
Abstract
Fragment-like natural products play a pivotal role in natural product research given their improved synthetic and computational tractability as well as commercial availability compared to more complex natural product structures. A multitude of computational tools have been developed to support the generation, analysis, and application of natural fragments for drug discovery and chemical biology research. In this contribution, the challenges and opportunities in such workflows are discussed and contextualized. Multiple successful applications and validations discussed herein attest to the relevance of natural fragments for drug discovery and the utility of machine learning and data science to support such endeavors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cheminformatics
- Data science
- Machine learning
- Substructure search
- Fragment-based drug discovery
- Natural products
- Retrosynthesis
- Scaffolds
1 Introduction
Natural products have been fueling drug discovery pipelines for decades [1, 2]. However, many challenging hurdles have hampered the straightforward application of complex natural product structures for drug discovery, such as a lack of synthetic accessibility for large-scale production [3, 4] as well as their unknown and insufficiently predictable polypharmacological properties [5, 6]. Conversely, natural product fragments overcome many of such shortcomings and provide privileged structures with ample opportunities for optimization and derivatization into synthetically accessible mimetics with known or predictable biological effects [4, 5, 7, 8]. It has been shown that natural product fragments can provide key substructures to bias a compound collection toward biological activity [9, 10], and it has been argued that fragment-sized natural products and natural fragments constitute some of the most relevant of natural products for drug discovery and development [11]. The biosynthesis of a large natural product often relies on the synthesis of fragment-like building blocks, such that these structures might present biologically motivated handles to further explore them as chemical probes and drug leads [12].
Furthermore, natural fragments provide innovative as well as structurally and spatially intricate molecular probes for fragment-based drug discovery [4, 8]. This is particularly relevant given the much smaller size of chemical fragment space: since there are orders of magnitude less possible fragment structures compared to the unfathomable larger number of possible organic molecules [10], a smaller fragment collection might be capable of spanning the complete chemical (fragment) space with sufficient resolution [13]. This implies that the high-throughput testing of fragments and their medicinal chemistry is more likely to enable the design of bioactive compounds with optimal activity profile [14]. Indeed, focusing on natural fragments benefits from the advantages of chemically diverse and complex natural product structures, while simultaneously harnessing fragment-based drug discovery for finding smaller structures that fit optimally into a binding pocket and can then be rationally further optimized [13]. The potential for such optimization is additionally attested to by orthogonal work on privileged fragments and scaffolds [5, 15], which are notorious for binding certain target classes and families. This advocates for the selection of molecular substructures that contribute most significantly to the desired activity of biologically active molecules—and natural product fragments constitute a prime resource to study these [16].
With these vital benefits of natural fragments in mind, it is important to realize that cheminformatic and bioinformatic approaches have been instrumental in generating and curating databases of natural product-derived fragments [4, 8, 17], analyzing and stratifying their properties [17, 18], as well as guiding their applications for drug discovery and chemical biology [5, 16, 19,20,21,22,23]. Many in silico tools exist that have been validated with impressive success in the context of natural fragments to support their generation, property and polypharmacology predictions, and derivatization, among others [9, 15, 24, 25]. This contribution discusses various challenges and opportunities occurring at different stages of computer-guided natural product fragment research, currently available computational tools to address these, as well as outstanding research questions and the prospective impact of computational workflows on natural product-based drug discovery and chemical biology.
2 Sources of Natural Product Fragments
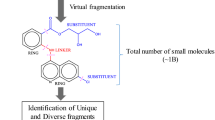
To utilize natural product fragments in drug discovery and pharmaceutical research, it is of utmost importance to source a large set of reliable and meaningful chemical structures for further analysis or screening. A common selection criterion for natural products and fragments is compound availability [4], and multiple chemical vendors now supply chemical structures of natural product fragments that they offer commercially for academic and industrial researchers to fuel natural fragment-based screening efforts [26,27,28,29]. However, the few currently available collections are limited in the number of included structures and are likely biased toward more readily accessible fragments or have undergone other external filtering criteria such as drug- or lead-likeness [30,31,32]. This can unknowingly distort the compound collection and thereby dramatically impact the trajectory of a given project. For cheminformatic analysis or downstream drug and chemical probe discovery, the automated extraction of fragments from vast natural product collections is a fruitful strategy to generate large datasets of natural product fragments. Multiple orthogonal strategies exist (Fig. 1) that enable rapid, chemically meaningful, and reproducible generation of natural fragment collections [4, 7,8,9, 17]. The strategy of choice will be determined by the desired goals of the specific project. This section outlines the most commonly used types of strategies and their various implementations.

Schematic on fragment generation. Shown is a natural product grouping consisting of proline, piperine, capsaicin, and dysidiolide. Fragment collections are shown that were generated relying on Murcko scaffolds [33], fragmentation through RECAP [15], and filtering (molecular weight <300). Asterisks indicate virtual attachment points generated through the in silico fragmentation. All procedures were implemented in RDKit [34]
2.1 Filtering
A straightforward approach to retrieve natural product fragments relies on filtering of natural product collections for their fragment-like sub-portion. Multiple studies have indicated that a large fraction of the currently known natural product space corresponds to low-molecular weight compounds [5, 17, 18, 35]. Thereby, molecular weight thresholds, commonly with a maximum allowed weight of around 300 Dalton, enable the rapid retrieval of the natural products that appear to be fragment-sized [8, 17]. More complex filtering strategies can ensure that the retrieved structures also fulfill other properties that are relevant for their concurrent application and thereby ensure the utility of the structures retrieved.
Indeed, many other definitions of fragment-likeness exist [36] and commonly other filters can be applied. A classic filter for fragment-like structures is the “Rule of Three” [37], a younger cousin of the Lipinski Rule of Five [32], which restricts fragments to a maximal weight of 300 Dalton, clogP < 3, up to three hydrogen bond donors (HBD), and up to three hydrogen bond acceptors (HBA). Generally speaking, the inclusion of pharmacophoric features (HBA and HBD, aromatic rings), measures of solubility (clogP, polar surface area), or measures of molecular complexity (number of rotatable bonds, number of rings, number of heteroatoms) can be rapidly derived from the molecular structures [38,39,40] and ensure that the extracted fragment collection fulfills the necessary criteria. Commonly, upper bounds on these properties ensure that the extracted structures are sufficiently simple and fragment-like [11]. It has also proven useful to establish lower bounds on properties such as the molecular weight, a minimum number of heavy atoms, or a sufficient molecular complexity to ensure that no trivial metabolites with limited pharmacophoric relevance are included in the natural fragment collection [17, 18].
Estimates of the fragment-like fraction of natural product databases range anywhere between 10 and 30% depending on both the investigated database and the exact definition of acceptable properties of the extracted fragments [5, 17, 18, 41]. While this number might be considered low, the vast size of many available natural product datasets [5, 26, 42, 43] usually still warrants a sufficient number of diverse fragment-sized structures for further applications [5, 8, 17]. Importantly, in absence of any computational structure generation or modification, this strategy promises to exclusively provide fragments that can be found in Nature and thereby might be available through isolation and, even more importantly, are more likely to constitute stable and biologically meaningful chemical matter [4].
