–553
Nelson CE et al (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351(6271):403–407
Tabebordbar M et al (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351(6271):407–411
Zetsche B et al (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163(3):759–771
Sahin U, Kariko K, Tureci O (2014) mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov 13(10):759–780
Stanton MG (2018) Current status of messenger RNA delivery systems. Nucleic Acid Ther 28(3):158–165
Pardi N et al (2018) mRNA vaccines – a new era in vaccinology. Nat Rev Drug Discov 17(4):261–279
Heiser A et al (2002) Autologous dendritic cells transfected with prostate-specific antigen RNA stimulate CTL responses against metastatic prostate tumors. J Clin Invest 109(3):409–417
Morse MA et al (2002) The feasibility and safety of immunotherapy with dendritic cells loaded with CEA mRNA following neoadjuvant chemoradiotherapy and resection of pancreatic cancer. Int J Gastrointest Cancer 32(1):1–6
Morse MA et al (2003) Immunotherapy with autologous, human dendritic cells transfected with carcinoembryonic antigen mRNA. Cancer Investig 21(3):341–349
Weide B et al (2009) Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunother 32(5):498–507
Rittig SM et al (2011) Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol Ther 19(5):990–999
Kübler, et al. (2011) Final analysis of a phase I/IIa study with CV9103, an intradermally administered prostate cancer immunotherapy based on self-adjuvanted mRNA. J Clin Oncol 29:4535
Sebastian M et al (2012) Messenger RNA vaccination and B cell responses in NSCLC patients. J Clin Oncol 30(15_suppl):2573–2573
Wilgenhof S et al (2013) A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol 24(10):2686–2693
Devoldere J et al (2016) Evading innate immunity in nonviral mRNA delivery: don’t shoot the messenger. Drug Discov Today 21(1):11–25
Granot Y, Peer D (2017) Delivering the right message: challenges and opportunities in lipid nanoparticles-mediated modified mRNA therapeutics-An innate immune system standpoint. Semin Immunol 34:68–77
Zangi L et al (2013) Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat Biotechnol 31(10):898–907
Baba M et al (2015) Treatment of neurological disorders by introducing mRNA in vivo using polyplex nanomicelles. J Control Release 201:41–48
Matsui A et al (2015) Messenger RNA-based therapeutics for the treatment of apoptosis-associated diseases. Sci Rep 5:15810
DeRosa F et al (2016) Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther 23(10):699–707
Ramaswamy S et al (2017) Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci USA 114(10):E1941–E1950
Kormann MS et al (2011) Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol 29(2):154–157
Wang Y et al (2013) Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol Ther 21(2):358–367
Schrom E et al (2017) Translation of angiotensin-converting enzyme 2 upon liver- and lung-targeted delivery of optimized chemically modified mRNA. Mol Ther Nucleic Acids 7:350–365
An D et al (2017) Systemic messenger RNA therapy as a treatment for methylmalonic acidemia. Cell Rep 21(12):3548–3558
Patel S et al (2017) Boosting intracellular delivery of lipid nanoparticle-encapsulated mRNA. Nano Lett 17(9):5711–5718
Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346(6287):818–822
Tuerk C, Gold L (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249(4968):505–510
Jayasena SD (1999) Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin Chem 45(9):1628–1650
Yu Y et al (2016) Molecular selection, modification and development of therapeutic oligonucleotide aptamers. Int J Mol Sci 17(3):358
Cruz-Toledo J et al (2012) Aptamer Base: a collaborative knowledge base to describe aptamers and SELEX experiments. Database (Oxford) 2012:bas006
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Coutinho, M.F., Matos, L., Santos, J.I., Alves, S. (2019). RNA Therapeutics: How Far Have We Gone?. In: Romão, L. (eds) The mRNA Metabolism in Human Disease. Advances in Experimental Medicine and Biology, vol 1157. Springer, Cham. https://doi.org/10.1007/978-3-030-19966-1_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-19966-1_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-19965-4
Online ISBN: 978-3-030-19966-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)