
Abstract
The definition of the mesothelioma genome is expected to have a great impact toward the development of new drugs and therapeutic approaches with a view to precision medicine. A few studies reported that the malignant pleural mesothelioma (MPM) genomic landscape is characterized by a much greater number of genomic losses than point mutations. Inactivating gene fusions, copy losses, and protein-truncating variants (PTVs) mostly affect tumor suppressor genes, above all BAP1, NF2, CDKN2A, and SETD2. Some of them may be exploited to design novel therapeutic strategies. Germline mutations in some of these genes represent MPM-predisposing risk factors. These mutations require a second hit, i.e., asbestos exposure, to induce carcinogenesis. The most studied of these genes is BAP1. Germline mutations in other tumor suppressor genes, mostly involved in DNA repair, have also been identified in about 10% of MPM patients. These patients are more sensitive to asbestos exposure than those who do not carry such mutations and may benefit of specific treatments. Additionally, epigenetic mechanisms, such as methylation or miRNA alterations, may modify gene expression and drive carcinogenesis. The same abnormalities may be used as disease biomarkers or therapeutic targets.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Malignant mesothelioma
- BAP1
- DNA repair genes
- Tumor suppressor genes
- Genetic risk factor
- Germline mutation
- Somatic mutation
4.1 Introduction
The definition of the malignant mesothelioma (MM) genome may have important endpoints, both in terms of pathobiology and translation to clinical practice. Generally, the identification of DNA changes within a tumor genome is useful to identify the molecular events that lead to carcinogenesis or tumor progression, i.e., the driver mutations. Early studies focused on the analysis of single genes, especially TP53. Looking at melanoma and lung cancer genomes, these studies achieved the important milestone of deciphering the mutational profile (signature) generated by two carcinogens, i.e., UV radiation and smoke carcinogens, respectively [1, 2]. The advent of next generation sequencing (NGS) and novel bioinformatic approaches allowed to explore systematically a large number of tumor types. The seminal studies by Stratton and co-workers allowed to identify several signatures, each associated with exposure to a specific carcinogen or due to key events in carcinogenesis, such as inactivation of specific DNA repair mechanisms or activation of deamination enzymes [2].
The identification of abnormalities in specific pathways shed light on shared carcinogenetic pathways in tumors with or without the same histological origin, paving the road to the creation of pathway-specific targeted drugs. In addition, tumor classification may be supported by looking at the tumor genome and transcriptome.
Furthermore, it is important to consider that the individual germline genome can modulate the response to carcinogens and hence transformation. Genetic risk factors are well known for several tumors and may have important translational output. For example, individuals carrying such risk factors may benefit from the implementation of screening programs aimed at early diagnosis of tumors. Additionally, the same risk factor may modify specific carcinogenic pathways and response to specific therapies.
Finally, it is well known that tumor suppressor genes may also be inactivated by epigenetic mechanisms. The term “epigenetic” refers to heritable and reversible changes in the mechanisms that regulate gene activity without altering the genomic sequence. In recent years, there is increasing evidence of the major role of epigenetic mechanisms in tumorigenesis, as well as in drug-response. Much attention is also devoted to epigenetic changes as biomarkers of early disease detection, prognosis, and response to therapy.
In this review, different patterns of genetic and epigenetic signatures of the malignant pleural mesothelioma (MPM) genomes will be discussed, together with peculiar aspects of genetic predisposition and gene/environment interactions. The potential use of these genetic/epigenetic signatures for the development of future therapeutics will also be addressed.
4.2 Genetic Risk Factors of Mesothelioma
MPM carcinogenesis is caused in the large majority of cases by asbestos or asbestos-like fibers exposure. It is well known that the level of asbestos exposure directly correlates with the risk of MM ([3], more details are given in a different chapter of this book), but several epidemiological studies suggested that different individuals may respond differently to this carcinogen. An important observation is that only about 10% of the workers heavily exposed to asbestos develop MPM [4]. Additionally, several papers reported familial aggregations of MPM [5]. These observations suggested the hypothesis of an inherited predisposition that modifies the carcinogenic effect of asbestos.
Generally, inherited predisposition factors are DNA variants that occur in the germline genome and modify the function of a specific gene. They are divided into three classes, depending on the relative risk (RR) they carry: low-, moderate-, and high-risk factors.
Low-risk factors are DNA variants that subtly modify the function of a gene or a biochemical pathway. In this case, a single DNA variant does not have any substantial effect on human phenotypes, but many DNA variants affecting the same biochemical pathway may alter its functions, favoring disease development. Therefore, the disease risk does not follow the rules of Mendelian heredity, because each variant is inherited independently from the others.
Risk factors are identified using genome wide association studies (GWAS) on thousands of patients and controls [6]. Large numbers are required to obtain statistically significant results, because each variant confers a low risk. The aim of these studies is to identify DNA variants that are differently represented in patients versus controls. These studies are expected to increase the knowledge of asbestos carcinogenesis and improve risk evaluation.
So far, only two GWAS on MPM have been performed, both including several hundreds of patients and controls, but not enough to obtain statistically significant results [7, 8]. However, both studies identified a region associated with the MPM status, that included FOXK1, encoding for an interactor of BAP1 (BRCA1-associated protein 1), a well-known high-risk factor for MPM.
BAP1 codes for a tumor suppressor that is frequently deleted in the genomes of several tumors, including cutaneous melanoma, uveal melanoma, mesothelioma, and others [9].
Germline variants in BAP1 characterize the BAP1-tumor predisposition syndrome (BAP1-TPDS, MIM#614327) [10]. Tumor predisposition syndromes are due to germline mutations in tumor suppressor genes and are inherited with an autosomal dominant pattern. The patients with these syndromes show a high or moderate risk for specific tumors during their whole life. Often they develop several independent tumors.
Individuals with BAP1-TPDS show a high risk of developing mesothelioma, cutaneous and uveal melanoma, clear cell renal carcinoma, and basal cell carcinoma [10]. Moreover, they develop peculiar nonmalignant skin tumors, called atypical Spitz tumors or MBAITs (BAP1-mutated atypical intradermal tumors) or bapomas [10, 11].
Patients with BAP1-TPDS and uveal melanoma have a poor prognosis [10, 12], whereas those with mesothelioma seem to have a longer survival than those without BAP1-TPDS [13].
Ninety-seven families with BAP1-TPDS have been identified so far, 48 of them included patients with MM; thus, this syndrome is indeed very rare [11, 14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38]. Age at onset of mesothelioma in patients with BAP1-TPDS is earlier than that in patients without this syndrome [13, 26]. Most of the MM are MPM and show an epithelioid histotype, while peritoneal mesothelioma (PM) has been rarely reported [10]. The prevalence of BAP1-TPDS among patients with familial MPM varied between 6% (9/153) and 7.7% (3/39) [26, 31] and was higher than the prevalence observed in sporadic cases [23, 39, 40].
Other tumors have been reported in patients with BAP1-TPDS, i.e., breast cancer [12, 14, 21, 22], cholangiocarcinoma [12, 22, 41], meningioma [18, 25, 38, 41], neuroendocrine tumors [18, 19], non-small cell lung cancer (NSCLC) [12, 18, 19, 42], thyroid carcinoma [21, 43], and mucoepidermoid carcinoma of the tongue [23].
BAP1 (#MIM 603089) is located on 3p21.1 and encodes for a ubiquitin carboxy-terminal hydrolase, a nuclear enzyme that catalyzes the cleavage of a ubiquitin residue from its target proteins. The product of the gene, BAP1, has three domains: the ubiquitin C-terminal hydrolase domain and two nuclear localization sequences. The BAP1 protein together with FOXK1, HCFC1, ASXL1/2, and OGT [44] forms a multiprotein complex.
BAP1 has been implicated in DNA repair, chromatin modulation, transcriptional regulation, cell proliferation, cell death, and glucidic metabolism [45,46,47,48,49]. The mechanism of BAP1-dependent carcinogenesis is not known, but these functions are not mutually exclusive. BAP1 is involved in DNA repair by the HRR (homologous recombination repair) pathway [49].
Bap1 (+/−) mice are more sensitive to asbestos compared with wild-type mice [50, 51]. Quantification of asbestos exposure has been reported only for four individuals with MPM and BAP1-TPDS: all showed very low exposure [31, 52].