2.2 Virtual Fragmentation
Instead of relying on smaller, fragment-like natural products already contained in natural product resources, in silico procedures to generate fragments can be harnessed to split larger natural products into smaller, fragment-like entities [7, 8, 15, 35]. Different strategies exist to virtually fragmentize larger structures into fragments, relying on distinct algorithms to ensure splitting of specific bonds or generating fragments of a specific size and character. Thereby, multiple fragments per natural product are generated, resulting in a potentially large set of virtual fragments that inherently contain natural product-like substructures [44]. A straightforward implementation of such fragmentation schemes can involve splitting of natural products at certain types of bonds, for example, all acyclic rotatable bonds [45]. Similarly, approaches to split up compounds into substructures of a specific size have been developed and applied to natural products [46, 47]. Following such strategies, exhaustive sets of substructures can be generated rapidly simply through analysis of the molecular graph and its partitions. However, many such generated fragments might be of limited chemical or biological relevance given their potential artificial nature and the lack of chemical reasoning for such partitions.
More advanced fragmentation schemes have been developed to ensure synthesis tractability, chemical stability, or other chemical and biological properties of the fragments. To this end, strategies have been implemented to virtually break bonds while considering their chemical context. Most prominently, the RECAP algorithm has implemented retrosynthetic fragmentation rules according to domain knowledge regarding bonds that can be easily and efficiently (re-)generated using established organic chemistry protocols [15]. In spite of not being specifically designed for natural product structures, the RECAP algorithm has been employed successfully to multiple natural product databases for the generation of large sets of natural fragments [7, 35, 41, 48]. Many publicly or commercially available cheminformatic software collections such as RDKit [34] and the Molecular Operating Environment [49] have re-implemented the RECAP algorithm with sufficient performance to process large natural product collections [7].
Many of the state-of-the-art de novo design implementations utilize virtual reactions. These construct novel compounds from commercially available building blocks according to hard-coded reaction schemes [50]. Pivoting this idea for fragmentation, reaction-based cleavage strategies have been implemented for natural products, for example, virtually performing reactions such as hydrolysis and ozonolysis [8]. These approaches might further the context-dependent analysis of cleavable chemical bonds and thereby generate virtual natural fragment-like collections with even further improved chemical relevance and a synthesis blueprint for their generation. Similarly, implementations of chemical reactions to introduce modifications in natural fragments can further diversify or stabilize the fragments of a virtual collection [4, 8]. While most of these approaches currently rely on hard-coded reaction schemes, novel approaches are emerging that can automatically extract such rules and further the applicability of computational fragmentation [51, 52].
Irrespective of the details of the implemented strategy, such in silico fragmentation approaches can rapidly and reproducibly provide large sets of natural fragments with novel chemical structures [7, 8, 48]. If virtual chemical reactions are utilized for fragment generation, these reaction schemes might provide blueprints for researchers to generate fragment-like molecules from a larger natural product serving as the template in the fragmentation. Even more importantly, virtual fragmentation is often implemented in such a way that it generates smaller structures with reactive handles that can serve subsequently as attachment points for further derivation and optimization [4] (cf. Fig. 1).
2.3 Scaffolds
Scaffold extraction can be regarded as a special case of fragmentation, whereas the molecular graph is not partitioned into smaller substructures but instead stripped into its central core structure by removing acyclic substituents [9]. While generally only one scaffold can be extracted per compound, the exact scaffold definitions and thereby the extracted molecular framework vary. The differences in the definitions concern whether the type of atoms or bonds are considered and whether adjacently connected heteroatoms are included in the scaffold definition [53, 54].
At the most abstract end of this spectrum are reduced molecular frameworks that ignore atom and bond types and even the size and types of rings within a scaffold [55, 56]. Molecular frameworks offer a mid-level of abstraction, representing full molecular graph-like structures utilizing all chemical bonds without atom and bond-type information. On the other end of this spectrum are decorated molecular substructures with fully defined atoms and bonds and the inclusion of specific adjacent heteroatoms [57].
Natural products, with their more common occurrence of fused-ring systems, will generate different reduced frameworks compared to synthetic compounds [57,58,59]. Full appreciation of the complexity and novelty of natural product scaffolds can only be achieved when including information on the distinct heteroatoms included or preservation of stereochemistry [60]. It is therefore not surprising that a large number of successful projects extract natural product scaffolds that consider atom-types, shortened side-chains, bond order, or chirality in their scaffold definitions [4, 9, 16].
A pioneering augmentation of scaffold generation and analysis is their hierarchical clustering based on substructure relationships to provide a structural classification of natural products (SCONP) [9]. Such hierarchical graphs of natural fragments enable tracking of chemical substructures and their impact on biological activity. This can aid significantly in the identification of lead fragments for further biological optimization [16]. Therefore, this concept has been implemented in the open-source software tool Scaffold Hunter to enable researchers to perform their own scaffold analysis [19]. Such approaches attest to scaffolds as some of the most useful natural fragment resource for further downstream analysis or as starting points for focused collection development (Table 1) [2, 9, 16].
3 Properties of Natural Product Fragments
Although the exact properties of natural fragments vary widely and are influenced by the approach taken to extract them (Fig. 2), there are some general trends observable for natural fragments that render them a particularly useful and unique resource of chemical matter.

Principal component analysis of standardized physicochemical properties of fragment-sized natural products (orange), scaffolds (red), and RECAP fragments (gray) from the TCM database. [43] Properties were calculated in RDKit [34], analyzed in KNIME [65], and visualized using Python and Inkscape (arbitrary units)
3.1 Chemical and Physical Properties
As expected, natural product fragments are more three-dimensional compared to flat synthetic fragments [4, 8] and rich in sp3-configured and chiral centers [5, 8, 66]. They also differ in their chemical composition and contain more oxygen, less nitrogen, and more aliphatic rings compared to synthetic screening collections [4, 12, 66], with an average number of three rings per fragment [9]. Some studies have investigated other properties relevant for biological activity and have shown a higher propensity for pharmacophoric features such as HBD and HBA and lower numbers of rotatable bonds [17], which has supported their perception as privileged structures with an increased potential to interact with a wider range of different biological targets compared to synthetic compound collections and their larger, complex natural counterparts [5, 7].
Chemography of natural products, their fragments, and drugs allows charting the general differences between compounds from these sources in terms of their physicochemical properties or chemical structures [67,68,69,70]. Generally speaking, researchers have found that natural product-derived fragments using various fragmentation sources generate good representative structures of the larger natural product collections in terms of fully spanning the natural product space [4, 57] while they show disparate placement compared to synthetic fragments [4] in terms of physicochemical properties. Pharmacophoric and structural assessments have placed natural fragments at the interface between synthetic bioactive compounds and complex natural products [7, 58].
Accordingly, less “Rule of Five” [32] violations are observed for natural fragments compared to natural products [18, 71], which might not be too surprising given that these rules include thresholds that depend heavily on the size of the investigated molecule and therefore are commonly conformed to by natural product fragments. Fascinatingly, natural products in general often violate the “Rule of Five” [12, 72], such that fragmentation might be regarded as a transformation of natural product space into “Rule of Five” compliant areas [73]. This is fully in line with the observation that the regions of physicochemical space that are populated exclusively by natural products but not drugs are mostly spanned by larger, more complex natural product structures [7, 57]. Thereby, fragments enable a more drug-like handle to natural product space [2]. This can be further utilized since their decoration with classic medicinal chemistry side chains enables populating drug-like spaces but with innovative scaffolds [57] with potentially superior properties such as higher three-dimensionality [8].
