
Abstract
Innate immune cells and mediators play important roles in recognizing molecular patterns of potential pathogens and unlike adaptive immune cells, the innate immune response is not antigen-specific. However, innate immune cells are critical for the early control of infection and act rapidly to alert the immune system and initiate adaptive immunity. Macrophages and neutrophils quickly respond to infectious pathogens, mediating their destruction via phagocytosis, and producing cytokines that recruit and activate immune cells. Dendritic cells play a vital role in innate and adaptive crosstalk to initiate the adaptive immune response to fight pathogens. Natural killer cells play a critical role in the control of viral infections and cancers and the modulation of macrophages and dendritic cells. Mast cells release proinflammatory mediators that induce vascular permeability and promote leukocyte recruitment. Lastly, both mast cells and granulocytes such as eosinophils and basophils defend against parasites and participate in the modulation of immune responses. While these innate immune cells and mediators are crucial in the early response to infection, the inappropriate response of the innate immune system, however, can play a role in the pathophysiology of conditions in which chronic inflammation contributes to disease symptoms and progression. In this chapter, the normal physiological role of the innate immune system and its components are discussed. Many of these mechanisms can be targeted by several drug classes which aim to suppress cytokines as well as block leukocyte recruitment in disease states in which the immune system may overreact to target self-antigens (i.e. autoimmunity).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Complement system
- Toll-like receptors
- Eosinophils
- Macrophages
- Monocytes
- Basophils
- Natural killer cells
- Neutrophils
- Acute-phase proteins
- Cytokines
- Dendritic cells
-
1.
Identify the mechanisms by which components of the innate immune system protect the body from infection.
-
2.
Discuss the events involved in the inflammatory response.
-
3.
Identify innate immune receptors and cytokines.
-
4.
Recognize pathogen-associated molecular patterns (PAMPs) and predict how the immune system will recognize them.
-
5.
Recognize the biological structure of the TNF-α inhibitors.
-
6.
Discuss the pharmacology of the TNF-α, IL-1, IL-6, and IL-12 inhibitors, including mechanism of action, clinical uses, and adverse effects.
-
7.
Discuss the pharmacology of the integrin inhibitors, including mechanism of action, clinical uses, and adverse effects.
-
8.
Discuss the pharmacology of interferon therapies including mechanism of action, clinical uses, and adverse effects.
Drugs discussed in this chapter
Drug | Classification |
|---|---|
Adalimumab (Humira®) | Anti-TNF-α monoclonal antibody |
Anakinra (Kineret®) | Recombinant IL-1R antagonist |
Canakinumab (Ilaris®) | Anti-IL-1β antibody |
Certolizumab pegol (Cimzia®) | Anti-TNF-α antibody fragment |
Etanercept (Enbrel®) | Anti-TNF-α fusion protein |
Golimumab (Simponi®) | Anti-TNF-α monoclonal antibody |
Infliximab (Remicade®) | Anti-TNF-α monoclonal antibody |
Interferon alfa-2a, 2b (Pegasys®, Pegintron®) | Interferon |
Interferon beta-1a (Avonex®) | Interferon |
Natalizumab (Tysabri®) | Anti-α4β1 integrin monoclonal antibody |
Rilonacept (Arcalyst®) | Anti-IL-1 fusion protein |
Sarilumab (Kevzara®) | Anti-IL-6 receptor monoclonal antibody |
Siltuximab (Sylvant®) | Anti-IL-6 monoclonal antibody |
Tocilizumab (Actemra®) | Anti-IL-6 receptor monoclonal antibody |
Ustekinumab (Stelara®) | Anti-IL-12/23 monoclonal antibody |
Vedolizumab (Entyvio®) | Anti-α4β7 integrin monoclonal antibody |
Introduction
As the immune system’s first defense against foreign invaders, innate immune cells and mediators play important roles in keeping the body free from infection. Because innate immune cells and their receptors are only equipped to recognize molecular patterns of potential pathogens and not specific antigens, their job is said to be non-specific. In some instances, however, the inappropriate response of the innate immune system can play a role in the pathophysiology of conditions in which chronic inflammation contributes to disease symptoms and progression. Here, we will discuss and outline the normal physiological role of the innate immune system and its components, and then highlight these as potential drug targets in disease states in which the immune system may be over-activated (i.e. autoimmunity).
First Barriers to Infection
Non-cellular Defenses
The body’s first lines of defense against infection by foreign invaders includes its physical barriers. These barriers include the skin and epithelia of the digestive, respiratory, and urogenital tracts—all areas of the body that are able to have first contact with the outside environment. Some of these barriers are colonized with the body’s microbiome, or microorganisms that have a commensalistic relationship with the host, creating a type of microbial antagonism that makes it difficult for disease-causing pathogens to penetrate and cause infection. Infants begin to be colonized with commensal microorganisms at birth. Besides these microbiological barriers, mechanical and chemical barriers also exist to prevent colonization by potential pathogens. Examples of these barriers can be found in Table 2.1.
If a pathogen penetrates any of the physical barriers, plasma proteins in the form of what is called the complement system are an immediate non-cellular defense against bacteria and virus particles. The complement system is a cascade of approximately 30 serine proteases that cleave subsequent proteases in the pathway, one of the most important being C3, and induce the removal or destruction of pathogens. The net effect of the activities of individual complement proteins in the cascade is to induce either the opsonization or lysis of pathogens and the recruitment of inflammatory cells to infectious sites. The complement cascades are arranged into three different pathways: Alternative, Classical, and Mannose-Binding Lectin (MBL) (Fig. 2.1). The alternative pathway is the first to be activated during initial infection and involves the protease C3 which is made in the liver and released into the blood. When C3 is hydrolyzed, especially near the surface of a pathogen, it becomes subsequently cleaved by an enzyme, C3 convertase into C3a and C3b. C3b is then able to bind covalently to the surface of the pathogen. This creates a positive feedback loop in which the deposition of more C3b results in further cleavage of C3 by C3 convertase, resulting in a coating of the pathogen by C3b. This C3b “coating” is then able to interact with complement receptors on phagocytes, resulting in a more efficient phagocytosis. The process of coating with complement for phagocytosis is known as opsonization.

Activation and roles of the three complement pathways. Activation of complement results in recruitment of leukocytes, opsonization of pathogens to promote phagocytosis, and formation of the membrane attack complex to aid is destruction of microbes via perforation of cell membranes
The MBL and classical pathways are activated later during infection, but result in the same effect, which is the deposition of C3b on the surfaces of pathogens. The MBL pathway is initiated when mannose-binding lectin, an acute phase protein produced by the liver during infection, binds mannose residues on the surface of pathogens. This activates the complement cascade via the lectin pathway. Similarly, the classical pathway is activated when C-reactive protein, another acute phase protein, binds phosphorylcholine on pathogen surfaces and triggers complement proteins. It is also activated by antibodies that are produced during the adaptive immune response.
While opsonization is one of the most important effects of complement, products beyond C3b are also integral for pathogen killing. Complement C5 is structurally similar to C3 and can be cleaved by C5 convertase into C5a and C5b. C5b initiates the forming of the membrane attack complex (MAC) which is composed of C5, C6, C7, and C9 interactions with the bacterial cell membrane. These, in effect, poke holes in the cell membranes of bacterial cells, causing them to lyse and die.
Lastly, while C3b and C5b contribute to important effector mechanisms of complement, the smaller components of C3 and C5 cleavage, C3a and C5a, sometimes referred to as anaphylatoxins for their ability to induce anaphylaxis, can help recruit leukocytes to the site of infection. Specific activities include the degranulation of mast cells and basophils with the consequent release of histamine, and a vasodilating effect on blood vessels that allows for easier passage of leukocytes out of the blood and into the site of infection. C5a in particular can act as a chemoattractant to help neutrophils and monocytes adhere to blood vessel walls and get to the site of infection where C3b molecules have already opsonized pathogens.
Defensins are usually found at mucosal surfaces in an effort to initially protect the epithelium from infection. Defensins are antimicrobial peptides; their main role in the immediate response to infection is to penetrate and disrupt the integrity of bacterial and fungal cell membranes, as well as the envelope of enveloped viruses. Defensins can be divided into two classes—α-defensins and β-defensins 6 and 4 There are six types of α-defensins. α-defensins HD5 and HD6, for example, are secreted by special epithelial cells within the crypts of the small intestine called Paneth cells. α-defensins can also be found in the granules of neutrophils to help kill microbes that have been phagocytosed. The four different types of β-defensins are typically produced by epithelial cells of the skin, respiratory, and urogenital tracts but can also be produced by monocytes, macrophages, and dendritic cells.