BAP1 germline mutations cause loss of function, and only ten of the different mutations have been identified in patients within apparently non-consanguineous families [24]. Recurrent mutations could be due to mutable hot spots, such as CpG dinucleotides.
Eleven other genes were reported to confer predisposition to MPM: CDKN2A, PALB2, BRCA1, FANCI, ATM, SLX4, BRCA2, FANCC, FANCF, PMS1, and XPC [32, 53] (Table 4.1). All these genes but PMS1 are tumor suppressors, responsible for cancer predisposition syndromes with specific tumor spectra. In particular, BRCA1, BRCA2, ATM, SLX4, and PALB2 can predispose women to breast and ovarian cancer whereas BRCA1 and BRCA2 also to prostate and pancreatic carcinomas [61]; CDKN2A to melanoma and pancreatic cancer [54]; and XPC to basal cell carcinoma, squamous cell carcinoma, and melanoma [62]. PMS1 is involved in MMR (DNA mismatch repair) and possibly in cancer predisposition [63, 64].
Homozygous germline variants in BRCA1 (also called FANCS); BRCA2 (FANCD1); FANCC, FANCI, FANCF, and SLX4 (FANCP); and PALB2 (FANCN) are found in patients with Fanconi anemia, a recessive disease that predisposes to a variety of hematological and solid tumors. This disorder can be caused by at least 20 different genes [65], all acting in a specific signaling pathway activated in response to cross-linking agents.
Mutations in XPC cause the recessive disease xeroderma pigmentosum (MIM# 278720). XPC is involved in the NER (nucleotide excision repair) pathway, a DNA repair system that removes the pyrimidine dimers induced by exposure to ultraviolet radiation.
In most cases, the loss of the wild-type allele, due to a further acquired mutation, induces carcinogenesis in the target tissues of patients with a germline variant. Except for CDKN2A, which is involved in the control of cell proliferation, all these genes have a role in DNA repair.
Anecdotal studies allow to include two more genes involved in cancer predisposition syndromes, NF2 and TP53, because MPM was reported in patients with neurofibromatosis Type 2 or Li-Fraumeni syndrome, due to germline variants in NF2 or TP53 [66, 67], respectively.
Interestingly, some of these genes are often somatically mutated in MPM, i.e., BAP1, CDKN2A, NF2, and TP53 [55, 68, 69].
The involvement of DNA repair genes in MPM risk has been confirmed by others [70] and is in accordance with the observation that 12% of patients with different types of metastatic tumors were reported to carry germline variants, 75% of which in DNA repair genes [71].
Most probably, the development of a specific tumor type in patients with these germline mutations depends on the carcinogen to which they are exposed. If the carcinogen is asbestos, the tumor is likely MPM. Analysis of the genomic signature of the different cancers affecting these patients may confirm this hypothesis.
4.3 The Mesothelioma Genome
Deciphering tumor genomes is important both to gather information about the processes that induce carcinogenesis and to identify druggable pathways in the landscape of precision oncology.
Different methodologies are required to identify point mutations or large rearrangements and copy number variants (CNVs). Ideally, rearrangements and CNVs are studied on the whole genome by using CGH (comparative genomic hybridization) arrays, SNP (single nucleotide polymorphism) arrays, or whole genome sequencing. These methods simultaneously identify all copy gains and copy losses in a genome. Point mutations (also called single nucleotide variants, SNVs) are detected by NGS. Different approaches may be used. Targeted resequencing screens hundreds of known cancer genes that are usually analyzed in the regions corresponding to exons (panel NGS analysis). Exome analysis has the advantage of studying all the genes of the human genome, with a focus on exons. Using appropriate bioinformatic tools, CNVs and rearrangements may be identified in exomes, but not those affecting noncoding regions.
Whole genome analysis addresses the entire genome and could theoretically identify all variants, but management of big data may be time-consuming. In addition, the role of the majority of the genome noncoding regions is not known, so the functional interpretation of variants is difficult.
Usually the cancer and the blood cell genomes are sequenced at the same time to distinguish somatic from germline variants. It should be considered that a very large amount of mutations are generated in each tumor cell at every cell division because of its genetic instability. Therefore, most of these variants are passenger (neutral) variants; only a small number are driver mutations, those that confer a selective advantage to the cell. It has been calculated that only half of the driver mutations in tumors are located in known cancer genes, whereas the others reside in genes or regions whose effect on carcinogenesis is still unknown [72].
The first studies reporting copy gains and copy losses in the mesothelioma genome were published 20 years ago (Table 4.2) [9, 55, 56, 69, 73,74,75,76,77,78,79,80], but point mutations in mesothelioma have been addressed only after the implementation of NGS strategies (Table 4.3) [9, 55,56,57,58,59, 68, 69, 74, 76, 78, 79, 81,82,83,84,85]. Most studies are focused on MPM and show that MPM genomes include a large number of chromosomal abnormalities, such as CNVs and chromosomal translocations often leading to gene fusion, but a relatively low number of protein altering mutations compared with most tumors [60]. These alterations involve mostly tumor suppressor genes. A great inter-individual heterogeneity is also typical.
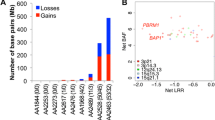
A recent study on CNVs in MPM was performed by Hylebos et al. [77]. They used information obtained using CGH arrays on 85 MPM patients and stored within The Cancer Genome Atlas (TCGA). Data were validated on a panel of 21 patients using low-pass whole genome sequencing. Both datasets showed losses on chromosomes 1, 3, 4, 6, 9, 13, and 22 in 25% of tumors. These losses included CDKN2A, NF2, BAP1, EP300, SETD2, and PBRM1. Copy number gains were less represented compared to losses. They were located on chromosomes 1, 5, 7, and 17 and occurred in 15% of tumors. Genes affected by these gains were TERT, FCGR2B, CD79B, and PRKAR1A. In conclusion, recurrent CNVs were detected in both datasets, occurring in regions harboring known MPM-associated genes and genes not previously linked to MPM.
The first studies addressing the MPM mutational landscape were reported by Lo Iacono et al. and Guo et al., independently in 2015, using different NGS approaches [55, 68]. A limit of both studies is that they included patients who had been subjected to chemotherapy; thus, it is possible that a portion of the mutations was due to the mutagenic effect of the drugs [60]. Lo Iacono et al. investigated 52 cancer genes in FFPE (formalin-fixed, paraffin-embedded) tumor samples of 123 MPM patients [68]. Mutated genes included TP53, SMARCB1, BAP1, PDGFRA, KIT, KDR, HRAS, PIK3CA, STK11, and NF2. The most represented pathways were the p53/DNA repair and the phosphatidylinositol 3-kinase-AKT. Guo et al. performed whole exome sequencing in fresh tumor samples from 22 patients [55]. These samples showed frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1. The MAPK and the Wnt signaling pathway frequently carried alterations.
Bueno et al. reported data on 216 MPM genomes, 99 of which were studied by whole exome and 103 by panel sequencing (344 genes) [69]. These data were paralleled by RNAseq, an approach that investigates all the RNA species transcribed and allows to validate the functional effect of genetic anomalies. They identified the following genes that are often mutated or lost in MPM: BAP1, NF2, TP53, SETD2, DDX3X, ULK2, RYR2, CFAP45, SETDB1, DDX51, TRAF7, and SF3B1. The pathways that were more frequently affected were Hippo, mTOR, histone methylation, RNA helicase, and p53 signaling [69].
De Rienzo et al. performed whole genome sequencing of 10 MPM patients [56]. The identified mutations and copy number aberrations were validated by targeted resequencing of 9 genes in 147 additional samples (BAP1, NF2, TP53, MYH9, MYH6, MYH10, PIK3C2A, RHOA, TNFRSF1A). A further 136 patients were analyzed for TP53, BAP1, NF2, and CDKN2A, which were the most frequently mutated genes. TP53 variants were more often found in women. Interestingly, three patients showed germline PTVs (protein-truncating variants) in BAP1 [56].
Exome NGS was also performed on cells from pleural effusions from 27 patients with MPM. Mutations in BAP1, CDKN2A, and NF2 and loss of TRAF7, LATS2, SETD2, and TP53 were identified [76], suggesting that analysis of pleural effusions might be used to monitor the MPM molecular evolution.
Looking at 61 primary mesothelioma cultures, Tranchant et al. identified a subgroup of tumors harboring both LATS2 and NF2 mutations [84]. Co-occurring mutations in these genes were associated with a poor prognosis. These cell lines showed abnormalities both in the Hippo signaling pathway and mTOR protein expression suggesting specific therapeutic strategies.