3.2 Spatial Properties
To assess the size and shape of natural product fragments, the three-dimensional conformation can be predicted from the two-dimensional molecular graph [74]. Conformation prediction of natural products is often challenging given their large size, complex structures, and common occurrence of chiral centers and macrocycles [75, 76]. Therefore, natural product research often harnesses advanced methods relying on experimental methods such as nuclear magnetic resonance measurements or machine learning predictions [77, 78], thereby limiting the application to only select natural products with available experimental data or sufficient interest to warrant the necessary experimental or computational resources. Since fragment-like natural products are smaller and populate a more restricted conformational search space, their conformations might be estimated more rapidly and more accurately [36]. This suggests that natural product fragments and their derivatives might enable researchers to study natural products in three dimensions with reduced need for advanced conformation estimations to identify targets via pharmacophore searching or rationalizing binding modes through docking [9, 21, 22, 79, 80]. On a more general level, such three-dimensional structure prediction tools have also been employed to whole collections of natural product fragments to calculate the distribution of their volumes: this has charted most natural fragments within the range of 100 to 500 A3. This renders them similar in size compared to volumes calculated for approved drugs and currently explored protein pocket cavities [9, 16, 35], further attesting to their utility for drug discovery. Similarly, the shape of natural product fragments has been investigated through their principal moments of inertia [81] and it was concluded that natural product fragments provide a wide range of different shapes and, most notably, are less “flat” compared to other compound databases, including the complex natural products stored in the Dictionary of Natural Products [42] as well as various synthetic fragment collections [8, 17].
3.3 Natural Fragments from Different Sources
Natural products and their fragments from different origins can vary drastically in their chemical structures and properties [12]. By selectively analyzing natural products from different sources, their properties can be compared to identify potentially helpful trends in physical or chemical differences between structures produced by distinct organisms or within specific environments or locations [35, 41, 66, 82]. A handful of studies have started to chart and compare natural products from different sources. For example, marine products seem to offer a larger variety of different substructures, while terrestrial products appear to borrow more frequently from similar substructures [35]. It has been suggested that natural products originating from fungi might represent distinct natural product properties without deviating too much from the drug-like space in terms of physicochemical descriptions [57]. Ertl and Schuffenhauer specifically investigated unusual chemical structures populating natural products from different sources and found that plant-based natural products contain more fused carbocycles compared to natural products from other sources [83]. Other studies have found that arenes, while dominating plant- and marine-derived natural products, seem to be almost completely absent from bacterial organisms [66]. Bacterial metabolites may also be sulfur-containing natural products [83] and marine natural products contain more oxygen compared to terrestrial natural products [35]. Isolated studies have started to draw conclusions from observed chemical structures and the implications for their occurrence in certain natural products: for example, marine natural product repositories contain more hydrophobic compounds and a lower number of ester bonds [35]. This could point potentially at evolutionary forces selecting for organisms that are more adapted to their marine environment by producing compounds with lower risks of losing metabolites to the aqueous environment surrounding them as well as spontaneous hydrolysis [35, 83]. Such insights are transferable into the fragment space of natural products and can be helpful in compound collection design when certain physicochemical properties are of importance. Relying on selected organisms or origins might enable compound pools to be steered in the desired direction. Furthermore, through such in-depth analysis, cheminformatic research potentially may assist fundamental research in metabolomics to better understand specific organisms or microenvironments through their small molecular armamentarium [84, 85].
3.4 Commercial Availability and Synthetic Tractability
Although fragment-sized natural products make up only around a third of the known natural product structures [5], they constitute the bulk of commercially available and hence easily-accessible natural compounds for drug discovery and biotechnology applications [4, 26]. Natural product fragments are often easier to synthesize compared to their complex, larger counterparts [4, 73]. Computational assessments of synthesizability as well as computational retrosynthesis planning potentially can aid at prioritizing natural fragments and synthesis pathways [15, 51, 86]. However, most of these tools were not designed to be specifically applicable to natural products and therefore might need to be augmented to enable their straightforward application to natural fragments. It has also been shown that, even in cases where the fragments are not readily available or synthesizable, they can often be represented through commercially available or easily synthesizable analogs [4] (Fig. 3). This can be achieved by employing classical ligand-based similarity and virtual screening approaches to search for similar fragments [87, 89]. Alternatively, clustering of fragment spaces enables partitioning the chemical fragment universe into regions of high similarity to substitute critical fragments with other co-clustered representatives [4, 7]. Extending this concept even further and relying on hierarchical clustering techniques, Koch et al. showed that constructing graphs through chemical substructure relationships can build data structures to enable such simplifications [16]. Applying this concept to scaffolds, such a graph can be built iteratively by populating the network with all possible scaffolds. Subsequently, two nodes are connected if the respective scaffolds can be transformed into each other by either removing or adding one ring structure. Thereby, a graph is generated based on a special case of a substructure relationship. Traversing this graph [19] can inform chemical derivatization and simplifications into smaller scaffolds with lower complexity and sufficient similarity to enable tackling of the same biological target [90]. If a substructure of the scaffold does not warrant a sufficient simplification, such graphs also enable “brachiating” into neighboring branches with potentially simplified chemical structures but retained biological activity [9]. In such advanced simplification approaches, it is important to keep in mind that too aggressive derivatization or simplification can lead to loss of the biological activity or other desired properties of the investigated natural product fragment [9, 91]. Prediction of the biological and physicochemical properties of the in silico-derivatized structures can enable researchers to monitor the expected behavior of the novel structures and guide the structural modifications [5, 22, 25, 92,93,94,95]. Conversely, the originally investigated fragments might be unstable or contain reactive substructures such as enamines or Michael acceptors, and slight modifications can easily eliminate such structures [4, 8, 20]. The prediction of reactive substructure or other liabilities can assist in identifying problematic fragments and speed up this process [96, 97].

Identifying accessible derivatives of natural products, e.g., through molecular similarity assessments, using, for example, the WHALES descriptor [87] (a), or through clustering and subsequent selection of representative structures via chemical structure similarity (b) [4]. Alternatively, hierarchical graphs of scaffolds can be utilized to identify related structures following the SCONP concept (c) [9, 88]
3.5 Problematic Natural Fragments
An increasing body of literature has served to warn of false-positive assay results, i.e., compounds that elicit a positive readout in spite of not showing the actually desired biological activity [97, 98]. This behavior can originate from various underlying causes, many of which relate to physicochemical properties of the investigated compound such as reactivity, quenching, membrane interactions, fluorescence, or colloidal aggregation [97,98,99]. Natural products and their fragments can potentially contain substructures that can elicit such effects in vitro even if such effects might be masked in their natural, biological (micro)environments [12]. This hints at the necessity to identify problematic structures in natural product collections and for fragment-like hit compounds to avoid hunting artifactual biological in vitro results [99].