Pathogen damage to host tissue, usually by secreted proteases can also initiate the release of plasma proteins known as α2-macroglobulins which are structurally similar to C3 but can aid in the inactivation of microbial proteases by chemically inducing a conformational change. The covalent binding of the α2-macroglobulin to the protease forms a complex that can then be cleared by macrophages which recognize this complex via a receptor present on the surface.
The Acute Phase Response
The innate immune response that occurs soon after the start of an infection and involves the production and systemic release of acute phase proteins by the liver is termed as the acute phase response (APR). This is mediated in response to trauma, infection, or inflammatory stimuli and usually occurs when immediate immune barriers have been breached. Acute phase proteins (APPs) are made in the liver in response to release of proinflammatory cytokines including IL-1, IL-6, and TNF-α from monocytes and macrophages at the site of inflammation or infection. APPs have many roles including activation of complement, pathogen elimination, and modulating the immune response. During the APR, when fever is being initiated, leukocyte numbers are increasing, and proinflammatory cytokines are being released, Kupffer cells in the liver are also producing IL-6, the major stimulus for release of APPs by hepatocytes. There are several APPs which provide for different immunomodulatory functions. Positive APPs include C-reactive protein (CRP ), fibrinogen, D-dimer protein, and mannose-binding protein which binds to mannose on the surface of pathogens and helps to activate complement in the MBL pathway. C-reactive protein also binds to bacterial surfaces, acting to opsonize the bacteria for phagocytosis as well as activate more complement via the classical pathway.
Innate Immune Cells
The innate immune system is made up of leukocytes that have various overlapping roles in ridding the body of potential pathogens. If the body’s primary borders are breached, innate immune cells are one of the next lines of defense (with nonspecific receptors for foreign invaders) to rid the body of the invader. These cells include macrophages, neutrophils, dendritic cells, eosinophils, basophils (Fig. 2.2) mast cells, and NK cells. As will be discussed, these cells release mediators (cytokines and chemokines) that further stimulate the innate response, so the infection is controlled while the adaptive arm of the immune system is mounting a response.

Morphology of cells of the innate immune system
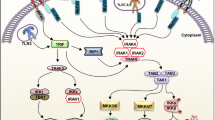
Many innate immune cells use receptors to detect infection. Bacterial components, such as lipopolysaccharide (LPS) signal to innate immune cells that an infection is present. LPS is found in the cell walls of Gram-negative bacteria, of which many human pathogens exist. Toll-like receptors (TLRs), of which there are at least ten, recognize a broad spectrum of pathogen components, and are usually found on mast cells, macrophages, and dendritic cells. TLRs are transmembrane proteins consisting of approximately 700–1100 amino acids with extracellular, transmembrane, as well as cytoplasmic domains, with the extracellular portion containing the site for ligand binding. TLRs 1, 2, 4, and 5 are expressed in the cell membrane and usually interact with extracellular components of pathogens. As is the case with LPS (recognized by TLR4), many ligands are from bacterial cell walls and membranes, including lipoteichoic acids and lipoproteins (recognized by TLR 1/2). Bacterial flagellin can be recognized by TLR5. Some TLRs (3, 7, 8, and 9) exist intracellularly in the endosomes and lysosomes. After a pathogen has been phagocytosed, it will end up in one of these organelles to be broken down. At that point, microbial DNA and RNA are exposed and can be recognized by these internal TLRs. TLRs 3, 7, and 8 are typically responsible for recognizing viral RNA while TLR9 recognizes bacterial DNA. Figure 2.3 illustrates Toll-like receptors and their ligands.

Toll-like receptors and the ligands they recognize. Some TLRs exist on the cell surface while others are internalized
After binding to their ligand, TLRs convert the signal to a response by the cell via a signal transduction pathway. Protein kinases which phosphorylate other proteins to activate them, are also a part of the TLR pathways. Some commonly studied messengers in the TLR pathways include IRAKs (IL-1 receptor-associated kinases) and MAPKs (mitogen-activated protein kinases). Kinases in this pathway are possible drug targets for mitigation of certain conditions in which inflammation contributes to the pathophysiology of the disease. The goal of the TLR pathway is to transcribe genes responsible for production of inflammatory mediators in response to the pathogen that was detected. The mediators, generally cytokines and chemokines, are released by the cell to activate other immune cells to help fight the infection.
The activation of the transcription factor nuclear factor -kB (NF-kB) plays a paramount role in the induction of many immune genes including genes for pro-inflammatory cytokines TNF-α, IL-1 and IL-6, and others that play a prominent role in immune responses. Engagement of a TLR such as TLR4 by a pathogenic component like LPS results in the activation of a signaling cascade that includes the recruitment of adapter proteins such as MyD88 and the phosphorylation of various tyrosine kinases. Inactive NF-κB is bound in the cytoplasm by IκB, which prevents its translocation to the nucleus. Activation of the TLR pathway leads to phosphorylation of IκB by IKK or I kappa kinase, resulting in the degradation of IκB and the release of NF-κB, which can now enter the nucleus. Activated NF-κB now induces the transcription of genes for proinflammatory cytokines (Fig. 2.4).

TLR4 recognizes bacterial LPS. Activation of this signaling pathway results in translocation of the transcription factor NF-kB to the nucleus, leading to transcription of pro-inflammatory cytokines that aid in activation and recruitment of other immune cells to help fight the infection
Agranular Leukocytes
Monocytes are large mononuclear cells. This cell type was discovered in the late 1800s by the famed immunologist Ilya Metchnikoff who used starfish to identify phagocytes after injury with a rose thorn. Since the 1960s, classical research has suggested that after spending 1–2 days in the circulation, inflammation causes monocytes to enter the tissues, which where they differentiate into macrophages. However, more recent findings have suggested that some macrophages already exist in tissues (tissue-resident macrophages), and probably arose during embryonic development. Both monocytes and macrophages can recognize, phagocytose, and destroy cellular debris, cells that have undergone apoptosis, and many types of pathogens without any help. Macrophages are covered in cell surface receptors, including TLRs, to recognize pathogen associated molecular patterns (PAMPs ). Upon recognition of a pathogen, they release cytokines to signal to other cells that there is an infection present. They are also considered to be one of the three types of antigen-presenting cells that can help coordinate the innate and adaptive immune responses.
Macrophages are derived from common myeloid progenitors in the bone marrow, where their development is controlled by macrophage colony-stimulating factor (M-CSF) produced by stromal cells. This growth factor drives the differentiation of progenitor cells to become monoblasts, which are committed to the monocyte lineage and eventually differentiate into macrophages in the tissues. Macrophages within tissues (both those derived from monocytes and tissue-resident macrophages) may be referred to by different names (Table 2.2).
On their surfaces, macrophages express several different receptors to help them recognize pathogens or phagocytose pathogens that have been “tagged” by other components of the immune system for removal. These include TLRs but also complement receptors and scavenger receptors whose ligands are diverse microbial proteins. In addition to the recognition of pathogens, TLRs also induce intracellular signaling that leads to macrophage activation. Complement receptors and scavenger receptors help macrophages recognize complexes targeted for phagocytosis. This binding usually results in an irreversible “capture” of the pathogen by the macrophage and results in receptor-mediated endocytosis, or engulfment of the target into a phagosome. The phagosome combines with the lysosome to form a phagolysosome wherein destructive enzymes in the membrane-bound vesicle mediate pathogen killing.
Macrophages may be activated based on stimuli produced by innate immune cells during early tissue damage or infection and thus can have different effects on the resulting physiology of the responding macrophages. Classically activated macrophages become active as a result of the cell-mediated immune response and their major function is the removal of pathogens during infection. These macrophages tend to be primed and activated by IFN-γ produced by innate or adaptive cells during the immune response and release TNF-α for enhanced microbicidal activity. NK cells are a major source of IFN-γ during the innate immune response. During the adaptive immune response, TH1 cells helps maintain this classically activated macrophage population. Interactions between macrophages and T cells will be explored further in Chap. 3. Upon activation, macrophages release a host of pro-inflammatory cytokines, superoxide, oxygen radicals, and nitric oxide. These mediators serve to destroy the infectious organism and alert the rest of the immune system. Cytokines produced by macrophages include a number of pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α, as well as those that induce that the recruitment of neutrophils (such as CXCL8) and those that activate NK cells (such as IL-12). In addition to IL-1, and IL-6, other cytokines released include IL-23 which aid in the development of IL-17-producing TH17 cells. These cells induce the activation of neutrophils to extracellular pathogens and are also implicated in autoimmunity. While classically activated macrophages are essential for host defense against infection, their effects must be highly regulated to prevent tissue damage. In the absence of such regulation, they can become highly pathogenic as occurs during certain autoimmune disorders such as inflammatory bowel disease or rheumatoid arthritis.