FFPE portions from 11 patients (7 MPM and 4 PM) were studied by Ugurluer et al. using a NGS panel including 236 cancer genes [78]. In MPM samples the mutations most commonly found were in BAP1, CDKN2A/B, and NF2. Other PTVs were found in PTCH1, SETD2, STK11, KDM6A, ASXL1, and BRIP1.
Two PM reported by Ugurluer et al. showed mutations in BAP1 or NF2, whereas the other two did not show PTVs. The whole genome of two PM was reported by Sheffield et al. in 2015 [58]. The two patients reported different histology and different response to chemotherapy. The first had an epithelioid histology, a high disease burden, and did not respond to chemotherapy, whereas the second showed minimal clinical symptoms; histology was poor-prognosis sarcomatoid MM but responded well to treatment. The two tumors shared PTVs in NF2 but were elsewhere very different. The first had only 18 variants, whereas the second had more than 260 variants in each of the 2 samples that were studied, corresponding to a status called somatic hypermutation. Another study focused on 12 patients with PM [59]. They used copy number analysis and exome sequencing and targeted sequencing and found a low number of CNVs (mostly losses) and SNVs. The gene that was more frequently affected was BAP1, whereas NF2 and CDKN2A were not affected. One of the patients carried a nonsense germline variant paired to gene loss in the tumor; thus, he had BAP1-TPDS.
Overall, PM seems to have a mutation rate lower than MPM, but driver mutations in PM seem to affect the same genes that are often involved in MPM.
A limit of these studies is that they do not generally consider the hypothesis of intra-tumor heterogeneity, which may be an important issue in mesothelioma considering that there are hints of a polyclonal origin of carcinogenesis [86]. The paper by Zhang et al. focused on testicular MM is a good example of intra-tumor heterogeneity and rapid molecular evolution [87]. They performed whole genome sequencing using DNA obtained from FFPE samples of four successive tumors from a single patient. The first sample was obtained from the primary tumor, whereas the other samples were from a local recurrent tumor, an inguinal lymph node metastasis and a recurrent tumor from the same localization. This study evaluated the tumor progression looking at molecular events. The signature of molecular lesions and also the mutated genes were different from those reported for MPM. Other patients should be studied to evaluate whether this testicular MM is different from the other MM [87].
Tumor exome sequencing may give important information about carcinogenesis in individuals who develop multiple independent tumors. This approach was followed in the case of a 73-year-old male who developed two independent lung cancers (adenocarcinoma and squamous cell carcinoma) and a malignant PM with an epithelioid histology. The patient was a heavy smoker and did not report asbestos exposure. The somatic mutational signatures of the two lung tumors were in agreement with the smoking carcinogen effect, and the mutated genes corresponded to those reported for the tumor types. Conversely, the PM showed a very low number of somatic events, including one PTV in BAP1 and one in SETD2. Several low-risk variants in DNA repair genes could account for the PM predisposition in this patient.
The mutation types prevalent in the tumor genome may be identified in large studies [69]. In particular, Bueno et al. analyzed the mesothelioma exome for transitions (C > T, T > C) and transversions (C > A, C > G, T > A, T > G), taking into account the flanking base immediately 5′ and 3′ of the somatic base (so-called triplets). They identified five distinct signatures (S1, S2, S4, S5, and S6) that are operative in MPMs, two of them being the most represented (S1 and S2). The patterns of contribution of these signatures were different between MPM and lung cancers, in agreement with epidemiological studies that revealed that MPM is not related to smoking like lung cancer. For example, signature S3, characterized by C > A transversions, caused by bulky adducts, is not shown by MPM but is typical of cigarette smoking, an exposure that is not epidemiologically associated with MPM.
The S1 signature is characterized by no predominant transition or transversion and is considered indicative of a base-agnostic mutagen such as reactive oxygen species (ROS) [88, 89]. The S2 signature is represented by C > T transitions at NpCpG trinucleotides and is attributed to an endogenous mechanism, the deamination of 5-methylcytosine to thymine in CpG dinucleotides. The S4 signature is characterized by C > T transitions and is typical of repair errors at UV-induced pyrimidine dimer sites observed in melanoma. Signature S5 shows C > T transitions or C > G transversions at TpCpN nucleotides, considered as indicative of the function of APOBEC enzymes responsible for cytidine deamination and frequently activated in cancer [88, 89].
In conclusion, the study of Bueno et al. identified a mutational pattern concordant with the effect of asbestos exposure (i.e., S1 signature) [69]. The authors did not observe a significant difference of this signature in samples with (n = 69) or without (17) asbestos exposure, but this may depend on the fact that asbestos fiber quantification in the lung was available only for 64/217 patients, whereas asbestos exposure of the other patients was reported, but not quantified.
Overall it is expected that asbestos causes DNA damage in two ways, first by inducing chromosomal breaks by interfering with spindle fibers during cell division and second by inducing inflammation and ROS production. The first mechanism may explain some of the chromosomal rearrangements whereas the second some of the point mutations.
4.4 Translation to the Clinics: Druggable Targets
The identification of driver mutations in mesothelioma is expected to pave the way to precision oncology. In general, this task may be particularly difficult in MPM, considering the wide inter-individual and possibly intra-tumor heterogeneity. Moreover, MPM driver mutations in protein-coding genes are rarer than in other tumors [72]. On the other hand, it is important to note that all these studies reported a frequent involvement of BAP1, NF2, CDKN2A, and SETD2.
A thorough evaluation of possible translational steps is beyond the scope of this review, and we refer to other chapters of this book and specific literature [90, 91]. We only mention that PARP or EZH2 inhibitor drugs have been considered for tumors characterized by BAP1 loss, CDK4/6 or PRMT5 inhibitors for tumors with CDKN2A mutations, FAK inhibitors for tumors with NF2 mutations, and PI3K-AKT inhibitors for tumors with PI3K-AKT abnormalities [90]. More in detail, a phase II clinical trial in BAP1-deficient patients with the EZH2 inhibitor, tazemetostat, was recently opened to accrual (NCTO02860286); a phase II clinical trial to evaluate the CDK4/6 inhibitor, ribociclib, in solid tumors carrying relevant CDK4/6, cyclinD1/3, or p16INK4A aberrations, including MPMs, has been designed (NCT02187783); while after a randomized switch maintenance, clinical trial (NCT01870609) with the FAK inhibitor defactinib (VS-6063) versus placebo was discontinued in late 2015, in 2016 a new single-center clinical trial tested defactinib before surgery for MPM (NCT02004028); at last, the modest response obtained in a phase I study of apitolisib (GDC-0980), dual phosphatidylinositol-3-kinase, and mammalian target of rapamycin kinase inhibitor (NCT00854152) indicated that combination regimens must be explored.
Conversely, predisposing factors may also give some therapeutic opportunities to the patients that carry them. Patients with ovarian cancer and germline variants in BRCA1 or BRCA2 respond to PARP1 inhibitors drugs, through a mechanism called synthetic lethality [92, 93]. This mechanism is induced when two (or more) variants are not lethal singularly but are lethal when both are present in a cell [94]. PARP1 is a nuclear enzyme that functions in three DNA repair systems, i.e., SSBs (single-strand breaks), BER (base excision repair), and alt-NHEJ (alternative nonhomologous end joining) [95]. PARP1 binds to SSBs and causes the formation of polymers of ADP-ribose (PAR) on its target proteins (this phenomenon is called PARylation). PARs are required for the recruitment of SSBs repair scaffolding proteins. PARP1 auto-PARylation is followed by its release from DNA and inactivation [94]. PARP1inhibitors traps PARP1 to the site of DNA damage and interfere with the progression of the replication fork causing the accumulation of SSBs that evolve to DSBs (double-strand breaks), following replication fork collapse. Both HRR and NHEJ (nonhomologous end joining) are used to repair DSBs and restart replication forks stalled by PARP1 inhibitors. When HRR is deficient, because of loss of BRCA1 or BRCA2, the damage cannot be repaired by alt-NHEJ, because this system requires PARP1. If these systems are not functional, cells can only use classical NHEJ, which causes chromosomal anomalies, genomic instability, and cell death [96].