Cheminformatics efforts to design automated pattern-recognition systems to filter such potentially problematic compounds have led to the development of multiple substructure-based filtering lists that flag chemistry with motifs associated with false-positive results for subsequent validation or elimination [97, 100, 101]. Although such methods have been designed based on screening data or with synthetic molecular probes in mind, it has been shown that such false-positive behavior can occur among natural products in general and their fragment-like portion in particular [101, 102]. False-positive results from natural products might be common, as indicated by multiple case studies and in-depth analysis of commonly used natural fragments [101, 102]. For example, the discovery of viable inhibitors of indoleamine 2,3-dioxygenase 1 (IDO1) has been fueled by fragment-like natural products such as β-carboline or galanal, but it has been suggested that potential false-positive readouts might be at play for such compounds [103]. Similar arguments have been made for fragments such as thymoquinone in the context of anti-protozoal agents [104] and plumbagin as a histone acetyltransferase inhibitor [105]. On a larger scale, statistical data analysis of natural product assay data has been conducted and suggests that a large fraction of the acquired positive assay results might stem from causes other than useful biological activity [99].
To counter such effects, researchers have designed taboo lists of chemical substructures that are linked to various artifactual readouts [97]. Many natural products contain such critical chemical substructures [12] recognized by filtering rules such as PAINS [2, 99, 101] or ALARM NMR [100, 106, 107]. In fact, it appears that natural products are particularly prone to contain such critical substructures recognized by automated filtering rules compared to synthetic compounds and approved drugs [5]. For example, around 40% of natural products currently studied in the context of their antiprotozoal activity contain PAINS substructures [104] and up to 65% of natural products from the commercial MicroSource collection are flagged according to the ALARM NMR filters [107]. While, to the best of our knowledge, no large-scale analysis on flagging of natural fragments has been conducted, it is fair to assume that fragmentation or selection of potentially problematic natural structures would transfer such liabilities into fragment-based collections and pipelines [108]. Furthermore, specific investigations of selected fragment-sized natural products have highlighted cases of such naturally occurring structures to be flagged by multiple different false-positive detection methods (Table 2) [103].
Colloidal aggregation has been suggested as the single largest reason for a compound to elicit artifactual, false-positive assay readouts in screening assays [97, 109]. Indeed, many fragment-sized natural products such as physcion and equol have been shown to form colloidal aggregates that can sequester proteins and thereby interfere with biochemical assay readouts [99, 102, 110]. A high logP for many natural products [58, 60] is an indicator that natural products and their fragments might possess such aggregation propensity [109]. More accurate and fine-grained computational prediction models exist that anticipate whether a compound aggregates from its molecular structure and physicochemical properties [109, 111, 112]. Such models might be applied fruitfully to get more accurate estimates on which natural product fragments form colloidal aggregates. However, given that such models rely on molecular data mostly derived from synthetic screening compounds [112], the discrepancy between natural compounds and the training data in terms of molecular properties and structures hint at natural products potentially lying outside of the applicability domain of such models [7, 113, 114]. An in-depth evaluation will be necessary to understand whether the colloidal aggregation of natural products can be studied by relying on data derived from synthetic screening compounds or whether specifically tailored machine learning tools will be necessary to accurately delineate the potentially common aggregation behavior of natural products [102].
Furthermore, fragment-like natural compounds such as genistein and capsaicin potentially can interact with and modulate lipid bilayer properties and thereby cause false-positive readouts in cell-based screening assays [115]. To rapidly decode such effects and flag compounds that potentially exert such behavior, computationally accessible properties such as lipophilicity [116], charge [117], and amphiphilicity [115, 118] can enable model development to predict the ability of natural fragments to interact with or modulate lipid bilayer properties. Molecular dynamic simulations are another tool that can potentially anticipate such effects and identify the natural fragments that can cause this type of behavior [115, 117, 119].
Not all natural fragments that are flagged by such methods need to be blindly eliminated [120]. Many liabilities will only be relevant in specific screening contexts [121, 122], which has led to major criticism against the blind application of the aforementioned prediction models and flagging lists to eliminate compounds from screening collections [98, 120]. Indeed, many safe and clinically effective medications have been shown to be flagged by various computational false-positive detection methods [123, 124]. Therefore, if sufficient caution is taken and validations and counter-screens are established, even apparently problematic structures might fuel successful drug discovery and development pipelines. In the future, augmented prediction methodologies could enable more fine-grained analysis of contextual assay results of natural product fragments [106]. Other studies have shown how natural fragments containing problematic structures constituted initial hits and were subsequently derivatized during optimization to eliminate the liability for downstream validations [8] or how potentially pernicious substructures might not be liabilities in their specific natural product context [5, 17]. Utilizing computational prediction models to anticipate context-specific liabilities, as well as the establishment of automated molecular design for the derivation of natural product fragments will automate such processes in the future.
4 Applications of Natural Product Fragments
Given the aforementioned advantageous properties of natural fragments, they have been suggested as representing innovative starting points for drug discovery and chemical biology [4, 5, 13]. Importantly, such efforts are most productive if the polypharmacological properties of the natural product under investigation is known and derivatives can easily be made to fine-tune biological and physicochemical properties of the molecular probes. As described in this section, computational tools can assist in predicting these properties, generating focused collections of derivatives and mimetics of natural compounds, as well as utilizing them for statistical analysis of natural product-likeness to assess the utility of a compound or molecular collection.
4.1 Predicting Biomacromolecular Targets of Natural Fragments
Natural fragments with known polypharmacological profiles may be regarded as most useful starting points for drug discovery campaigns and chemical probe development since candidate structures will already possess the desired activity and potential known off-targets can be avoided [5]. Unfortunately, the biological effects of a vast majority of natural products and their fragments is currently not known [6, 7, 9]. In silico target-interference methods represent easily deployable prediction tools to anticipate the biomacromolecular receptors targeted by natural products [5]. However, classical target prediction methods usually underperform for natural product fragments given the stark difference in chemical structure of natural products and their fragments to the synthetic compounds that populate (training) databases of ligand-target interactions [4, 5, 18]. Therefore, with few exceptions [125, 126], most of the advanced and well-validated target prediction technologies, which are largely based on applying the chemical similarity principle [127] to chemical substructure descriptions [128, 129], underperform at identifying targets for natural product fragments compared to their impressive success reported for the target identification of drugs and synthetic compounds [95, 130,131,132,133]. Therefore, in the context of natural product fragments, researchers have employed or designed target prediction methods that generalize from the underlying chemical substructure and instead directly or indirectly quantify the pharmacophoric potential of natural product fragments [5].
For example, Rollinger et al. have used 2208 three-dimensional pharmacophore models to screen a collection of 16 fragment-like secondary metabolites isolated from Ruta graveolens and found between ten and 287 confident predictions per natural fragment [79]. In prospective experiments, arborinine was validated successfully as an acetylcholinesterase inhibitor (IC 50 = 34.7 ± 7.1 μM). Their model for binding the cannabinoid-2 receptor was based on five selective agonists. The only confident prediction of this model was rutamarin. Indeed, rutamarin was the only metabolite that showed ligand displacement with a K i of 7.4 ± 0.6 μM [79]. These data suggest that such models are not only able to correctly identify inhibitors but also are robust in recognizing true negatives—although further testing will need to statistically validate these results [79]. Follow-up research has led to the identification of acetylcholinesterase inhibitors among morphinans and isoquinolines [134] as well as partial agonists of proliferator-activated receptor gamma among neolignans [80, 135].