Other types of macrophages can also play a large role in wound healing and are necessary for tissue repair. In doing so they are activated by both innate and adaptive mediators. An early mediator released by mast cells and basophils is the cytokine IL-4, which is produced in response to tissue injury or even fungal infection. IL-4 is also produced during the adaptive response by TH2 cells, primarily at mucosal surfaces including the lung and intestines. IL-4 released during these responses (innate or adaptive) results in increased arginase activity in macrophages. Arginine is then converted to another amino acid (ornithine), which is a precursor of collagen and leads to production of extracellular matrix for tissue repair. While the response of wound healing macrophages is not aimed at pathogen removal, there is evidence to suggest that this subset of macrophages may play a role in antifungal and antiparasitic responses. Like classically-activated macrophages, a dysregulation of the matrix-building effects of wound healing macrophages may also be responsible for some of the pathological manifestations of autoimmunity.
Lastly, regulatory macrophages have anti-inflammatory activity and can be activated either innate or adaptive immune responses. These macrophages usually produce high levels of IL-10 and downregulate IL-12 and other pro-inflammatory cytokines. Later in the adaptive response, regulatory macrophages suppress the immune response and decrease inflammation.
Dendritic cells (DCs) are also derived from the myeloid lineage and are found at ports of microbial entry such as the skin, the respiratory tract, and the gastrointestinal/genitourinary (GI/GU) tracts. DCs are commonly recognized by their dendrite-like projections and are essential for innate/adaptive immune crosstalk. Myeloid DCs are central to this task, first engulfing the pathogen at the site of infection, then processing its antigens and displaying them on their cell surface for CD4 T cells to “see” on MHC II molecules in the secondary lymph nodes. When DCs themselves are infected by viruses or intracellular bacteria, processed antigens are similarly presented on the surface of MHC I molecules to CD8 T cells. This makes DCs one of the professional antigen-presenting cells (APCs ). Antigens that are picked up in the blood may be transported to the spleen, while antigens captured in the respiratory, GI, or reproductive tissues are transported through a draining lymphatic vessel to the closest lymph node. At the sites of infection or antigen capture, these DCs are referred to as immature DCs (iDCs). As they migrate toward the secondary lymphoid organ, they undergo several physiologic changes that make them better equipped for their interaction with naive T cells and are referred to as mature DCs.
Like macrophages, DCs also sense pathogens and undergo activation through TLR-mediated signaling. DCs express all the TLRs except TLR9. The induction of TLR signaling activates the DC and enhances its ability to engulf and process antigen for display on MHC II. It also increases surface expression of CCR7, which is a receptor for the chemokine CCL21 expressed within secondary lymphoid organs. This induces the migration of DCs to the secondary lymphoid organ, where they can prepare to interact with naïve T cells. Specific DC/T cell interactions will be elaborated on in Chap. 3.
Granular Leukocytes
Neutrophils are one of the major pathogen-fighting leukocytes recruited to the site of infection. They have a short lifespan of approximately 8–12 h in the circulation and 1–2 days in the tissue. They are formed in the bone marrow in response to GM-CSF and are generally found circulating in the bloodstream until they are called to a site of infection. Neutrophils account for 50–70% of circulating leukocytes, and as such much of the bone marrow is dedicated to their development. Neutrophil count is normally defined as Absolute Neutrophil Count (ANC) which is the portion of the total white blood cell count that is made up of neutrophils. It is defined as:
A normal ANC is approximately 1500–8000/mm3. A level less than 1000 is referred to as neutropenia. A higher than normal level is considered neutrophilia, which is most often indicative of infection, but can also occur as a result of any type of acute inflammation.
Two hematopoietic growth factors that are essential for neutrophil function are granulocyte colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF). G-CSF is a glycoprotein which acts on granulocyte precursors in the bone marrow to produce differentiated granulocytes and release them into the bloodstream. G-CSF can also enhance neutrophil function and stimulate the release of proinflammatory cytokines by neutrophils such as TNF-α. In contrast, GM-CSF can recruit and enhance the functions of both neutrophils and monocytes. Due to their ability to mobilize circulating neutrophils, these factors are often used therapeutically in immunosuppressed patients, most notably those who are neutropenic from undergoing chemotherapy.
To get to the site of infection, neutrophils must squeeze themselves out of the bloodstream, a process called diapedesis. The small cytokine/chemokine, CXCL8 (IL-8) is a potent chemoattractant molecule that helps call neutrophils from the circulation, through the tissue, and to the site of inflammation or injury. CXCL8 is a chemokine which binds to chemokine receptors CXCR1 and CXCR2, both of which are expressed on neutrophils. These receptors may also be expressed on epithelial and endothelial cells, as well as fibroblasts and neurons, but one of the main roles of CXCL8 is neutrophil recruitment.
Neutrophils are activated by LPS, TNF, chemokines, and growth factors. Once they reach the sites of infection, the vast repertoire of pattern recognition receptors expressed by neutrophils allow them to recognize various types of microbes and phagocytose opsonized pathogens. C-type lectin receptors like Dectin-1 recognize fungal β-glucan. The cytosolic microbial sensors NOD1 and NOD2 can recognize peptidoglycan molecules of Gram-negative and Gram-positive bacteria, respectively. Lastly, neutrophils also express a wide array of Toll-like receptors, including TLRs 1, 2, 4, 5, 6, 8, and 10 and complement receptors.
At the site of infection, neutrophils help macrophages in cleaning up the infection by recognizing, phagocytosing, and killing pathogens. The destruction of pathogens by neutrophils occurs within the phagolysosome. This internal microbial killing is aided by hydrogen peroxide, as well as several enzymes and antimicrobial peptides, which kill the ingested bacteria. When pathogens are engulfed into the phagosome, the activity of enzymes such as NADPH oxidase produces superoxide radicals that are rapidly converted to hydrogen peroxide. This raises the pH of the phagosome to 7.8–8.0, activating antimicrobial peptides and other enzymes that attack the engulfed pathogen. After about 10–15 min, the pH of the phagosome returns to 7.0, inducing the formation of the phagolysosome. Here the pathogen is completely degraded by acid hydrolases. The release of hydrogen peroxide and other superoxide radicals is referred to as the respiratory burst and has the potential to harm neighboring cells. Enzymes that inactive these molecules such as catalase are also produced during the respiratory burst and limit the damage to host cells.
The successful destruction of the pathogen through the release of granular contents results in the death of the neutrophil and its phagocytosis by nearby macrophages. In addition to the above-mentioned method of pathogen destruction, neutrophils can also kill pathogens during the process of dying themselves. This process called netosis produces neutrophil extracellular traps (NETs) which consists of nuclear contents, defensins, and other proteins that can trap and destroy pathogens.
Basophils and Mast cells Basophils, like the other leukocytes, are produced in the bone marrow and circulate in the peripheral blood. During a parasitic infection, they can be recruited to tissues, where they can be activated to release granular contents and various cytokines. Basophils make up less than approximately 1–2% of circulating leukocytes. They have a bilobed nucleus and contain many large cytoplasmic granules. These contain several preformed mediators including heparin and the vasodilator histamine. Basophils share many similarities and functional characteristics with their tissue-resident counterparts, the mast cells. They are the only circulating leukocytes that contain histamine, which is rapidly released when they are activated. In addition, they also release a number of cytokines including IL-4, IL-13 and GM-CSF and synthesize lipid mediators including the prostaglandins and leukotrienes as well as platelet activating factor. They also express receptors for various cytokines and chemokines, and Fc receptors for antibodies such as IgE.
In contrast to basophils, mast cells are tissue-resident immune cells that have a sentinel role in immune function and protect the host from parasitic infections; their precursors come from the bone marrow and circulate in the blood until they home to the tissues to mature, usually the blood vessels or epithelial surfaces. Mast cells are approximately 20 μm in diameter and contain an abundance of cytoplasmic granules compared to basophils. These contain a number of mediators including histamine, tryptases, heparin, and TNF-α among others. When activated, mast cells are also induced to release numerous cytokines and synthesize lipid mediators such as leukotrienes and prostaglandins.
Both mast cells and basophils constitutively express the high affinity receptor for IgE antibodies, FcεRI. During a parasitic infection, large amounts of parasite-specific IgE are made which bind the receptor on mast cells and basophils. When parasitic antigens bind these IgE molecules, they induce aggregation of the receptor, which induces degranulation of the mast cells and a FcεRI-mediated signaling cascade. This results in the release of preformed mediators such as TNFα, histamine, and proteoglycans and the de novo synthesis of cytokines, lipid mediators, and platelet activating factor. In addition to IgE-mediated activation, mast cells and basophils can also be activated via other means including TLRs. The binding of TLR3 on mast cells by dsRNA induces the release of IFN-γ.
Both mast cells and basophils are pathologic during the development of allergic reactions, including allergic rhinitis, urticaria, asthma, and anaphylaxis. Mast cells can also contribute to other conditions such as rheumatoid arthritis , osteoporosis, and cancers.