PARP1 inhibitors could inhibit growth of cells that have lost both BAP1 alleles either because of a germline and a somatic variant or because of two somatic variants. Tumor cells in patients with a germline variant in BAP1 have a very high likelihood of a second somatic variant on the wild-type allele. Thus, theoretically, in patients with a germline variant in BAP1 MPM, tumor tissue could have a more homogeneous BAP1 loss than in sporadic patients and may better respond to this treatment. Patients with germline mutations in other HRR genes may also show such behavior.
4.5 Tumor Epigenetics
The mechanisms underlying tumor initiation and progression involve also epigenome aberrations that share an intricate relationship with genetic instability in the tumor evolution process.
Epigenetic includes three main regulatory mechanisms: histone modifications, DNA methylation, and microRNA (miRNA)-mediated gene regulation.
Histones are members of a highly conserved family of proteins that associate with DNA to organize chromatin in the nucleus. Several posttranslational modifications may occur at N-terminal histone tails, including the addition or removal of methyl and acetyl residues. Histone modification is associated with the transcriptional regulation of genes, promoting the transition between open and close chromatin conformation.
DNA methylation consists in the addition of a methyl residue (–CH3) to the cytosine residues within the dinucleotide CpG. DNA methylation mainly occurs at the carbon-5 position of the cytosine ring [97], even though a small fraction (~2%) may occur at cytosines in any context of the genome, or also in a non-CpG context in embryonic stem cells [98]. CpGs DNA methylation may occur in gene promoters, where a high concentration of CpGs dinucleotides can be seen in the so-called CpG islands. Promoter DNA methylation is a well-known mechanism to repress gene transcription, leading to gene silencing through inhibition of transcription factor binding to DNA [99]. Deregulation of the DNA methylation levels may result in cell transformation. Diffuse genome-wide hypomethylation is frequently seen in cancer cells, together with site-specific hypermethylation [100, 101].
miRNAs are a class of small noncoding RNAs involved in gene silencing through a posttranscriptional mechanism that requires miRNA binding to 3′-UTR regions of mRNAs and leads to translation inhibition or mRNA degradation [102]. Dysregulation of miRNAs has been associated to cancer development [103,104,105], and they have been proposed as tools for cancer diagnosis, classification, prognosis, and treatment [106,107,108,109].
Epigenetic alterations may be critical determinants of malignant transformation of pleural mesothelial cells following asbestos exposure. The relationship between DNA methylation modifications and in vitro asbestos exposure in MeT5A mesothelial cell lines was recently described [110]. The authors report slight DNA methylation in MeT5A cells after both crocidolite and chrysotile treatments, mainly in genes involved in the regulation of cellular matrix and adhesion, which are mechanisms for mesothelial infiltration and injury, facilitating epithelial-to-mesenchymal transition (EMT) in MPM. This finding may suggest an involvement of methylation changes as potential modulators of asbestos-induced pleural injury.
Evidence of relationship between asbestos burden and promoter methylation of selected tumor suppressor genes (APC, CCND2, CDKN2A, CDKN2B, HPPBP1, and RASSF1) was also reported in lung tissue from MPM patients. Moreover, the increase in methylation of these genes correlates with asbestos body counts [111]. Inactivation of CDKN2A by methylation was also reported by Kobayashi et al. [112].
The examination of over 6000 CpG islands in MPM and lung adenocarcinomas showed that 387 genes (6.3%) and 544 genes (8.8%) were hypermethylated in MPM and adenocarcinoma, respectively, and that the two malignancies have characteristic DNA methylation patterns, likely a result of different pathologic processes [113]. Moreover, Goto et al. suggest that KAZALD1, MAPK13, and TMEM30B genes, which were specifically methylated only in MPM, could serve as potential diagnostic markers.
In a larger study of 158 mesothelioma specimens and 18 normal pleura samples, Christensen et al. reported that the DNA methylation profile of 803 cancer-associated genes was able to discriminate normal pleura from mesothelioma and was a predictor of shorter survival [114].
Aberrant promoter methylation of WIF-1 and SFRP1, 2, 4 genes was found in MPM tissue and mesothelioma cell lines [115]. The analysis of 52 MPM samples and 38 histologically non-tumor lung samples identified higher methylation levels of ESR1, SLC6A20, and SYK genes in MPM [116]. The combination of SLC6A20, SYK, and APC yielded a sensitivity of 92% and a specificity of 73% as positive markers for MPM. The inclusion of ESR1 methylation as a third positive marker increased sensitivity but reduced specificity.
Cheng et al. [117] reported downregulation of the ZIC1 gene via promoter methylation in MPM. This gene acts as a tumor suppressor, targeting apoptosis-related miRNAs. In particular, miR-23a and miR-27a are expressed at higher levels in epithelioid MPM patients with shorter survival. These studies highlight that epigenetic silencing through promoter hypermethylation is a frequent event in MPM.
Other studies looked for miRNAs involved in MPM development. Guled et al. [118] identified a number of miRNAs that were differentially expressed between MPM tissue and normal pericardium.
With an in vitro study, Pass et al. reported that miR-29c-5p may be a tumor suppressor in MPM and thus a potential therapeutic target [119].
Several miRNA-targeted therapeutics have reached clinical testing. For example, miR-16 is involved in a phase I clinical trial, MesomiR 1. The trial is based on the work by Reid et al. who reported the downregulation of miR-15-16 in MPM tissue and cell lines associated with increased levels of the target oncogenes CCND1 and Bcl-2 [120]. Restoring miRNA expression, cell growth is inhibited, and cells acquire sensitivity to gemcitabine and pemetrexed. miR-16 is also a regulator of programmed death ligand 1 (PD-L1) in MPM and may therefore contribute to immune system evasion [120].
In MPM, miR-34b/c and miR-126 are regulated by methylation and oxidative stress [121, 122]. Several studies showed that miR-34b/c is a regulator of C-MET and BCL-2 oncogenes, and its downregulation promotes transformation of mesothelial cells [122,123,124]. In vivo studies showed that during oxidative stress, miR-126 compromises mitochondrial function, induces autophagy by altering cell metabolism, and inhibits cell growth and tumor formation, showing that increased autophagy has a protective role in MPM [121, 125].
The identification of miRNA target genes is of paramount importance for understanding how these small noncoding RNAs regulate MPM cell function. A recent approach [126] identified miR-21-5p as a candidate regulator of MSLN (mesothelin). The increased expression of miR-21-5p reduced MSLN expression and inhibited MPM cell proliferation, uncovering a potential tumor suppressing miRNA in MPM.
A single miRNA can regulate many genes, and one gene may be targeted by many miRNAs. MCL-1 is overexpressed in MPM and is associated with the resistance to apoptosis and chemotherapy [127]. Khodayari et al. reported that the transfection of MPM cells with miR-302b reduced MCL-1 expression, decreasing cell and tumor growth and inducing apoptosis [128]. The same antitumor activity has been observed for miR-193a-3p, suggesting that miRNA replacement therapy to target MCL-1 may provide an effective treatment for MPM [129].
4.6 Epigenetic as a Potential Diagnostic Biomarker
Epigenetic markers are considered potential biomarkers for early diagnosis and prognosis in cancer research [130].
DNA methylation is rather stable but may change across time [131], and it can be modified by several factors during lifetime [132], such as lifestyle, environmental exposures, aging, and diseases [133, 134]. The DNA methylation asset of each individual is thus considered as an adaptive phenomenon potentially linking environmental factors and development of disease phenotypes [135]. Aberrant DNA methylation is found as an early event in tumor development and has been suggested as a tool for early cancer detection and prognosis [136, 137], including MPM [138].
Whereas tumor tissue DNA methylation is widely investigated in MPM, only few studies addressed the relationship between DNA methylation in blood-derived specimens and MPM.
With a targeted study focused on free serum DNA of mesothelioma patients, Fischer et al. [139] investigated the methylation status of the promoter region of nine candidate genes that were previously shown to be epigenetically altered in MPM tissue and cell lines. The authors reported hypermethylation in the promoter region of FHIT and the gene encoding for E-cadherin and to a lower extent ACP1A, RASSF1A, and DARK genes. Intermediate values were observed for CDKN2A, APC1, ARF, and RARβ [139]. The same study reported a correlation of the methylation levels of DAPK, RASSF1A, and RARβ genes with overall survival, though the effect was only seen in combination.