Instead of deriving a pharmacophore model for a protein target of interest, researchers have also successfully employed docking strategies to assess the potential of a natural fragment to bind a pocket of a target protein [136,137,138]. For example, Lanz and Riedl probed the S1′-binding site of matrix metalloproteinase 13 [22]. They identified uracil as a natural fragment with a unique binding mode that was further optimized into nanomolar inhibitors with impressive selectivity over other matrix metalloproteinase subtypes. In a broader screen against 400 proteins, Bernard and colleagues identified peroxisome proliferator-activated receptor gamma and cyclooxygenase-2 as targets of the coumarin derivative meranzin [139]. Both pharmacophore models and computational docking strategies have shown impressive results for identifying biomacromolecular targets of natural product fragments [5], but require good estimates of the confirmation space of natural fragments and their stereochemistry, which are not always available.
Other prediction technologies enable target identification for natural products while circumventing the challenges originating from the differences in chemical structure compared to synthetic screening collections or unknown conformations [5]. For example, researchers have employed productively biological fingerprints as an alternative strategy to compare molecules and predict their targets [140]. In brief, the underlying assumption is that if two compounds have shown similar activities for some biological targets, it is likely that they will behave similarly when tested against other targets. This can be employed for target prediction if one of the compounds has been tested against targets against which the other compound is yet to be tested. Wassermann et al. have shown how this approach predicts more targets for natural products compared to chemical fingerprint-based approaches and have used this concept to identify vascular endothelial growth factor receptor 2 as a target for fisetin with an IC 50 of 230 nM, which might aid explaining the antiangiogenic effects of this fragment-like flavonoid [141]. However, this successful strategy is exclusively applicable to natural products that have been screened for biological activity previously. A majority of natural product fragments have not been investigated yet or the results have not been made publicly available [6, 7], highlighting the need for additional technologies that can predict targets of natural fragments exclusively from their structures without the need for conformational sampling or previous biological screening.
In an effort to design a prediction technology specifically focusing on its ability to predict targets for novel chemical structures, Reker et al. have designed the SPiDER method [25]. The method circumvents the problem of predicting targets for chemicals with unusual or previously underexplored chemical substructures by explicitly employing “fuzzy” descriptors that enable relating chemicals through their two-dimensional graph structure via pharmacophore correlations (CATS2 descriptor) [142, 143] and physicochemical properties [39, 49]. The method relies on self-organizing maps as a clustering approach [144] to define local regions (Voronoi fields) of equivalent biological activity [145, 146]. Confidence scores for every prediction are derived from statistical interpretation of molecular similarities to enable prioritization of the most meaningful target hypotheses [25]. It was realized that this workflow not only is successful at identifying targets for novel, de novo-designed synthetic compounds [25, 147] but specifically excels at predicting targets of natural products [5, 7, 21, 148]. Natural fragments, in particular, appear to show the highest number of confident SPiDER predictions [5, 7], highlighting the ability of the SPiDER algorithm to predict new targets for this important compound class. Indeed, SPiDER has been utilized to identify biomacromolecular targets for various naturally occurring fragments such as β-lapachone [149], graveolinine, isomacroin, dl-goitrin [21], sparteine [5], valerenic acid, isopimaric acid, and dehydroabietic acid [20] (Fig. 4). While these and other studies [7, 148] highlight the power of ligand-based target prediction methods such as SPiDER to identify the targets of natural fragments, these studies also provide powerful insights into how these methods can be used in concert with molecular docking [21], molecular similarity assessments [20], and orthogonal machine learning technology [149] to fuse multiple prediction methodologies for further improved predictive confidence or to enable additional hit rationalization. Similarly, conceptually related target prediction methods such as TIGER show promise to further the computational toolset for polypharmacological prediction of natural product fragments such as resveratrol [150] that can be easily derivatized for further structure-activity relationship studies and in vivo applications [151].

Predicting biological targets of natural fragments. (a) Prediction of the binding mode of uracil in the S1′-binding site of matrix metalloproteinase 13 [22]. Orange, hydrogen bond acceptor; red, hydrogen bond donor. (b) Pharmacophore-based identification of rutamarin as cannabinoid-2 binder [76]. Orange, hydrogen bond acceptor, cyan, hydrophobic interaction. (c) Biological activity fingerprints enabled the identification of endothelial growth factor receptor 2 as a target of fisetin [133]. (d) Selection of different fragments for which the targets were identified through ligand-based target prediction technology [5, 20, 21, 149, 150]. The target prediction method utilized for the individual study is highlighted in the boxes underneath the structure and the identified targets are annotated at the bottom
Although multiple striking examples exist of utilizing advanced target prediction technology to predict targets of large and structurally intricate natural products [7, 125, 148, 152, 153], it seems that fragment-like natural products are more computationally relatable to the available screening data [2, 7, 57] and therefore lead to more confident predictions [5, 7, 21, 107]. Some investigations suggest that this trend might also be true for other target prediction methodologies such as computational docking, where fragment-like entities lead to higher scores or improved retrieval of correct binding modes [36], which might be connected to the problem of conformational sampling of complex natural product structures [78].
Taken together, it appears that fragment-like natural products are exquisitely positioned to provide starting points for drug and chemical tool development to modulate the activity of their anticipated targets [13]. For example, sparteine is a natural fragment that has been studied extensively and computational methods such as ligand-based target prediction and clustering-based diversity selection have identified biomacromolecular targets spanning different protein families such as p38α MAP kinase [4], muscarinic and nicotinic receptors [5, 154], and the kappa opioid receptor [5]. Such initial hits can then be further optimized by derivatizing the fragment and adding additional chemical functionality: sparteine derivatives were further functionalized with a primary amine that enabled improved p38α MAP kinase activity from additional polar interactions as indicated by molecular modeling [4].
4.2 Collection Design
Natural product fragments are believed to coalesce the advantages of fragment-based drug discovery and biologically privileged natural product structures [13]. Therefore, general-purpose screening collections of natural fragments with diverse structures have been harnessed to generate compound collections that could provide novel hits at improved rates with better selectivity compared to classical synthetic fragment sets [8, 11, 17, 155]. For example, Over et al. [4] used scaffold-based fragmentation on the Dictionary of Natural Products [42] and subsequently filtered for fragment-like structures without reactive groups. Clustering-based diversity selection [156] and identification of commercially available cluster representatives (cf. Fig. 3) lead to the assembly of a screening collection that was employed successfully to identify novel, allosteric ligands of p38α MAP kinase as well as phosphatase inhibitors [4]. Similarly, Quinn and colleagues [17] used filtering directly on the Dictionary of Natural Products [42] to arrive at a small, diverse collection of naturally occurring, three-dimensional fragments that were successfully isolated or purchased. This compound collection was then screened in parallel for their potential to bind to multiple malarial protein targets as well as phenotypically for their activity against asexual intraerythrocytic blood stage Plasmodium falciparum 3D7 parasites. Analyzing these data in parallel enabled the target identification of 31 relevant antimalarial targets as well as the generation of 79 innovative hit structures for further optimization [17]. Such efforts attest to the enormous potential of diverse, target-agnostic screening collections composed of natural product fragments to fuel fragment-based discovery efforts against targets from various protein families using various assay technology as well as validating their utility in phenotypic screens.