Eosinophils Like mast cells and basophils, eosinophils are also involved in host defense against parasitic worm infections. They are also involved in the pathogenesis of conditions such as allergic asthma. Eosinophils have a bilobed nucleus and release two main types of granules—specific granules and primary granules. The specific granules contain major basic protein, eosinophil peroxidase, eosinophil cationic protein, and eosinophil-derived neurotoxin.
The development of eosinophils is mediated by cytokines such as IL-5, IL-3, and GM-CSF. IL-5 released at the site of helminth infection promotes the generation and differentiation of eosinophils from bone marrow progenitors, after which they enter the circulation and home to the tissues. Many eosinophils that are located in the tissues exist in the gastrointestinal tract mucosal surfaces during homeostasis, and at TH2-dominated sites of inflammation where IL-4 and IL-13 are released. Here, the upregulation of eotaxin and cell adhesion molecules by various immune cells induces the recruitment of eosinophils. On their surfaces, eosinophils express Fc receptors for antibodies including IgE, IgA, and IgG. The receptor for IgE is induced during infection. Eosinophils also express other receptors including complement receptors, cytokine receptors, and chemokine receptors. They also express adhesion molecules, leukotriene and prostaglandin receptors, and TLRs 7 and 8. Upon activation by the cross-linking of Fc receptors, eosinophils release pro-inflammatory mediators including granule-stored cationic proteins, newly-synthesized eicosanoids, and cytokines. While a normal level of eosinophils in the peripheral blood is up to 500/mm3, an elevated level (eosinophilia) may indicate atopic asthma (usually mild eosinophilia in blood), a drug reaction, or helminth infection. With atopic asthma, eosinophilia is more likely to be found in nasal secretions, sputum, or the bronchoalveolar lavage (BAL).
Natural Killer (NK) cells play a prominent role in the initiation and maintenance of antiviral immunity. They are also extremely effective against cancerous cells. Under the microscope, NK cells look like T and B cells, deriving from the common lymphoid progenitor and the lymphoid lineage. They are a highly diverse cell type consisting of heterogeneous subsets that can carry out cytotoxic function or release inflammatory cytokines. In humans they are identified on the basis of the adhesion molecule CD56, as well as other NK cell markers. Similarly, in mice they can be identified using markers such as NK1.1 and DX5. These populations can be further divided into those that perform cytotoxic activity (CD56 dim) and those that release cytokines (CD56 bright). They are distinguished from T cells and other invariant T cells such as NKT cells based on the absence of CD3.
NK cells play a vital role in the induction of immune responses to viruses and tumors. Their activation and cytotoxic functions are governed by an intricate balance between the functions of various surface receptors that promote either activation or inhibition of cell function. These receptors are stochastically expressed on various cell subsets and include inhibitory receptors such as the lectin CD94-NKG2 and KIR receptors and activating receptors such as NKG2D. The inhibitory receptors bind HLA-E molecules on the surface of endogenous cells which prevents the activation of NK cells. Infection or other inflammatory conditions can induce loss of MHC class I and induction of stress proteins such as MICA and MICB. This results in disengagement of the inhibitory MHC I receptor and engagement of activating receptors such as NKG2D with its ligand (MICA/B), inducing activation and NK cell-mediated cytotoxicity.
Activation of NK cells can occur in response to several stimuli, predominantly the release of type I interferons by infected cells (Fig. 2.5). Soon after viral entry, both infected cells and neighboring epithelial cells produce large amounts of IFN-α and IFN-β. In addition, plasmacytoid dendritic cells, also called interferon producing cells, are potent producers of type I interferons. The release of type I interferons results in the induction of the interferon response, leading to resistance to viral infection and the activation of NK cells. This results in the killing of infected cells by activated NK cells. In addition to type I interferons, NK cells can also be activated via cross-talk with macrophages and DCs. These cells produce cytokines such as IL-12 and IL-15, which can promote their activation and function.

Antiviral immunity and the roles of NK cells. Host cells infected with a virus begin releasing Type I IFNs (IFN-αβ) which signal to neighboring uninfected cells to begin making antiviral proteins. Type I IFNs also activate NK cells to release lytic granules which will kill virally-infected cells. This also stimulates NK cells to release IFN-γ to initiate the adaptive response by promoting DC processing of viral antigen for presentation to CD4 T cells
Activated NK cells produce large amounts of IFN-γ. This cytokine not only has the ability to promote anti-viral effects, but also has other effects on the immune system such as the activation of the immunoproteasome and the upregulation of MHC II on APCs. In addition, NK cells are known to produce other cytokines including TNF-α and type II cytokines such as IL-13 and IL-5.
The cytotoxic functions of NK cells are mediated via the induction of caspases and programmed cell death. They involve the release of perforin and granzymes from granular contents. While perforin and other proteins such as granulysin poke holes into the target cell membrane, granzymes initiate proteolytic cleavage reactions resulting in the induction of apoptosis and death of the infected cell.
NK cells can also be activated to perform antibody dependent cellular cytotoxicity or ADCC. This occurs when antigen-specific IgG antibodies engage the FcγRIII or CD16 receptor on NK cells, inducing their activation. Activated NK cells then kill target cells expressing the antigen. ADCC is a common mechanism of action of many therapeutic drugs such as rituximab .
Inflammation
Inflammation is the characteristic response to infection (non-self) or the presence of danger. The process of immunity begins when normal host defenses are breached and when the immune system senses a threat to the host. This can occur as a result of pathogen sensing by TLRs or the production of alarmin cytokines by epithelial and other sentinel cells indicating the presence of injury or danger. The type of immune response that is initiated ultimately depends on the type of pathogen and the offended target tissue. Various strategies are employed by the immune system depending on whether the pathogen is extracellular or intracellular, or depending on the type of injury or danger to tissue. The initial response is targeted towards the presence of molecular patterns such as LPS, while the latter adaptive response is more specific and antigenic in nature. Extracellular offenders include extracellular bacteria, some fungi, virions in the extracellular space and allergens. Intracellular offenders include viruses, intracellular bacteria and some parasites.
A ubiquitous initial response to the entry of extracellular bacteria is the induction of the complement system. As described earlier, this complex system involves the activation of a number of heat-labile serine proteases that are constitutively produced by the liver. These are classified as C1-C9, depending on when they were discovered and the functions they perform. Several complement proteins can be further cleaved into smaller components by enzymes referred to as convertases. The most important complement component is C3. This protein is normally present in the inactive state, but in the presence of infection or on engagement of complement components by ligands such as mannose-binding protein, C-reactive protein (CRP ) or antibody, it can be hydrolyzed or cleaved into a larger C3b molecule and a smaller C3a molecule. C3b tags the extracellular pathogen, initiating the process of complement fixation. In the case of some bacteria such as Neisseria, the deposition of C3b on the bacterial surface can lead to further events in the complement cascade, resulting in the cleavage of C5 and the formation of the membrane attack complex (MAC ), which perforates pathogen cell membranes and induces their lysis. Pathogen-bound C3b itself is detected by complement receptors on cells such as macrophages, resulting in the induction of phagocytosis. Other complement components, such as C3a and C5a induce the recruitment of neutrophils and induce mast cell and basophil activation. Thus, activation of complement has the net effect of inducing phagocyte recruitment, opsonizing macrophages and neutrophils for phagocytosis, and lysing bacterial cell membranes. All of these result in death of the pathogen.
In addition to complement activation, several other non-specific mechanisms may also contribute to innate defense including the production of antimicrobial peptides such as defensins, the activation of the kinin system and the production of α-macroglobulins.
The induction of infection or the presence of injury also results in the secretion of a number of cytokines and immune mediators by injured cells. Depending on the type of injury or infection, cells such as epithelial cells can release a vast number of mediators including cytokines such as TNF-α, IL-33, IL-25, thymic stromal lymphopoietin (TSLP) and type I interferons, various chemokines, and antimicrobial peptides. This has the net effect of recruiting and activating various innate cells such as macrophages, dendritic cells, innate lymphoid cells, mast cells, and NK cells.
Pathogen sensing by macrophages using either scavenger receptors or the TLRs results in the activation of NF-κB, and the induction of genes for various cytokines. Some of the first cytokines to be produced by macrophages are the proinflammatory cytokines TNF-α, IL-1 and IL-6. TNF-α is a potent cytokine that has pleiotropic effects on the immune system. It induces vasodilation increasing the flow of blood within blood vessels and causes vascular permeability, allowing cells and molecules to leak out of blood vessels. It also promotes the expression of adhesion molecules on blood vessels to enable leukocyte migration. Mast cells in tissues can also be activated resulting in the production of histamine which can also contribute to vasodilation.