A recent study [140] investigated for the first time the whole genome DNA methylation levels in peripheral blood cells to assess the potentiality of DNA methylation profiles in blood to discriminate MPM cases from asbestos-exposed controls without MPM. The authors report significant case/control differential DNA methylation (>800 CpG sites) with consistent hypomethylation in MPM cases with respect to controls. Moreover, a small panel of seven differentially methylated CpGs was able to significantly increase discrimination between cases and controls (AUC = 0.81 vs AUC = 0.89) when considering DNA methylation together with asbestos exposure vs asbestos exposure alone.
miRNAs have been also suggested as promising candidates for the development of noninvasive techniques for early cancer detection and as therapeutic targets [141, 142]. Specific miRNA profiles have been suggested as diagnostic/prognostic biomarkers also for MPM [143,144,145,146]. Aberrant miRNA profiles have been already described in MPM tissue and biological fluids [145, 146]. Weber at al. [147], in a pilot study, identified miR-103a-3p in peripheral blood cells as a potential marker for the discrimination of mesothelioma patients from both asbestos-exposed controls and general population. The use of miR-103a-3p improved the discrimination power of serum mesothelin, reaching a sensitivity of 95% and a specificity of 81% when the two biomarkers were combined [147].
More recently, Cavalleri et al. further validated the suitability of miR-103a-3p as a MPM biomarker. A miR-103a-3p/miR-30e-3p signature of plasma-derived extracellular vesicles distinguished MPM patients from subjects reporting a past asbestos exposure with a sensitivity of 95.5% and a specificity of 80.0% [148]. While miR-103a-3p is a potential biomarker, several other studies that investigated miRNA deregulation in plasma/serum yielded heterogeneous and inconclusive results.
miR-200 family members have been suggested as potential candidates for discriminating MPM from lung cancer [144, 145, 149, 150]. Gee et al. reported downregulated miRNAs as potential biomarkers to distinguish MPM and lung adenocarcinoma [149]. Also Benjamin et al. identified a panel of three deregulated miRNAs (miR-193-3p, miR-200c, miR-192) reaching a sensitivity of 100% and a specificity of 94% to discriminate MPM from carcinoma of epithelial origin that may invade the pleura [145, 150]. High diagnostic accuracy was also reached by using a panel of four miRNAs (miR-126, miR-143, miR-145, and miR-652) that were significantly downregulated in MPM compared with nonneoplastic pleura [151]. Santarelli et al. quantified the levels of 88 miRNAs reported to be associated with cancer in 10 samples of MPM and 1 sample of healthy mesothelial tissue using a customized PCR Array [146]. The study identified three miRNAs (miR-335, miR-126, and miR-32), but only miR-126 replicated in 27 FFPE MPM samples and 27 adjacent healthy pleural tissues. Limits of these studies were the small number of miRNA investigated and the different methods used to preserve samples (RNA later in discovery and FFPE in replication phase).
The downregulation of miR-126 is also a significant prognostic factor associated with poor survival [152]. Andersen et al. showed an epigenetic downregulation of miR-126 and its host gene EGFL7. Silencing of EGFL7 is associated with a poor clinical outcome in epithelioid subtype [152]. Understanding DNA hypermethylation of EGFL7 and miR-126 may provide potential avenue for therapeutic intervention.
The first study suggesting that miRNA can be used to predict survival outcomes identified miR-29c-5p as an independent prognostic factor for time to disease progression [119]. Pass et al. identified a signature as a potential tool for predicting survival, based on the expression of let-7c-5p and miR-151a-5p in 52 MPM tumors [153].
4.7 Conclusions and Future Developments
The identification of driver mutations in MPM is a prerequisite for precision medicine, and the results are expected in the long run. The presence of germline predisposing mutations in tumor suppressor genes may be useful to identify the driver genes in cancers and address their specific therapy. miRNAs are also attractive therapeutic targets because of their powerful regulatory functions.
Additionally, different epigenetic profiles, which include miRNA and DNA methylation, in peripheral blood might be a useful tool to monitor exposed subjects.
References
Pfeifer GP. Environmental exposures and mutational patterns of cancer genomes. Genome Med. 2010;2:54.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21.
Ferrante D, Mirabelli D, Tunesi S, Terracini B, Magnani C. Pleural mesothelioma and occupational and non-occupational asbestos exposure: a case-control study with quantitative risk assessment. Occup Environ Med. 2016;73:147–53.
Hodgson JT, Darnton A. The quantitative risks of mesothelioma and lung cancer in relation to asbestos exposure. Ann Occup Hyg. 2000;44:565–601.
Ugolini D, Neri M, Ceppi M, Cesario A, Dianzani I, Filiberti R, et al. Genetic susceptibility to malignant mesothelioma and exposure to asbestos: the influence of the familial factor. Mutat Res. 2008;658:162–71.
Sud A, Kinnersley B, Houlston RS. Genome-wide association studies of cancer: current insights and future perspectives. Nat Rev Cancer. 2017;17:692–704.
Matullo G, Guarrera S, Betti M, Fiorito G, Ferrante D, Voglino F, et al. Genetic variants associated with increased risk of malignant pleural mesothelioma: a genome-wide association study. PLoS One. 2013;8:e61253.
Cadby G, Mukherjee S, Musk AWB, Reid A, Garlepp M, Dick I, et al. A genome-wide association study for malignant mesothelioma risk. Lung Cancer. 2013;82:1–8.
Nasu M, Emi M, Pastorino S, Tanji M, Powers A, Luk H, et al. High incidence of somatic BAP1 alterations in sporadic malignant mesothelioma. J Thorac Oncol. 2015;10:565–76.
Pilarski R, Rai K, Cebulla C, Abdel-Rahman M. BAP1 tumor predisposition syndrome. Seattle: University of Washington; 2016.
Carbone M, Ferris LK, Baumann F, Napolitano A, Lum CA, Flores EG, et al. BAP1 cancer syndrome: malignant mesothelioma, uveal and cutaneous melanoma, and MBAITs. J Transl Med. 2012;10:179.
Njauw C-NJ, Kim I, Piris A, Gabree M, Taylor M, Lane AM, et al. Germline BAP1 inactivation is preferentially associated with metastatic ocular melanoma and cutaneous-ocular melanoma families. PLoS One. 2012;7:e35295.
Baumann F, Flores E, Napolitano A, Kanodia S, Taioli E, Pass H, et al. Mesothelioma patients with germline BAP1 mutations have 7-fold improved long-term survival. Carcinogenesis. 2015;36:76–81.
Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–5.
Haugh AM, Njauw C-N, Bubley JA, Verzì AE, Zhang B, Kudalkar E, et al. Genotypic and phenotypic features of BAP1 Cancer syndrome: a report of 8 new families and review of cases in the literature. JAMA Dermatol. 2017;153:999–1006.
Rai K, Pilarski R, Boru G, Rehman M, Saqr AH, Massengill JB, et al. Germline BAP1 alterations in familial uveal melanoma. Genes Chromosomes Cancer. 2017;56:168–74.
O’Shea SJ, Robles-Espinoza CD, McLellan L, Harrigan J, Jacq X, Hewinson J, et al. A population-based analysis of germline BAP1 mutations in melanoma. Hum Mol Genet. 2017;26:717–28.
Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856–9.
Wadt K, Choi J, Chung J-Y, Kiilgaard J, Heegaard S, Drzewiecki KT, et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012;25:815–8.
Wiesner T, Fried I, Ulz P, Stacher E, Popper H, Murali R, et al. Toward an improved definition of the tumor spectrum associated with BAP1 germline mutations. J Clin Oncol. 2012;30:e337–40.
Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d’Enghien C, et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet. 2013;92:974–80.
Pilarski R, Cebulla CM, Massengill JB, Rai K, Rich T, Strong L, et al. Expanding the clinical phenotype of hereditary BAP1 cancer predisposition syndrome, reporting three new cases. Genes Chromosomes Cancer. 2014;53:177–82.
Betti M, Casalone E, Ferrante D, Romanelli A, Grosso F, Guarrera S, et al. Inference on germline BAP1 mutations and asbestos exposure from the analysis of familial and sporadic mesothelioma in a high-risk area. Genes Chromosomes Cancer. 2015;54:51–62.
Rai K, Pilarski R, Cebulla CM, Abdel-Rahman MH. Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet. 2016;89:285–94.
Cheung M, Kadariya Y, Talarchek J, Pei J, Ohar JA, Kayaleh OR, et al. Germline BAP1 mutation in a family with high incidence of multiple primary cancers and a potential gene-environment interaction. Cancer Lett. 2015;369:261–5.