Instead of generating natural fragment sets from whole natural product collections [4, 17], molecular series derived from one specific natural product core can provide focused collections with privileged and novel structures [12, 16]. Especially when the biomacromolecular targets of the template natural product are known or predicted [9, 149, 157], the derived structures often inherit the template’s polypharmacological profile—leading to dramatically increased hit rates for the focused natural fragment collections on the target of interest [9, 158]. Furthermore, if the template fragments were generated in silico through virtual fragmentation and (retro)synthesis approaches, their attachment points might constitute useful chemical handles to add side chains with additional pharmacophore functionality while preserving the original scaffold and shape [4] (cf. Fig. 1). Thereby, potentially inaccessible natural products can serve as templates for synthetically tractable sets of derivatives [91]. Waldmann and colleagues have pioneered and validated this concept as biologically oriented synthesis (BIOS) [16]. Relying on and derivatizing core structures originating from fragments that were computationally extracted from natural ligands of an enzyme of interest or other known or predicted inhibitors, focused screening collections can be generated with high hit rates and, even more importantly, an often improved selectivity to structurally related protein targets compared to other screening approaches [9, 90, 158].
Fascinatingly, such efforts can be performed phenotypically without necessarily understanding the exact mechanism of action of the template compounds. For example, in an effort to identify molecular tools with neurotrophic activity, Schröder et al. relied on BIOS to simplify N-deoxymilitarinone A (Fig. 5), a fungal metabolite causing neurite outgrowth in PC-12 cells [90]. Previous research had shown that this effect is relatively robust toward simplifying the natural product and its side chains have minor impact on its neurotrophic activity [159, 160], suggesting a pivotal role of the scaffold for activity. This motivated the generation of 59 compounds around the 4-hydroxy-2-pyridone scaffold and its 2,4-dimethoxypyridine derivative [90]. The most active compound 11e showed 74% neurite growth at 10 μM compared to control. Interestingly, activity in the phenotypic screen of the focused collection could be correlated with MAP 4K4 activity, which potentially links this kinase to neurotrophic effects and proposes it as a target to combat neurodegenerative diseases [90].

Collection design using biology-oriented synthesis (BIOS). Natural ligands with known biological activities, such as N-deoxymilitarinone or glycyrrhetinic acid, can be simplified into smaller scaffolds through traversing the structural classification of natural products (SCONP) graph. This leads to the identification of a core scaffold that can be used to generate focused collections of derivatives with a desired biological activity [9, 90]
An earlier study by Koch et al. set out to identify novel 11β-hydroxysteroid dehydrogenase 1 inhibitors from the natural ligand glycyrrhetinic acid [9]. The complex pentacyclic scaffold was simplified into a two-ring system following the SCONP hierarchical clustering of scaffold structures [9]. In an additional step of collection development, the scaffold was subsequently substituted by a more stable derivative with endocyclic double bond through “brachiation” within the scaffold tree [161]. Such a horizontal shift from one arm of the scaffold tree into another can enable transformations of target fragments into more suitable structures in terms of advantageous physicochemical properties or improved chemical tractability for derivation and collection design. However, this comes at the risk of losing the associated pharmacological effect of the original natural product through deviating too much from the privileged scaffold arrangement [9, 91]. In this specific case, further confidence in the applied modification was drawn from the fact that the aspired fragment corresponds to the scaffold of the natural product dysidiolide, which is an inhibitor of Cdc25A phosphatase [162]. Since Cdc25A phosphatase is structurally related to the 11β-hydroxysteroid dehydrogenase 1 target protein according to the protein structure similarity clustering (PSSC) approach [158], it is likely that they share common inhibitors and that therefore scaffolds targeting Cdc25A phosphatase might also warrant 11β-hydroxysteroid dehydrogenase 1 inhibitors. Indeed, a collection of 162 compounds derived from the octahydronaphthalene scaffold afforded 30 inhibitors of 11β-hydroxysteroid dehydrogenase 1 and some of the hits revealed remarkable selectivity against 11β-hydroxysteroid dehydrogenase 1 over 2 [9], further hinting at the potential of natural fragments to serve as starting points for highly selective lead structures and probes [5].
In these and other examples [16], BIOS has specifically excelled at addressing challenging targets where out-of-the-box fragment or screening collections might give unsatisfactory results [4]. This has been associated with the ability of such approaches to generate focused sets of compounds that inherit relevant physicochemical properties and pharmacophores from the template natural product [4, 9, 16]. However, notwithstanding the impressive success rate of the BIOS approach, this hypothesis is not always correct, and depending on the chemistry employed, compounds with vastly different properties can emerge [9, 91]. In a fascinating meta-analysis, Pascolutti and Quinn [163] investigated the distribution of molecular weight and logP as well as the number of HBD, HBA, rotatable bonds, and rings in collections generated through derivatizing natural product templates. They found that such generated collections can show markedly different ranges of properties and, most importantly, could be drastically different from the original natural product that was used as the template structure [163]. A close monitoring of the properties of collections generated, including their potential to trigger false-positive assay readouts (cf. Sect. 3.5) potentially can improve the quality of such generated collections and even further enhance success rates [5].
The impressive success of the BIOS approach is possible due to advanced chemical knowledge and manual labor to delineate stable core structures and suitable chemical routes for their derivation [16]. In an orthogonal and automated approach, computational de novo design can be installed to generate large collections of natural product mimetics autonomously [20, 58, 92, 164]. To this end, natural product fragments with known biological activities can fuel ligand-based de novo design algorithms [50] to derivatize them and build small collections of novel chemical structures with similar chemical features and biological activities [92].
Schneider and colleagues have pioneered both established as well as novel de novo design methods for the generation of collections of synthetically accessible natural product mimetics from natural product fragments [20, 92]. For example, the DOGS software [24] was harnessed to create a collection of synthetically accessible natural product mimetics with potential inhibitory effect against the retinoid X receptor [20]. DOGS implements a virtual synthesis algorithm that connects 25,144 commercially available chemical building blocks according to 58 chemical reaction principles to generate novel chemical matter with a suggested protocol for its synthesis [24]. Since the connection of all building blocks would lead to a combinatorial explosion of possibilities, the DOGS designs are iteratively guided by a graph kernel similarity towards a template structure [165]. The DOGS designs have been validated extensively to afford novel chemical matter with the desired activity against (patho)biologically relevant protein targets [166,167,168,169,170,171,172] and were most recently validated in the context of fragment-based drug discovery [173] and were used to identify mimetics of large, complex natural products [164]. In a consequent next step, Schneider and colleagues applied the concept to natural fragments as retinoid X receptor modulators, utilizing small natural compounds with known retinoid X receptor activity such as honokiol, drupanin, valerenic acid, isopimaric acid, and dehydroabietic acid as template structures (Fig. 6) [20]. These designs were prioritized further using a consensus of a CATS2 descriptor-based similarity assessment [142] as well as SPiDER target predictions [25]. This workflow led to the synthesis of six de novo-generated natural product mimetics, of which five showed the desired retinoid X receptor activity [20].