IL-1 and IL-6 are pyrogenic and raise the body temperature, making it difficult for pathogens to survive. IL-6 also acts on hepatic cells inducing the acute phase response, by leading to the increased production of the acute phase proteins MBP and CRP. These acute phase proteins further act to activate the lectin and the classical pathways of the complement cascade.
In addition to these cytokines, macrophages also produce the chemokine CXCL8, which is a chemotactic migratory factor for neutrophils, drawing in neutrophils from blood vessels. Neutrophils act as first responders, quickly phagocytosing bacteria and producing cytokines. The phagocytic response is dependent on the release of several antibacterial substances in neutrophil granules and the induction of a respiratory burst, resulting in the release of superoxides that damage the bacteria. Eventually, acid hydrolases within the neutrophil granules completely digest the bacteria, leading to death of the neutrophil and the formation of pus.
Similarly, when infection occurs due to intracellular pathogens such as intracellular bacteria or viral infection, macrophages can produce additional cytokines such as IL-12. The concomitant production of type I interferons by virally infected cells has the net effect of activating NK cells and inducing their recruitment to the infected tissue.
The series of physiologic events give rise to the four main hallmarks of inflammation described since ancient times which include redness (rubor), pain (dolor), heat (calor), and swelling (tumor). Vasodilation results in increased blood volume contributing to redness; vascular permeability leads to leakage of blood vessel constituents such as cells and molecules into tissues resulting in swelling or edema; the swelling has the effect of pinching on associated nerves resulting in pain; and the cumulative effects of vasodilation and cytokine function make the inflamed area warm to touch.
Innate Immune Deficiencies
Genetic mutations leading to dysfunction in one or more components in the innate immune system can often manifest in an immunodeficiency disorder characterized by frequent and recurrent infections. Some of these disorders are outlined in Table 2.3.
Therapeutic Inhibitors Which Target the Innate Immune Response
Phagocyte recruitment
Targeting cell adhesion molecules (CAMs) to reduce leukocyte recruitment may be an effective method of reducing inflammation in certain disease states in which leukocyte migration contributes to inflammation. CAMs allow cells to bind and interact with their extracellular environment, including other cells. Certain CAMs allow leukocytes to migrate out of general circulation, “roll” along the surface of blood vessels, and permeate through tissue to reach tissues where they contribute to inflammation and injury (Fig. 2.6). Leukocytes expressing a specific CAM, α4β7 integrin, have been implicated in the pathogenesis of inflammatory bowel diseases such as ulcerative colitis and Crohn’s disease. Cells expressing α4β7 integrin have been shown to exhibit preferential binding to endothelial surfaces of the GI tract.

Leukocyte recruitment to the site of inflammation is initiated by stimuli at the site of infection, i.e. cytokines and chemotactic factors. Neighboring endothelial cells begin expressing selections and adhesion molecules which bind to integrins on the surface of neutrophils, causing them to slow down and “stick” to the CAM-expressing endothelial cells. As neutrophils “roll” they adhere to the endothelium more firmly and extravasate out of the blood vessel into the inflamed tissue where they sense chemokines and other inflammatory mediators
Vedolizumab (Entyvio®) is a relatively new antibody that targets the α4β7 integrin. It inhibits leukocyte binding to the GI tract, significantly decreases the symptoms of inflammatory bowel disease (IBD) and is approved for adult ulcerative colitis and Crohn’s disease. Adverse effects of vedolizumab include infusion-related reactions, nasopharyngitis, headache, arthralgia, and upper respiratory tract infection.
Natalizumab (Tysabri®) is another humanized monoclonal antibody with a similar mechanism of action in that it is an α4β1 integrin inhibitor used for Crohn’s disease and multiple sclerosis. The most common adverse effects include headache, pain in the arms and legs, abdominal pain, fatigue, joint pain, vaginitis, urinary tract infection, and lung infection. Taking natalizumab increases a patient’s risk of developing a rare but severe brain infection called progressive multifocal leukencephalopathy (PML ). The risk increases with long-term use (greater than 2 years), infection with John Cunningham (JC) virus, or taking other immunosuppressant drugs before beginning natalizumab. Patients should be tested periodically for JC virus, and because of the PML risk, natalizumab is only distributed through the TOUCH® prescribing program to help patients understand the risk of PML and if necessary, diagnose it earlier if it occurs.
Pro-inflammatory Cytokines
While it is important to understand the pivotal roles innate immune cells play in the immune response, it is also important to know that many of their activities are mediated by cell to cell communication through cytokines. Thus, cytokines represent an important therapeutic target in several inflammatory diseases, in which in the case of inappropriate inflammation, one can intervene with pharmacotherapy in an effort to “turn off” or suppress intercellular crosstalk to reduce symptoms.
Cytokines are small proteins that are released in response to a stimulus and induce responses by binding to their specific receptor. There are three main families of cytokines, the larger families being the Hematopoietin and TNF families, as well as the smaller IFN family (Table 2.4).
The three main pyrogenic (fever-inducing) cytokines are IL-1 , TNF-α, and IL-6. Acting on the hypothalamus to increase the temperature of the body, fever allows for decreased viral and bacterial replication, increased antigen processing, and a more efficient adaptive immune response. The effects of these pyrogenic cytokines are outlined in Fig. 2.7.

Effects of pyrogenic cytokines IL-1, IL-6, and TNF-α on tissues
TNF Inhibition
Tumor Necrosis Factor -α (TNF-α) was first described in 1975 by the laboratory of Dr. Lloyd Old at the Sloan Kettering Memorial Cancer Center in New York. Dr. Old’s team observed that a cytotoxic factor produced during infection with endotoxins could cause the necrosis of tumors. Previously in 1968 Gale Granger from the University of California and Nancy Ruddle from Yale University had described another TNF family member, which they called lymphotoxin (TNF-β).
The hypothesis that infections can trigger the regression of tumors had been around since the late nineteenth century, when the surgeon William Coley used therapeutic dead bacteria (Coley’s toxins) to treat sarcomas. This treatment was based on a few reports of erysipelas and tumor regression. Not surprisingly, Coley’s toxins induced fever and chills and patients but without true infection. Subsequently, in 1943, scientists at the National Cancer Institute isolated lipopolysaccharide (LPS ) from Gram-negative bacteria, and in turn used it to induce the necrosis of tumors. Subsequent research demonstrated that animals injected with LPS produced mediators which they considered to be anti-tumor factors, and accordingly named these mediators “tumor necrosis factor.”
Macrophages are the main cell type that can secrete TNF, but it can also be produced by neutrophils, NK cells, mast cells, eosinophils, and CD4 T cells. TNF-α is an endogenous pyrogen, meaning it acts on the hypothalamus to induce fever to slow down pathogen replication in the body. It can also induce apoptosis through death receptor TNFR1. As a signal transducer, TNF activates MAPK pathways which can result in cellular differentiation and proliferation, as well as activates transcription factor NF-κB which is generally proinflammatory and anti-apoptotic. Thus, the activation of NF-κB often masks the pro-apoptotic effects of TNF through TNFR1 signaling. As a chemoattractant, TNF recruits neutrophils not only by encouraging the bone marrow to release new neutrophils, but also by promoting the expression of adhesion molecules on the endothelium, and acting as a direct chemoattractant. In the liver, it can stimulate the acute phase response and the release of C-reactive protein and complement for opsonization of bacteria. In macrophages, it helps to stimulate phagocytosis and PGE2. In other tissues, it can promote insulin resistance. Its many effects on various tissues are outlined in Fig. 2.7.
TNF-α can exist in two forms: soluble TNF and transmembrane TNF, with macrophages being the biggest producers. Two receptors exist which have different outcomes upon receptor ligation. TNF receptor 1 (TNFR1) is widely expressed on most cell types (with the exception of erythrocytes) and as mentioned, plays a role in apoptosis and induction of acute inflammation. It binds both soluble and transmembrane TNF. With regard to TNFR2, several studies have suggested that signaling through the TNFR2 mediates cell survival. TNF-α plays a variety of roles in the immune response. It has been reported in humans that after immune encounter with Gram-negative LPS, TNF becomes elevated in the blood 60–90 min later, and is associated with fever and flu-like symptoms including chills and myalgia. TNF-β is very similar to TNF-α in the response it elicits, binds to the same receptor, and has 30% homology. The main difference is that TNFβ is produced primarily by T cells and is thus often referred to as lymphotoxin.
The first TNF inhibitor biologic drugs were approved by the FDA in 1998 for rheumatoid arthritis (RA) and Crohn’s disease. Most of the TNF inhibitors are monoclonal antibodies (except for the etanercept fusion protein) and have indications for gastroenterological, rheumatological, and dermatological conditions (Table 2.5). Nomenclature of monoclonal antibodies will be further described in Chap. 3.