Ohar JA, Cheung M, Talarchek J, Howard SE, Howard TD, Hesdorffer M, et al. Germline BAP1 mutational landscape of Asbestos-exposed malignant mesothelioma patients with family history of Cancer. Cancer Res. 2016;76:206–15.
Abdel-Rahman MH, Rai K, Pilarski R, Davidorf FH, Cebulla CM. Germline BAP1 mutations misreported as somatic based on tumor-only testing. Familial Cancer. 2016;15:327–30.
Ribeiro C, Campelos S, Moura CS, Machado JC, Justino A, Parente B. Well-differentiated papillary mesothelioma: clustering in a Portuguese family with a germline BAP1 mutation. Ann Oncol. 2013;24:2147–50.
Cheung M, Talarchek J, Schindeler K, Saraiva E, Penney LS, Ludman M, et al. Further evidence for germline BAP1 mutations predisposing to melanoma and malignant mesothelioma. Cancer Genet. 2013;206:206–10.
Turunen JA, Markkinen S, Wilska R, Saarinen S, Raivio V, Täll M, et al. BAP1 germline mutations in finnish patients with uveal melanoma. Ophthalmology. 2016;123:1112–7.
Betti M, Aspesi A, Ferrante D, Sculco M, Righi L, Mirabelli D, et al. Sensitivity to asbestos is increased in patients with mesothelioma and pathogenic germline variants in BAP1 or other DNA repair genes. Genes Chromosomes Cancer. 2018;57(11):573–83.
Betti M, Aspesi A, Biasi A, Casalone E, Ferrante D, Ogliara P, et al. CDKN2A and BAP1 germline mutations predispose to melanoma and mesothelioma. Cancer Lett. 2016;378:120–30.
Harbour JW, Onken MD, Roberson EDO, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–3.
Höiom V, Edsgärd D, Helgadottir H, Eriksson H, All-Ericsson C, Tuominen R, et al. Hereditary uveal melanoma: a report of a germline mutation in BAP1. Genes Chromosomes Cancer. 2013;52:378–84.
Busam KJ, Wanna M, Wiesner T. Multiple epithelioid spitz nevi or tumors with loss of BAP1 expression. JAMA Dermatol. 2013;149:335.
Gupta MP, Lane AM, DeAngelis MM, Mayne K, Crabtree M, Gragoudas ES, et al. Clinical characteristics of uveal melanoma in patients with germline BAP1 mutations. JAMA Ophthalmol. 2015;133:881–7.
Gerami P, Yélamos O, Lee CY, Obregon R, Yazdan P, Sholl LM, et al. Multiple cutaneous melanomas and clinically atypical moles in a patient with a novel germline BAP1 mutation. JAMA Dermatol. 2015;151:1235–9.
de la Fouchardière A, Cabaret O, Savin L, Combemale P, Schvartz H, Penet C, et al. Germline BAP1 mutations predispose also to multiple basal cell carcinomas. Clin Genet. 2015;88:273–7.
Rusch A, Ziltener G, Nackaerts K, Weder W, Stahel RA, Felley-Bosco E. Prevalence of BRCA-1 associated protein 1 germline mutation in sporadic malignant pleural mesothelioma cases. Lung Cancer. 2015;87:77–9.
Sneddon S, Leon JS, Dick IM, Cadby G, Olsen N, Brims F, et al. Absence of germline mutations in BAP1 in sporadic cases of malignant mesothelioma. Gene. 2015;563:103–5.
Wadt KAW, Aoude LG, Johansson P, Solinas A, Pritchard A, Crainic O, et al. A recurrent germline BAP1 mutation and extension of the BAP1 tumor predisposition spectrum to include basal cell carcinoma. Clin Genet. 2015;88:267–72.
Aoude LG, Wadt K, Bojesen A, Crüger D, Borg A, Trent JM, et al. A BAP1 mutation in a Danish family predisposes to uveal melanoma and other cancers. PLoS One. 2013;8:e72144.
McDonnell KJ, Gallanis GT, Heller KA, Melas M, Idos GE, Culver JO, et al. A novel BAP1 mutation is associated with melanocytic neoplasms and thyroid cancer. Cancer Genet. 2016;209:75–81.
White AE, Harper JW. Cancer. Emerging anatomy of the BAP1 tumor suppressor system. Science. 2012;337:1463–4.
Daou S, Hammond-Martel I, Mashtalir N, Barbour H, Gagnon J, Iannantuono NVG, et al. The BAP1/ASXL2 histone H2A deubiquitinase complex regulates cell proliferation and is disrupted in cancer. J Biol Chem. 2015;290:28643–63.
Yu H, Pak H, Hammond-Martel I, Ghram M, Rodrigue A, Daou S, et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc Natl Acad Sci U S A. 2014;111:285–90.
Ji Z, Mohammed H, Webber A, Ridsdale J, Han N, Carroll JS, et al. The forkhead transcription factor FOXK2 acts as a chromatin targeting factor for the BAP1-containing histone deubiquitinase complex. Nucleic Acids Res. 2014;42:6232–42.
Bononi A, Yang H, Giorgi C, Patergnani S, Pellegrini L, Su M, et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017;24:1694–704.
Ismail IH, Davidson R, Gagné J-P, Xu ZZ, Poirier GG, Hendzel MJ. Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res. 2014;74:4282–94.
Napolitano A, Pellegrini L, Dey A, Larson D, Tanji M, Flores EG, et al. Minimal asbestos exposure in germline BAP1 heterozygous mice is associated with deregulated inflammatory response and increased risk of mesothelioma. Oncogene. 2016;35:1996–2002.
Xu J, Kadariya Y, Cheung M, Pei J, Talarchek J, Sementino E, et al. Germline mutation of Bap1 accelerates development of asbestos-induced malignant mesothelioma. Cancer Res. 2014;74:4388–97.
Betti M, Aspesi A, Sculco M, Matullo G, Magnani C, Dianzani I. Genetic predisposition for malignant mesothelioma: a concise review. Mutat Res. 2019;781:1–10.
Betti M, Casalone E, Ferrante D, Aspesi A, Morleo G, Biasi A, et al. Germline mutations in DNA repair genes predispose asbestos-exposed patients to malignant pleural mesothelioma. Cancer Lett. 2017;405:38–45.
Aoude LG, Wadt KAW, Pritchard AL, Hayward NK. Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res. 2015;28:148–60.
Guo G, Chmielecki J, Goparaju C, Heguy A, Dolgalev I, Carbone M, et al. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015;75:264–9.
De Rienzo A, Archer MA, Yeap BY, Dao N, Sciaranghella D, Sideris AC, et al. Gender-specific molecular and clinical features underlie malignant pleural mesothelioma. Cancer Res. 2016;76:319–28.
Vanni I, Coco S, Bonfiglio S, Cittaro D, Genova C, Biello F, et al. Whole exome sequencing of independent lung adenocarcinoma, lung squamous cell carcinoma, and malignant peritoneal mesothelioma. Medicine (Baltimore). 2016;95:e5447.
Sheffield BS, Tinker AV, Shen Y, Hwang H, Li-Chang HH, Pleasance E, et al. Personalized oncogenomics: clinical experience with malignant peritoneal mesothelioma using whole genome sequencing. PLoS One. 2015;10:e0119689.
Alakus H, Yost SE, Woo B, French R, Lin GY, Jepsen K, et al. BAP1 mutation is a frequent somatic event in peritoneal malignant mesothelioma. J Transl Med. 2015;13:122.
Carbone M, Gaudino G, Yang H. Recent insights emerging from malignant mesothelioma genome sequencing. J Thorac Oncol. 2015;10:409–11.
Petrucelli N, Daly MB, Pal T. BRCA1- and BRCA2-associated hereditary breast and ovarian cancer. Seattle: University of Washington; 2016.
Kraemer KH, DiGiovanna JJ. Xeroderma pigmentosum. Seattle: University of Washington; 2016.
Peltomäki P. Lynch syndrome genes. Familial Cancer. 2005;4:227–32.
Wang Y, Zhou X, Song Y, Ji X, Zhang A, Zhang G, et al. The mismatch repair gene hPMS1 (human postmeiotic segregation 1) is down regulated in oral squamous cell carcinoma. Gene. 2013;524:28–34.