De novo design of natural product-derived fragments for the generation of synthetically accessible mimetics. The natural fragments and inhibitors of retinoid X receptor, honokiol, drupanin, valerenic acid, isopimaric acid, bigelovin, and dehydroabietic acid can be coupled to ligand-based de novo design software such as DOGS [24] or deep learning-based generative models [174] to identify novel chemical entities with natural product-likeness [44] and a desired biological activity [20, 92]
In a second study against the same target, the team added bigelovin to the natural product templates for a novel, generative deep-learning campaign [92]. Generative deep neural networks are the newest addition to the molecular design toolbox, teaching a machine molecular structure constraints by providing tens of thousands of valid chemical structures in text representation such as SMILES formats [174,175,176]. These neural networks can then sample novel text representations of molecules that translate into compounds with desired properties. To this end, the neural network is first trained to produce chemically meaningful text representations through a large corpus of chemical structures. Providing the six natural fragments as data for an additional round of training enables the fine-tuning of the model and bias the generation of chemical structures to natural mimetics with potential for the desired biological activity (“transfer learning”). Indeed, the fine-tuned model was able to create hundreds of chemically valid and novel structures that exhibited the desired natural product-likeness [44]. Around half of these designs were predicted as ligands of retinoid X receptor by orthogonal target prediction methodology relying on SPiDER predictions [25]. Further filtering of these positively predicted designs using WHALES molecular similarity assessment [87] and visual inspection for synthesizability lead to four chosen designs, of which two possessed retinoid X receptor activity with varying subtype selectivity [92].
Such de novo design campaigns are complementary to the previously mentioned BIOS approaches. While BIOS harnesses chemical expert knowledge to generate structurally related compound series employing the same or a similar privileged scaffold, automated molecular design can create large sets of novel structures that are chemically more different from the template but inherit natural product-likeness [44] and crucial pharmacological features from the template natural fragments [73, 92]. Given their impressive success in previous studies, the herein discussed and orthogonal tools are likely to become essential parts of the drug and chemical probe discovery toolbox to provide novel and privileged compound collections from natural fragment templates [73].
4.3 Analysis of Natural Product-Likeness
Instead of using fragments directly as tools in computer-assisted drug discovery, researchers have utilized them as chemical patterns to quantify the similarity between two compounds or compound collections. Scaffolds in particular have been utilized as the indicators of structural novelty for collection design and compound development [18, 58, 132, 177]. Therefore, the concept of “scaffold hopping” has been coined by Schneider and colleagues to ascribe the capabilities of a method or the success of a project to identify new chemical entities [143, 167]. Conversely, in the context of natural product-inspired drug discovery research where scaffold similarity to a naturally occurring compound is desired, the detection of natural product fragments in a new lead structure or screening collection is a measure of desirability [44]. On a larger scale, fragmentation and structure matching can provide statistically quantifiable measures of “natural product-likeness” [5, 44]. For example, Hou and colleagues [35] analyzed the natural product-likeness of the Comprehensive Medicinal Chemistry Database and found that about 20% of the scaffolds in this dataset were present in the Terrestrial Natural Product Database [64] while only 10% could be found in the Dictionary of Marine Natural Products [63]. While this bias might be in parts explained through the smaller size of the Dictionary of Marine Natural Products (cf. Table 1), other effects such as the underrepresentation of marine natural products in historic drug discovery might help explain this effect as well [57].
To measure natural product-likeness of individual chemical structures, Rodrigues et al. have devised a scoring procedure that captures the count of natural product fragments occurring in one specific molecule normalized by its molecular weight [5]. Through this normalization, each score captures the relative frequency of natural product fragments found within one specific structure compared to the size of the molecule. Through applying this score to FDA-approved drugs, therapeutics that are more or less similar to natural products can be identified. Interestingly, while there were strong variations per year, a sustained occurrence of natural fragments in approved drugs was observed [5]. This is fully in line with previous observations of the relevance of natural products for drug discovery [3, 178].
Ertl et al. have devised the most commonly utilized score that specifically captures the occurrence of fragments from natural products that cannot be found in synthetic molecules [44]. Thereby, the score removes “background noise” fragments that can be found in either compound class. This score enabled the classification of natural products vs. synthetic molecules with higher enrichment compared to machine learning models based on physicochemical properties, thereby highlighting its utility to measure the natural product-likeness of a compound in terms of chemical fragments. In a striking experiment, they compared the scores for natural products, synthetic compounds, and approved drugs: while natural products and synthetic compounds show disparate distributions, approved drugs show higher natural product-likeness, further attesting to the utility of using natural product fragments in the design of new therapeutics [179].
Accordingly, such measures of natural product-likeness act not only as a seismograph to measure the relevance of natural products for drug discovery over time [5], but have also been used to guide screening efforts for specific collection design [71] or to assess the natural product-likeness of de novo-generated natural product derivatives and collections of mimetics [20, 155]. The success of these studies is a remarkable testimony to the relevance of natural product fragments for the development of novel and impactful chemical probes and drug discovery hits [73].
5 Concluding Remarks and Outlook
As highlighted in this contribution, multiple impressive projects have relied on natural fragments to efficiently discover novel and selective starting points for drug discovery and chemical biology with great potential for further optimization [7, 9, 19, 20, 22]. The success of such endeavors stems, at least in part, from the potential to benefit from fragment-based approaches [17, 180] for compound discovery and design. At the same time, such natural fragments provide innovative, three-dimensional molecular frameworks [8, 17] that are chemically [4, 8] and computationally [2, 5, 7, 57, 107, 169] more accessible compared to their complex counterparts. Computational workflows have been implemented that support this process at all stages. Generating novel fragments [4, 9], their property analysis [8, 18, 57], their biological applications [4, 5, 21, 22], and compound collection designs [16, 20] can be supported through in silico approaches. It is noteworthy, however, that most of the computational tools discussed herein were not designed specifically with an application to natural products or their fragments in mind. Some early studies have indicated that algorithmic tools specifically tailored to process natural products might even further increase the performance and success rate of such pipelines [7, 9]. This indicates that further studies to design computational tools specifically for natural fragments, optimizing currently available workflows, and their intelligent application to drug discovery and chemical biology promise multiple avenues for impactful research and innovative molecular matter through coalescing data science and natural product research.