Etanercept (Enbrel®) is a fusion protein (not an antibody) that binds to TNF in both its alpha and beta forms. TNFβ is also known as lymphotoxin-alpha (LT-α). Etanercept is indicated for the treatment of RA, polyarticular juvenile idiopathic arthritis (JIA), plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis. Etanercept is given as a subcutaneous injection.
Infliximab (Remicade®) is a human-mouse chimeric IgG monoclonal antibody with Human Fc region and murine variable regions. Due to this, it is generally considered to be more immunogenic than the other anti-TNF biologics. It can neutralize all forms of TNF-α (including extracellular, transmembrane, and receptor-bound TNF). Infliximab has several indications including Crohn’s disease, ulcerative colitis, RA, psoriatic arthritis, ankylosing spondylitis, and plaque psoriasis. Unlike the other TNF inhibitors, infliximab is usually given in the form of IV infusion.
Adalimumab (Humira®) gets its name from being a “Human Monoclonal Antibody in Rheumatoid Arthritis”. Thus, is a fully human IgG antibody targeting TNF. Besides its indication for RA, adalimumab is also used to treat juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, plaque psoriasis, hidradenitis suppurativa, and uveitis. Adalimumab is given as a subcutaneous injection.
Certolizumab pegol (Cimzia®) is a pegylated Fab fragment of a humanized anti-TNF monoclonal antibody and is indicated for moderate to severe RA, psoriatic arthritis, active ankylosing spondylitis, and Crohn’s disease. Unlike the other anti-TNF biologics, certolizumab does not have an Fc region. Its pegylation on the Fab fragment, however, increases its bioavailability, extends its half-life, and decreases its dosing frequency. Certolizumab is given as a subcutaneous injection.
Golimumab (Simponi®), like adalimumab is a fully human monoclonal antibody used for moderate to severe RA, psoriatic arthritis, active ankylosing spondylitis, and moderate to severe ulcerative colitis. Like most of the other TNF inhibitors, golimumab is also given as a subcutaneous injection. Another form of golimumab, Simponi Aria® is given via IV infusion.
The structures of the TNF inhibitors can be compared in Fig. 2.8.

Antibody structures of current TNF-α inhibitors
There are, however, several potential problems with TNF inhibitors, depending on the patient. One of the most common is increased risk of infection, particularly from intracellular pathogens. This is why the TNF inhibitors should not be started without first completing a tuberculin skin test to rule out latent tuberculosis. Increased risk of malignancy is also present while on one of these drugs. Other possible adverse events include hypersensitivity reactions, worsening of congestive heart failure or new onset of CHF, Hepatitis B reactivation, neurological reactions, and pancytopenia or anemia. Other possible issues with the TNF inhibitors include:
-
Primary non-response in approximately 25–50% of patients
-
Loss of response in some patients
-
Incomplete pain relief despite an indicated decrease in inflammation
-
Incomplete clinical response
-
High cost
IL-1 Inhibition
The interleukin-1 ( IL-1 ) family is a group of pro-inflammatory cytokines involved in the pathogenesis of many inflammatory diseases. Thus, the IL-1 family represents a potential therapeutic target. There are three forms of IL-1 that are known to exist: IL-1α, IL-1β, and IL-1Ra. IL-1 receptor antagonist (IL-1Ra) is a natural antagonist for IL-1 and can be produced synthetically to be used as a therapy. One of these is anakinra (Kineret®) which is a recombinant IL-1 receptor antagonist that competitively inhibits IL-1, specifically IL-1α and IL-1β, and is used for the treatment of RA in patients who have failed one or more disease modifying antirheumatic drugs (DMARDs ). It is also approved for the treatment of Neonatal Onset Multisystem Inflammatory Disease (NOMID ), a type of Cryopyrin Associated Periodic Syndromes (CAPS). RA patients who take anakinra experience a delayed progression of their physical symptoms, and those with NOMID experience improvement in symptoms and reduction in SAA and CRP levels.
Like TNF-α, IL-1β is an endogenous pyrogen and is mainly released by monocytes and macrophages, but also NK cells, dendritic cells, and epithelial cells early in the immune response. As mentioned earlier, the release of IL-1β and other endogenous pyrogens stimulates the release of APPs from the liver and acts on the hypothalamus to induce fever. It can also act as as a chemoattractant for granulocytes, and in mast cells it can induce histamine release, contributing to inflammation at the site of inflammation.
Some biologics have been designed to target IL-1 itself. Rilonacept (Arcalyst®) is a dimeric fusion protein that binds to and inhibits the actions of IL-1β and is approved for the treatment of CAPS, a group of rare, inherited auto-inflammatory diseases in which IL-1β is overproduced. IL-1β is a highly pro-inflammatory member of the IL-1 family. Rilonacept decreases the severity of symptoms associated with CAPS, and the levels of Serum Amyloid A (SAA) and C-Reactive Protein (CRP) which are indicators of inflammation that are generally elevated in patients with CAPS. Canakinumab (Ilaris®) is another IL-1β inhibitor and is used to treat Systemic Juvenile Idiopathic Arthritis (SJIA), and a number of Periodic Fever Syndromes. Periodic Fever Syndrome is a group of auto-inflammatory diseases characterized by cyclical fevers, which includes CAPS. Canakinumab is a human monoclonal IL-1β antibody that binds to and neutralizes IL-1β, which is overproduced in Periodic Fever Syndromes and SJIA. It also decreases CRP and SAA levels and improves symptoms of both of these conditions. In addition to increased risk of infection, the IL-1 inhibitors seem to be generally well-tolerated with the most common adverse effect being injection site reaction.
IL-6 Inhibition
As with TNF and IL-1, IL-6 is also an endogenous pyrogen and a pleiotropic cytokine with roles that can include both pro- and anti-inflammatory. As a pyrogen it acts on the nervous system to induce fever, as well as the liver to release acute phase proteins. Because IL-6 which is released by T cells and macrophages in response to infection and trauma also plays an important role in inflammation, studies have demonstrated that anti-IL-6 treatments, particularly in RA, are also efficacious. IL-6 transmits its pro-inflammatory signal through a soluble IL-6 receptor and a transmembrane protein called gp130 (CD130) which is ubiquitously expressed in all cells. When IL-6 binds to its receptor, gp130 dimerizes, leading to activation of JAK family kinases. This is known as trans signaling. These kinases will phosphorylate and activate STAT transcription factors, leading to the transcription of several more cytokines. This can lead to recruitment of monocytes to the site of inflammation, increased adhesion molecule expression on endothelial cells, as well as promotion of TH17 cells. The anti-inflammatory effects of IL-6 tend to occur through classical signaling at the IL-6 receptor, which is only expressed on macrophages, neutrophils, hepatocytes, and some T-lymphocytes. It is thought that this type of signaling inhibits endothelial cell apoptosis and promotes intestinal epithelial cell proliferation.
Tocilizumab (Actemra®) is a humanized anti-IL-6 monoclonal antibody that exerts its effects via inhibition of the IL-6 receptor (Fig. 2.9), which can be both membrane-bound and soluble. Tocilizumab is currently used for the treatment of severe RA.

Mechanisms of action of IL-6 and IL-12/23 monoclonal antibodies. (a) While tocilizumab binds to IL-6 receptor, siltuximab is able to bind to circulating IL-6. Both prevent IL-6 from binding to its receptor, thereby suppressing its anti-inflammatory effect. (b) Ustekinumab binds to the p40 subunit found on both IL-12 and IL-23, preventing both from binding to their receptors
Sarilumab (Kevzara®) is a recently approved human monoclonal antibody targeting the IL-6 receptor. Like tocilizumab, sarilumab was also approved for treatment of RA.
Siltuximab (Sylvant®) is a chimeric monoclonal antibody that binds to IL-6 itself (Fig. 2.9) and is currently approved for the treatment of Multicentric Castleman’s disease, a rare group of lymphoproliferative disorders characterized by excessive release of proinflammatory cytokines including IL-6. Siltuximab has shown some ability to promote tumor cell apoptosis and may be recommended as a cancer treatment.
The main adverse effects associated with the IL-6 inhibitors include increased risk of infection, increase in liver enzymes, neutropenia, infusion-related reactions, and perforation in the stomach or intestines. Several other IL-6 inhibitors, including olokizumab, and clazakizumab are currently undergoing clinical trials for RA and renal transplant rejection, respectively.