Mehta PA, Tolar J. Fanconi Anemia. Seattle: University of Washington; 2018.
Ceelen WP, Van Dalen T, Van Bockstal M, Libbrecht L, Sijmons RH. Malignant peritoneal mesothelioma in a patient with Li-Fraumeni syndrome. J Clin Oncol. 2011;29:e503–5.
Baser ME, Rai H, Wallace AJ, Evans DGR. Neurofibromatosis 2 (NF2) and malignant mesothelioma in a man with a constitutional NF2 missense mutation. Familial Cancer. 2005;4:321–2.
Lo Iacono M, Monica V, Righi L, Grosso F, Libener R, Vatrano S, et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: a retrospective study. J Thorac Oncol. 2015;10:492–9.
Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. 2016;48:407–16.
Panou V, Gadiraju M, Wolin A, Weipert CM, Skarda E, Husain AN, et al. Frequency of germline mutations in cancer susceptibility genes in malignant mesothelioma. J Clin Oncol. 2018;36:2863–71.
Robinson DR, Wu Y-M, Lonigro RJ, Vats P, Cobain E, Everett J, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548:297–303.
Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell. 2017;171:1029–1041.e21.
Lee WC, Testa JR. Somatic genetic alterations in human malignant mesothelioma (review). Int J Oncol. 1999;14:181–8.
Bott M, Brevet M, Taylor BS, Shimizu S, Ito T, Wang L, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–72.
Borczuk AC, Pei J, Taub RN, Levy B, Nahum O, Chen J, et al. Genome-wide analysis of abdominal and pleural malignant mesothelioma with DNA arrays reveals both common and distinct regions of copy number alteration. Cancer Biol Ther. 2016;17:328–35.
Sneddon S, Dick I, Lee YCG, Musk AWB, Patch A-M, Pearson JV, et al. Malignant cells from pleural fluids in malignant mesothelioma patients reveal novel mutations. Lung Cancer. 2018;119:64–70.
Hylebos M, Van Camp G, Vandeweyer G, Fransen E, Beyens M, Cornelissen R, et al. Large-scale copy number analysis reveals variations in genes not previously associated with malignant pleural mesothelioma. Oncotarget. 2017;8:113673–86.
Ugurluer G, Chang K, Gamez ME, Arnett AL, Jayakrishnan R, Miller RC, et al. Genome-based mutational analysis by next generation sequencing in patients with malignant pleural and peritoneal mesothelioma. Anticancer Res. 2016;36:2331–8.
Kim JE, Kim D, Hong YS, Kim K-P, Yoon YK, Lee DH, et al. Mutational profiling of malignant mesothelioma revealed potential therapeutic targets in EGFR and NRAS. Transl Oncol. 2018;11:268–74.
Bueno R, De Rienzo A, Dong L, Gordon GJ, Hercus CF, Richards WG, et al. Second generation sequencing of the mesothelioma tumor genome. PLoS One. 2010;5:e10612.
Tallet A, Nault J-C, Renier A, Hysi I, Galateau-Sallé F, Cazes A, et al. Overexpression and promoter mutation of the TERT gene in malignant pleural mesothelioma. Oncogene. 2014;33:3748–52.
Yoshikawa Y, Sato A, Tsujimura T, Emi M, Morinaga T, Fukuoka K, et al. Frequent inactivation of the BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci. 2012;103:868–74.
Zauderer MG, Bott M, McMillan R, Sima CS, Rusch V, Krug LM, et al. Clinical characteristics of patients with malignant pleural mesothelioma harboring somatic BAP1 mutations. J Thorac Oncol. 2013;8:1430–3.
Tranchant R, Quetel L, Tallet A, Meiller C, Renier A, de Koning L, et al. Co-occurring mutations of tumor suppressor genes, LATS2 and NF2, in malignant pleural mesothelioma. Clin Cancer Res. 2017;23:3191–202.
Kiyotani K, Park J-H, Inoue H, Husain A, Olugbile S, Zewde M, et al. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. 2017;6:e1278330.
Comertpay S, Pastorino S, Tanji M, Mezzapelle R, Strianese O, Napolitano A, et al. Evaluation of clonal origin of malignant mesothelioma. J Transl Med. 2014;12:301.
Zhang S, Zhang Q, Sun Q, Tang J, Chen J, Ji N, et al. Genome evolution analysis of recurrent testicular malignant mesothelioma by whole-genome sequencing. Cell Physiol Biochem. 2018;45:163–74.
Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–59.
Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93.
Yap TA, Aerts JG, Popat S, Fennell DA. Novel insights into mesothelioma biology and implications for therapy. Nat Rev Cancer. 2017;17:475–88.
McCambridge AJ, Napolitano A, Mansfield AS, Fennell DA, Sekido Y, Nowak AK, et al. Progress in the management of malignant pleural mesothelioma in 2017. J Thorac Oncol. 2018;13:606–23.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34.
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87.
Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–8.
Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7:20–37.
Ohmoto A, Yachida S. Current status of poly(ADP-ribose) polymerase inhibitors and future directions. Onco Targets Ther. 2017;10:5195–208.
Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer. 2011;2:607–17.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22.
Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22.
Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629–56.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
Macfarlane L-A, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010;11:537–61.
Romero-Cordoba SL, Salido-Guadarrama I, Rodriguez-Dorantes M. Hidalgo-Miranda A: miRNA biogenesis: biological impact in the development of cancer. Cancer Biol Ther. 2014;15:1444–55.
Hata A, Kashima R. Dysregulation of microRNA biogenesis machinery in cancer. Crit Rev Biochem Mol Biol. 2016;51:121–34.
Melo SA, Esteller M. Disruption of microRNA nuclear transport in human cancer. Semin Cancer Biol. 2014;27:46–51.
Steer CJ, Subramanian S. Circulating microRNAs as biomarkers: a new frontier in diagnostics. Liver Transpl. 2012;18:265–9.
Allegra A, Alonci A, Campo S, Penna G, Petrungaro A, Gerace D, et al. Circulating microRNAs: new biomarkers in diagnosis, prognosis and treatment of cancer (review). Int J Oncol. 2012;41:1897–912.
Wu W, Sun M, Zou G-M, Chen J. MicroRNA and cancer: current status and prospective. Int J Cancer. 2007;120:953–60.
Tutar L, Tutar E, Özgür A, Tutar Y. Therapeutic targeting of microRNAs in cancer: future perspectives. Drug Dev Res. 2015;76:382–8.
Casalone E, Allione A, Viberti C, Pardini B, Guarrera S, Betti M, et al. DNA methylation profiling of asbestos-treated MeT5A cell line reveals novel pathways implicated in asbestos response. Arch Toxicol. 2018;92(5):1785–95.
Christensen BC, Godleski JJ, Marsit CJ, Houseman EA, Lopez-Fagundo CY, Longacker JL, et al. Asbestos exposure predicts cell cycle control gene promoter methylation in pleural mesothelioma. Carcinogenesis. 2008;29:1555–9.
Kobayashi N, Toyooka S, Yanai H, Soh J, Fujimoto N, Yamamoto H, et al. Frequent p16 inactivation by homozygous deletion or methylation is associated with a poor prognosis in Japanese patients with pleural mesothelioma. Lung Cancer. 2008;62:120–5.
Goto Y, Shinjo K, Kondo Y, Shen L, Toyota M, Suzuki H, et al. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res. 2009;69:9073–82.
Christensen BC, Houseman EA, Godleski JJ, Marsit CJ, Longacker JL, Roelofs CR, et al. Epigenetic profiles distinguish pleural mesothelioma from normal pleura and predict lung asbestos burden and clinical outcome. Cancer Res. 2009;69:227–34.
Kohno H, Amatya VJ, Takeshima Y, Kushitani K, Hattori N, Kohno N, et al. Aberrant promoter methylation of WIF-1 and SFRP1, 2, 4 genes in mesothelioma. Oncol Rep. 2010;24:423–31.
Tsou JA, Galler JS, Wali A, Ye W, Siegmund KD, Groshen S, et al. DNA methylation profile of 28 potential marker loci in malignant mesothelioma. Lung Cancer. 2007;58:220–30.
Cheng YY, Kirschner MB, Cheng NC, Gattani S, Klebe S, Edelman JJB, et al. ZIC1 is silenced and has tumor suppressor function in malignant pleural mesothelioma. J Thorac Oncol. 2013;8:1317–28.