IL-12/IL-23 Inhibition
IL-12 is a heterodimeric cytokine known as p70 (made up of p35 and p40 subunits) produced mainly by antigen presenting cells, (B cells, macrophages, and dendritic cells) usually in response to TLR recognition of intracellular pathogens. While IL-12 may induce production of other cytokines, including IFN-γ and TNF-α from NK cells and T cells, it can also act to enhance the activity of these cells as well as act as a growth factor. Early in infection, IL-12 aids in activating phagocytes. Later in the immune response, IL-12 acts to bridge the innate response to the antigen-specific adaptive response, specifically enhancing the cytotoxic activities of NK and CD8 T cells. To promote adaptive immunity, IL-12 induces IFN-γ in favor of TH1 cell differentiation as well as cytotoxic T cell generation. IL-12 is also thought to be anti-angiogenic in that it upregulates the chemokine CXCL10 which inhibits new blood vessel formation in tumors. Because it activates CD8 T cells, it may also enhance CD8 T cell-induced apoptosis of certain tumor cells. Thus, IL-12 may have an important anticancer role. A related cytokine, IL-23 is similar to IL-12 in that it also uses the IL-12 p40 subunit in its signaling. Like IL-12, IL-23 also induces IFN-γ and activates T cells. IL-23 may also act to enhance IL-10 release and synthesis of IL-17.
Ustekinumab (Stelara®) is an anti-IL-12 monoclonal antibody approved for the treatment of psoriatic arthritis. Ustekinumab is a human antibody which binds to the p40 subunit of cytokines IL-12 and IL-23, preventing them from binding to their receptors (Fig. 2.9). The most common adverse effects include injection site reactions, cold symptoms, headache, fatigue, skin rash, and itching.
Type I Interferons
Interferons (IFN) play a role in antiviral immunity. For example, once a cell is infected with a virus, it begins releasing IFNs-α and -β. Since viruses are intracellular pathogens, they are recognized by several different internalized pattern recognition receptors (PRRs) including TLR9 which recognizes viral DNA, as well as TLR3, 7, and 8 which recognize various types of viral RNA. Activation of these receptors with their viral ligands initiates a downstream signaling cascade that results in the transcription of type I IFN genes. IFNs-α and -β released by the infected cell bind to IFN receptor on infected and nearby uninfected cells, resulting in JAK-STAT signaling. STATs will then activate transcription of several genes that inhibit viral replication including endoribonucleases that degrade viral RNA, as well as other proteins which inhibit viral replication. Additionally, type I IFNs increase the cellular immune response to infection, promote synthesis of MHC I at the cell surface so viral peptides can be presented to CD8 T cells, and activate NK cells to kill virus-infected cells.
Two IFN-α drugs include peginterferon alfa-2a (Pegasys®) and peginterferon alfa-2b (PegIntron®), both of which have been used in the treatment of Hepatitis C in combination with ribavirin. Depending on the HCV genotype, the patient sustained virologic response (SVR) rate is somewhere between 50 and 80%. Due to the development of newer Hepatitis C drugs such as sofosbuvir and ledipasvir which offer a much higher SVR rate of 96–98%, the use of the IFN drugs has declined. Both drugs have a 40 kDa polyethylene glycol chain added to increase their half-lives, making them long-acting. The dose of pegylated interferon alfa-2a is the same for all patients, regardless of weight or size, while the dosing of pegylated interferon alfa-2b is based on an individual’s weight.
Common adverse effects of interferon drugs include flu-like symptoms, depression or other neurological-related symptoms, or irritability. Many Hepatitis C patients on a ribavirin/interferon combination complain of a side effect they call “Riba Rage.” This depression, changes in mood or irritability is not a result of the ribavirin, but of the interferon.
Lastly, IFN-beta (Avonex®) is used mainly in Multiple Sclerosis to improve the integrity of the blood brain barrier. Like the interferon-α drugs, many patients also experience flu-like symptoms as an adverse effect of this drug.
From Bench to Bedside: Discovery of TNF Inhibitors
Initially, TNF inhibitors were developed for the treatment of Rheumatoid Arthritis (RA ). Sir Marc Feldman, an Australian immunologist published his hypothesis in 1983 on the role of pro-inflammatory cytokines in the mechanism of autoimmune diseases. In 1984, a collaboration with Ravinder Maini at the Kennedy Institute of Rheumatology highlighted RA in particular, finding that impacted joints contained more pro-inflammatory cytokines than normal joints, in particular TNF-α. In vitro studies demonstrated that blocking TNF-α could indeed reduce its levels. Subsequent studies of TNF blockade would be done in patients who had failed current RA treatments. The first of these trials took place in 1992 at Charing Cross Hospital in London, using infliximab.
In 1993, RA patients began taking the fusion inhibitor etanercept as part of a Phase I study. In 1998, etanercept became the first TNF inhibitor to be approved by the FDA for moderate to severe RA. It subsequently received FDA approval for juvenile rheumatoid arthritis, psoriatic arthritis, plaque psoriasis and ankylosing spondylitis. Clinical trials using TNF inhibitors were able to show that inhibiting a single cytokine is enough to inhibit and gain control of the immune activation that leads to inflammatory symptoms in certain disease states. Subsequently, infliximab was approved by the FDA in 1998 for the treatment of Crohn’s disease, particularly in patients where conventional therapies had failed, as well as RA. Several years later, adalimumab, the third TNF inhibitor, and first fully-human monoclonal antibody against TNF for RA was approved in 2002. It was branded Humira® (Human monoclonal antibody in RA). Two more TNF inhibitors were to follow. Certolizumab pegol was first approved in 2008 and is a recombinant humanized Fab fragment specific for TNF which was conjugated with a PEG molecule to enhance its half-life and bioavailability. One year later, golimumab (Simponi®) was approved for the treatment of severe RA, active psoriatic arthritis, and active ankylosing spondylitis. While golimumab may seem like another “me too” drug, it was prepared uniquely using transgenic mice with the human immunoglobulin locus. Among all of the TNF inhibitors, golimumab has the highest affinity for TNF, and lacks some of the immunogenicity of infliximab because, like adalimumab, it lacks mouse antibody fragments.
Certain biosimilars have also recently been gaining approval by the FDA. Etanercept-szzs (Erelzi®) was approved in 2016 and, like etanercept, is indicated for the treatment of RA, polyarticular juvenile idiopathic arthritis (JIA), psoriatic arthritis , and ankylosing spondylitis. Adalimumab-atto (Amjevita®) was also approved in 2016 for the treatment of seven inflammatory diseases, including plaque psoriasis, Crohn’s, and moderate-to-severe RA. Infliximab-dyyb (Inflectra®) is yet another biosimilar approved in 2016 for Crohn’s disease, RA, and several other inflammatory diseases. Due to patent litigation with the parent drugs and their manufacturers, only a couple of these biosimilars have launched onto the US market. Several more are already available on the European market.
For their work on TNF blockade and important contribution to the treatment of inflammatory disease, both Feldman and Maini received several awards including the Crafoord Prize, the Albert Lasker Award for Clinical Medical Research, and the Ernst Schering Prize, among others.
Summary
The primary purpose of the innate immune system is to mount an initial response to infection. Cells of the innate immune system are programmed to destroy invading pathogenic organisms, and thus, most routine infections are rapidly terminated by the innate immune response. In addition, innate immune cells also alert cells of the adaptive immune system to the presence of infection, initiating the development of adaptive immunity. During the initial response, and while the adaptive response is mounting, innate immune cells recognize nonspecific bacterial, viral, and fungal molecular patterns using Toll-like receptors (TLRs). When pattern recognition receptors such as the TLRs are bound to microbial products, they initiate downstream signaling pathways that activate transcription factors that result in production of pro-inflammatory cytokines and chemokines. These mediators recruit leukocytes to the sites of infection to phagocytose pathogens and kill pathogen-infected cells. Leukocyte recruitment is mediated by selectins and adhesion molecules expressed on both the leukocyte and the endothelial surface of blood vessels. This results in the slowing down of leukocytes and their exit from blood vessels into tissues. Natalizumab and vedolizumab are integrin inhibitors which prevent the inappropriate recruitment of leukocytes into tissues in certain inflammatory diseases. The development of inflammation during immune-mediated diseases may be targeted by inhibiting specific cytokines that contribute to the pathology of the disease. There are several TNF inhibitors that are used in the treatment of several inflammatory diseases in which TNF-α plays a role in the disease pathophysiology. Most of these are monoclonal antibodies, with the exception of etanercept, which is a fusion protein, and certolizumab pegol, which is a pegylated humanized anti-TNF Fab fragment. These drugs must be injected either subcutaneously or intravenously due to their biological properties. Because TNF inhibitors are immunosuppressive, they are associated with an increased risk of infection. Monoclonal antibodies may also be used to inhibit the deleterious effects of IL-1, IL-6, and IL-12 either by binding directly to the cytokine, or acting as a competitive inhibitor by binding to the receptor. Lastly, while IFN-α therapies have been used in the treatment of hepatitis to promote antiviral immunity, they are being used less often with the advent of newer hepatitis antivirals with higher response rates. IFN-β is still used for relapsing multiple sclerosis and is discussed in Chap. 7.