Guled M, Lahti L, Lindholm PM, Salmenkivi K, Bagwan I, Nicholson AG, et al. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma -A miRNA microarray analysis. Genes Chromosomes Cancer. 2009;48:615–23.
Pass HI, Goparaju C, Ivanov S, Donington J, Carbone M, Hoshen M, et al. hsa-miR-29c∗ is linked to the prognosis of malignant pleural mesothelioma. Cancer Res. 2010;70:1916–24.
Reid G, Pel ME, Kirschner MB, Cheng YY, Mugridge N, Weiss J, et al. Restoring expression of miR-16: a novel approach to therapy for malignant pleural mesothelioma. Ann Oncol. 2013;24:3128–35.
Tomasetti M, Nocchi L, Staffolani S, Manzella N, Amati M, Goodwin J, et al. MicroRNA-126 suppresses mesothelioma malignancy by targeting IRS1 and interfering with the mitochondrial function. Antioxid Redox Signal. 2014;21:2109–25.
Kubo T, Toyooka S, Tsukuda K, Sakaguchi M, Fukazawa T, Soh J, et al. Epigenetic silencing of microRNA-34b/c plays an important role in the pathogenesis of malignant pleural mesothelioma. Clin Cancer Res. 2011;17:4965–74.
Tanaka N, Toyooka S, Soh J, Tsukuda K, Shien K, Furukawa M, et al. Downregulation of microRNA-34 induces cell proliferation and invasion of human mesothelial cells. Oncol Rep. 2013;29:2169–74.
Maki Y, Asano H, Toyooka S, Soh J, Kubo T, Katsui K, et al. MicroRNA miR-34b/c enhances cellular radiosensitivity of malignant pleural mesothelioma cells. Anticancer Res. 2012;32:4871–5.
Tomasetti M, Monaco F, Manzella N, Rohlena J, Rohlenova K, Staffolani S, et al. MicroRNA-126 induces autophagy by altering cell metabolism in malignant mesothelioma. Oncotarget. 2016;7:36338–52.
Vencken S, Hassan T, McElvaney NG, Smith SGJ. Greene CM: miR-CATCH: microRNA capture affinity technology. Methods Mol Biol. 2015;1218:365–73.
Soini Y, Kinnula V, Kaarteenaho-Wiik R, Kurttila E, Linnainmaa K, Pääkkö P. Apoptosis and expression of apoptosis regulating proteins bcl-2, mcl-1, bcl-X, and bax in malignant mesothelioma. Clin Cancer Res. 1999;5:3508–15.
Khodayari N, Mohammed KA, Lee H, Kaye F, Nasreen N. MicroRNA-302b targets Mcl-1 and inhibits cell proliferation and induces apoptosis in malignant pleural mesothelioma cells. Am J Cancer Res. 2016;6:1996–2009.
Williams M, Kirschner MB, Cheng YY, Hanh J, Weiss J, Mugridge N, et al. miR-193a-3p is a potential tumor suppressor in malignant pleural mesothelioma. Oncotarget. 2015;6:23480–95.
Yang X, Dai W, Kwong DL, Szeto CYY, Wong EH, Ng WT, et al. Epigenetic markers for noninvasive early detection of nasopharyngeal carcinoma by methylation-sensitive high resolution melting. Int J Cancer. 2015;136:E127–35.
Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–83.
Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009;5:e1000602.
Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Front Cell Dev Biol. 2014;2:49.
Bell CG, Beck S. The epigenomic interface between genome and environment in common complex diseases. Brief Funct Genomics. 2010;9:477–85.
Marsit CJ. Influence of environmental exposure on human epigenetic regulation. J Exp Biol. 2015;218:71–9.
Shivapurkar N, Gazdar AF. DNA methylation based biomarkers in non-invasive cancer screening. Curr Mol Med. 2010;10:123–32.
Dong Y, Zhao H, Li H, Li X, Yang S. DNA methylation as an early diagnostic marker of cancer (review). Biomed Rep. 2014;2:326–30.
Vandermeers F, Neelature Sriramareddy S, Costa C, Hubaux R, Cosse J-P, Willems L. The role of epigenetics in malignant pleural mesothelioma. Lung Cancer. 2013;81:311–8.
Fischer JR, Ohnmacht U, Rieger N, Zemaitis M, Stoffregen C, Kostrzewa M, et al. Promoter methylation of RASSF1A, RARbeta and DAPK predict poor prognosis of patients with malignant mesothelioma. Lung Cancer. 2006;54:109–16.
Guarrera S, Viberti C, Cugliari G, Allione A, Casalone E, Betti M, et al. Peripheral blood DNA methylation as potential biomarker of malignant pleural mesothelioma in asbestos-exposed subjects. J Thorac Oncol. 2019;14:527–39.
Bolha L, Ravnik-Glavač M, Glavač D. Circular RNAs: biogenesis, function, and a role as possible cancer biomarkers. Int J Genomics. 2017;2017:6218353.
Di Leva G, Garofalo M, Croce CM. MicroRNAs in cancer. Annu Rev Pathol. 2014;9:287–314.
Ramírez-Salazar EG, Salinas-Silva LC, Vázquez-Manríquez ME, Gayosso-Gómez LV, Negrete-Garcia MC, Ramírez-Rodriguez SL, et al. Analysis of microRNA expression signatures in malignant pleural mesothelioma, pleural inflammation, and atypical mesothelial hyperplasia reveals common predictive tumorigenesis-related targets. Exp Mol Pathol. 2014;97:375–85.
Birnie KA, Prêle CM, Thompson PJ, Badrian B, Mutsaers SE. Targeting microRNA to improve diagnostic and therapeutic approaches for malignant mesothelioma. Oncotarget. 2017;8:78193–207.
Kirschner MB, Cheng YY, Badrian B, Kao SC, Creaney J, Edelman JJB, et al. Increased circulating miR-625-3p: a potential biomarker for patients with malignant pleural mesothelioma. J Thorac Oncol. 2012;7:1184–91.
Santarelli L, Strafella E, Staffolani S, Amati M, Emanuelli M, Sartini D, et al. Association of MiR-126 with soluble mesothelin-related peptides, a marker for malignant mesothelioma. PLoS One. 2011;6:e18232.
Weber DG, Casjens S, Johnen G, Bryk O, Raiko I, Pesch B, et al. Combination of MiR-103a-3p and mesothelin improves the biomarker performance of malignant mesothelioma diagnosis. PLoS One. 2014;9:e114483.
Cavalleri T, Angelici L, Favero C, Dioni L, Mensi C, Bareggi C, et al. Plasmatic extracellular vesicle microRNAs in malignant pleural mesothelioma and asbestos-exposed subjects suggest a 2-miRNA signature as potential biomarker of disease. PLoS One. 2017;12:e0176680.
Gee GV, Koestler DC, Christensen BC, Sugarbaker DJ, Ugolini D, Ivaldi GP, et al. Downregulated microRNAs in the differential diagnosis of malignant pleural mesothelioma. Int J Cancer. 2010;127:2859–69.
Benjamin H, Lebanony D, Rosenwald S, Cohen L, Gibori H, Barabash N, et al. A diagnostic assay based on microRNA expression accurately identifies malignant pleural mesothelioma. J Mol Diagn. 2010;12:771–9.
Andersen M, Grauslund M, Ravn J, Sørensen JB, Andersen CB, Santoni-Rugiu E. Diagnostic potential of miR-126, miR-143, miR-145, and miR-652 in malignant pleural mesothelioma. J Mol Diagn. 2014;16:418–30.
Andersen M, Trapani D, Ravn J, Sørensen JB, Andersen CB, Grauslund M, et al. Methylation-associated silencing of microRNA-126 and its host gene EGFL7 in malignant pleural mesothelioma. Anticancer Res. 2015;35:6223–9.
De Santi C, Melaiu O, Bonotti A, Cascione L, Di Leva G, Foddis R, et al. Deregulation of miRNAs in malignant pleural mesothelioma is associated with prognosis and suggests an alteration of cell metabolism. Sci Rep. 2017;7:3140.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Aspesi, A. et al. (2019). Genetics and Epigenetics of Mesothelioma. In: Ceresoli, G., Bombardieri, E., D'Incalci, M. (eds) Mesothelioma. Springer, Cham. https://doi.org/10.1007/978-3-030-16884-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-16884-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16883-4
Online ISBN: 978-3-030-16884-1
eBook Packages: MedicineMedicine (R0)