Practice Questions
-
1.
The increased metabolic rate of cells in an injured area can speed up healing if the _________is increased.
-
(a)
heat
-
(b)
pain
-
(c)
vasodilation
-
(d)
swelling
-
(a)
-
2.
Which of the following correctly describes the structure of infliximab?
-
(a)
It is a fully human antibody
-
(b)
It is a chimeric human/mouse antibody
-
(c)
It is a humanized antibody
-
(d)
It is a fusion protein
-
(a)
-
3.
Which of the following conditions can be treated with etanercept?
-
(a)
Lupus
-
(b)
C. diff colitis
-
(c)
Rheumatoid arthritis
-
(d)
Crohn’s disease
-
(a)
-
4.
Which of these is a fully human anti-TNF-α monoclonal antibody?
-
(a)
Golimumab
-
(b)
Etanercept
-
(c)
Certolizumab
-
(d)
Infliximab
-
(a)
-
5.
Patient MF is about to be put on infliximab for his psoriatic arthritis. Knowing the mechanism of action of this drug, what should be done prior to beginning this therapy?
-
(a)
MF should learn how to inject himself with this drug at home.
-
(b)
MF should be tested for latent TB infection.
-
(c)
MF should have a total WBC count done.
-
(d)
MF should be tested for Type I hypersensitivity for this drug.
-
(a)
-
6.
Which of the following is true concerning ustekinumab?
-
(a)
It binds to the IL-6 receptor
-
(b)
It binds to both IL-12 and IL-23 receptors.
-
(c)
It is a biosimilar for adalimumab.
-
(d)
It is an IL-1 receptor antagonist.
-
(a)
-
7.
Interferon-beta is used for which of the following conditions?
-
(a)
Crohn’s disease
-
(b)
Chronic granulomatous disease
-
(c)
Hepatitis C
-
(d)
Multiple Sclerosis
-
(a)
-
8.
Which of the following statements is true concerning Type I interferons?
-
(a)
They signal to CD4 T lymphocytes for perforin-mediated killing of the virally-infected cell
-
(b)
They signal through Toll-like receptors to activate downstream transcription of genes for cell death
-
(c)
They signal directly to the adaptive immune response for antigen presentation and clonal expansion
-
(d)
They signal through the JAK/STAT pathway to activate transcription of genes that inhibit viral replication
-
(a)
-
9.
Which of these inhibits IL-6 by binding to its receptor?
-
(a)
Siltuximab
-
(b)
Ustekinumab
-
(c)
Rilonacept
-
(d)
Tocilizumab
-
(a)
-
10.
Which of the following describes the mechanism of action of vedolizumab?
-
(a)
Anti-IL-12 monoclonal antibody
-
(b)
Inhibitor of α4β7 integrin
-
(c)
Inhibitor of IL-6 receptor
-
(d)
IL-1 receptor antagonist
-
(a)
-
11.
Which of the following cytokines are released by virally-infected host cells to activate NK cells to aid in killing virally-infected cells? (Select all that apply)
-
(a)
IFN-α
-
(b)
IFN-β
-
(c)
IL-8
-
(d)
IL-12
-
(a)
-
12.
Which of the following TNF inhibitors does not contain an Fc region and also contains a pegol group?
-
(a)
Remicade®
-
(b)
Simponi®
-
(c)
Humira®
-
(d)
Cimzia®
-
(a)
Suggested Reading
Abreu MT. Anti-TNF failures in Crohn’s disease. Gastroenterol Hepatol. 2011;7(1):37–9.
Bendtzen K. Immunogenicity of anti-TNF-α biotherapies: I. individualized medicine based on immunopharmacological evidence. Front Immunol. 2015;6:152.
Brennan FM, Chantry D, Jackson A, Maini RN, Feldmann M. Inhibitory effect of TNF-alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;334:244–7.
Campa M, Mansouri B, Warren R, Menter A. A review of biologic therapies targeting IL-23 and IL-17 for use in moderate-to-severe plaque psoriasis. Dermatol Ther (Heidelb). 2016;6(1):1–12.
Cessak G, Kuzawinska O, Burda A, Lis K, Wojnar M, Mirowska-Guzel D, et al. TNF inhibitors—mechanisms of action, approved and off-label indications. Pharmacol Rep. 2014;66(5):836–44.
Chen R, Chen B. Siltuximab (CNTO 328): a promising option for human malignancies. Drug Des Devel Ther. 2015;9:3455–8.
Christmas P. Toll-like receptors: sensors that detect infection. Nat Educ. 2010;3(9):85.
De Paepe B, Creus KK, De Bleecker JL. The tumor necrosis factor superfamily of cytokines in the inflammatory myopathies: potential targets for therapy. Clin Dev Immunol. 2011;2012:369432.
Dinarello CA, van der Meer JWM. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25(6):469–84.
Doss GP, Agoramoorthy G, Chakraborty C. TNF/TNFR: drug target for autoimmune diseases and immune-mediated inflammatory diseases. Front Biosci. 2014;19:1028–40.
Dubé PE, Punit S, Polk DB. Redeeming an old foe: protective as well as pathophysiological roles for tumor necrosis factor in inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2015;308(3):G161–70.
Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, et al. Randomised double blind comparison of a chimaeric monoclonal antibody to tumour necrosis factor-alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. 1994;344:1105–10.
Feldmann M, Maini RN. Anti-TNF-alpha therapy of rheumatoid arthritis: what have we learned? Ann Rev Immunol. 2001;19:163–96.
Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Ann Rev Immunol. 1996;14:397–440.
Giancane G, Minoia F, Davì S, Bracciolini G, Consolaro A, Ravelli A. IL-1 inhibition in systemic juvenile idiopathic arthritis. Front Pharmacol. 2016;7:467.
Goldback-Mansky R. Blocking interleukin-1 in rheumatic diseases. Ann N Y Acad Sci. 2009;1182:111–23.
Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I IFNs. Nat Rev Immunol. 2012;12:125–35.
Haanstra KG, Hofman SO, Lopes Estêvão DM, Blezer EL, Bauer J, Yang LL. Antagonizing the α4β1 integrin, but not α4β7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol. 2013;190(5):1961–73.
Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signaling through the gp130/JAK/STAT pathway. Biochem J. 1998;334:297–314.
Hennigan S, Kavanaugh A. Interleukin-6 inhibitors in the treatment of rheumatoid arthritis. Ther Clin Risk Manag. 2008;4(4):767–75.
Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheum. 2016;12:49–62.
Kim GW, Lee NR, Pi RH, Lim YS, Lee YM, Lee JM. IL-6 inhibitors for treatment of rheumatoid arthritis: past, present, and future. Arch Pharm Res. 2015;38(5):575–84.
Kishimoto J, Akira S, Taga T. IL-6 receptor mechanism of signal transduction. Int J Immunopharmacol. 1992;14:431–8.
Lukina GV, Sigidin Ya A. Certolizumab in therapy of rheumatoid arthritis. Sovrem Revmatol. 2012;2:44–9.
Mayadas TN, Culler X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. 2014;9:181–218.
Mclean LP, Shea-Donahue T, Cross RK. Vedolizumab for the treatment of ulcerative colitis and Crohn’s disease. Immunotherapy. 2012;4(9):883–98.
Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin Sci. 2012;122(4):143–59.
Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69.
Orrock JE, Ilowite NT. Canakinumab for the treatment of active systemic juvenile idiopathic arthritis. Expert Rev Clin Pharmacol. 2016;9(8):1015–24.
Perry AK. The host type I interferon response to viral and bacterial infections. Nat Cell Res. 2005;15(6):407–22.
Shealy D, Cai A, Staquet K, Baker A, Lacy ER, Johns L, et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. MAbs. 2010;2:428–39.
Spooner CE, Markowitz NP, Saravolatz LD. The role of tumor necrosis factor in sepsis. Clin Immunol Immunopathol. 1992;62(1 Pt 2):S11–7.
Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S73–80.
Taylor P. Developing anti-TNF and biology agents. The past, the present, and the future. Rheumatology. 2011;50(8):1351–3.
Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;12:251–76.
Wajant H, et al. Tumor necrosis factor signaling. Cell Death Differ. 2003;10(1):45–65.
Zhang JM, An J. Cytokines, inflammation and pain. Int Anesthes Clin. 2007;45(2):27–37.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Szollosi, D.E., Mathias, C.B. (2020). Modulation of the Innate Immune System. In: Mathias, C., McAleer, J., Szollosi, D. (eds) Pharmacology of Immunotherapeutic Drugs. Springer, Cham. https://doi.org/10.1007/978-3-030-19922-7_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-19922-7_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-19921-0
Online ISBN: 978-3-030-19922-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)