References
Ganesan A (2008) The impact of natural products upon modern drug discovery. Curr Opin Chem Biol 12:306
Barnes EC, Kumar R, Davis RA (2016) The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat Prod Rep 33:372
Harvey AL (2008) Natural products in drug discovery. Drug Discov Today 13:894
Over B, Wetzel S, Grütter C, Nakai Y, Renner S, Rauh D, Waldmann H (2013) Natural-product-derived fragments for fragment-based ligand discovery. Nat Chem 5:21
Rodrigues T, Reker D, Schneider P, Schneider G (2016) Counting on natural products for drug design. Nat Chem 8:531
Rollinger JM, Stuppner H, Langer T (2008) Virtual screening for the discovery of bioactive natural products. In: Petersen F, Amstutz R (eds) Natural compounds as drugs, vol I. Birkhäuser, Basel, p 211
Reker D, Perna AM, Rodrigues T, Schneider P, Reutlinger M, Mönch B, Koeberle A, Lamers C, Gabler M, Steinmetz H, Müller R, Schubert-Zsilavecz M, Werz O, Schneider G (2014) Revealing the macromolecular targets of complex natural products. Nat Chem 6:1072
Prescher H, Koch G, Schuhmann T, Ertl P, Bussenault A, Glick M, Dix I, Petersen F, Lizos DE (2017) Construction of a 3D-shaped, natural product like fragment library by fragmentation and diversification of natural products. Bioorg Med Chem Lett 25:921
Koch MA, Schuffenhauer A, Scheck M, Wetzel S, Caaulta M, Odermatt A, Ertl P, Waldmann H (2005) Charting biologically relevant chemical space: a structural classification of natural products (SCONP). Proc Natl Acad Sci U S A 102:17272
Dobson CM (2004) Chemical space and biology. Nature 432:824
Irwin JJ (2006) How good is your screening library? Curr Opin Chem Biol 10:352
Clardy J, Walsh C (2004) Lessons from natural molecules. Nature 432:829
Shoichet BK (2013) Nature’s pieces. Nat Chem 5:9
Koeberle A, Werz O (2015) Multi-target approach for natural products in inflammation. Drug Discov Today 19:1871
Lewell XQ, Judd DB, Watson SP, Hann MM (1998) RECAP – retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry. J Chem Inf Comput Sci 38:511
Wetzel S, Bon RS, Kumar K, Waldmann H (2011) Biology-oriented synthesis. Angew Chem Int Ed 50:10800
Vu H, Pedro L, Mak T, McCormick B, Rowley J, Liu M, Di Capua A, Williams-Noonan B, Pham NB, Rouwer R, Nguyen B, Andrew KT, Skinnner-Adams T, Kim J, Hol WGJ, Hui R, Crowther GJ, Van Voorhis WC, Quinn RJ (2018) Fragment-based screening of a natural product library against 62 potential malaria drug targets employing native mass spectrometry. ACS Infect Dis 4:431
Pascolutti M, Campitelli M, Nguyen B, Pham N, Gorse A-D, Quinn RJ (2015) Capturing Nature’s diversity. PLoS One 10:e0120942
Wetzel S, Klein K, Renner S, Rauh D, Oprea TI, Mutzel P, Waldmann H (2009) Interactive exploration of chemical space with Scaffold Hunter. Nat Chem Biol 5:581
Merk D, Grisoni F, Friedrich L, Gelzinyte E, Schneider G (2018) Computer-assisted discovery of retinoid X receptor modulating natural products and isofunctional mimetics. J Med Chem 61:5442
Rodrigues T, Reker D, Kunze J, Schneider P, Schneider G (2015) Revealing the macromolecular targets of fragment-like natural products. Angew Chem Int Ed 54:10516
Lanz J, Riedl R (2015) Merging allosteric and active site binding motifs: de novo generation of target selectivity and potency via natural-product-derived fragments. ChemMedChem 10:451
Rollinger JM, Hornick A, Langer T, Stuppner H, Prast H (2004) Acetylcholinesterase inhibitory activity of scopolin and scopoletin discovered by virtual screening of natural products. J Med Chem 47:6248
Hartenfeller M, Zettl H, Walter M, Rupp M, Reisen F, Proschak E, Weggen S, Stark H, Schneider G (2012) DOGS: reaction-driven de novo design of bioactive compounds. PLoS Comp Biol 8:e1002380
Reker D, Rodrigues T, Schneider P, Schneider G (2014) Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc Natl Acad Sci U S A 111:4067
Chen Y, de Bruyn Kops C, Kirchmair J (2017) Data resources for the computer-guided discovery of bioactive natural products. J Chem Inf Model 57:2099
Natural Fragment Library|Pierre Fabre (2018) https://www.pierre-fabre.com/en/natural-fragment-library
BIONET Fragments from Nature (2019) https://www.keyorganics.net/downloads-bionet-databases/
Life chemicals – natural product-like fragment library (2019) https://lifechemicals.com/screening-libraries/fragment-libraries#natural-lib
Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL (2012) Quantifying the chemical beauty of drugs. Nat Chem 4:90
Lipinski C, Hopkins A (2004) Navigating chemical space for biology and medicine. Nature 432:855
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3
Bemis GW, Murcko MA (1996) The properties of known drugs 1. Molecular frameworks. J Med Chem 39:2887
Landrum G (2012) RDKit: open-source cheminformatics. https://www.rdkit.org
Shang J, Hu B, Wang J, Zhu F, Kang Y, Li D, Sun H, Kong D-X, Hou T (2018) A cheminformatic insight into the differences between terrestrial and marine originated natural products. J Chem Inf Model 58:1182
Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD (2003) Improved protein-ligand docking using GOLD. Proteins 52:609
Congreve M, Carr R, Murray C, Jhoti H (2003) A “rule of three” for fragment-based lead discovery? Drug Discov Today 8:876
Todeschini R, Consonni V (2008) Handbook of molecular descriptors. Wiley-VCH, Weinheim
Labute P (2000) A widely applicable set of descriptors. J Mol Graphics Model 18:464
Allu TK, Oprea TI (2005) Rapid evaluation of synthetic and molecular complexity for in silico chemistry. J Chem Inf Model 45:1237
Ntie-Kang F, Onguéné PA, Scharfe M, Owono Owono LC, Megnassan E, Mbaze LM, Sippl W, Efange SMN (2014) ConMedNP: a natural product library from Central African medicinal plants for drug discovery. RSC Adv 4:409
Hall C & The Chapmann & Hall/CRC Chemical Database Dictionary of Natural Products. Chapman and Hall/CRC, available at dnp.chemnetbase.com
Chen CY-C (2011) TCM database@Taiwan: the world’s largest traditional Chinese medicine database for drug screening in silico. PLoS One 6:e15939
Ertl P, Roggo S, Schuffenhauer A (2008) Natural product-likeness score and its application for prioritization of compound libraries. J Chem Inf Model 48:68
Jain AN (2004) Ligand-based structural hypotheses for virtual screening. J Med Chem 47:947
Klopman G (1984) Artificial intelligence approach to structure-activity studies. Computer automated structure evaluation of biological activity of organic molecules. J Am Chem Soc 106:7315
Rosenkranz HS, Liu M, Cunningham A, Klopman G (1996) Application of structural concepts to evaluate the potential carcinogenicity of natural products. SAR QSAR Environ Res 5:79
Schreyer A, Blundell T (2009) CREDO: A protein-ligand interaction database for drug discovery. Chem Biol Drug Des 73:157
ChemicalComputingGroup (2011) Molecular operating environment (MOE) 2011.10. 1010 Sherbooke St. West, Suite #910, Montreal QC, Canada, H3A 2R7
Schneider G (2013) De novo molecular design. Wiley-VCH, Weinheim
Coley CW, Rogers L, Green WH, Jensen KF (2017) Computer-assisted retrosynthesis based on molecular similarity. ACS Cent Sci 3:1237
Coley CW, Green WH, Jensen KF (2018) Machine learning in computer-aided synthesis planning. Acc Chem Res 51:1281
Langdon SR, Ertl P, Brown N (2010) Bioisosteric replacement and scaffold hopping in lead generation and optimization. Mol Inf 29:366
Hu Y, Stumpfe D, Bajorath J (2016) Computational exploration of molecular scaffolds in medicinal chemistry. J Med Chem 59:4062
Schneider G, Schneider P, Renner S (2006) Scaffold-hopping: how far can you jump? QSAR Comb Sci 25:1162
Kontijevskis A (2017) Mapping of drug-like chemical universe with reduced complexity molecular frameworks. J Chem Inf Model 57:680
Grabowski K, Baringhaus K-H, Schneider G (2008) Scaffold diversity of natural products: inspiration for combinatorial library design. Nat Prod Rep 25:892
Lee M-L, Schneider G (2001) Scaffold architecture and pharmacophoric properties of natural products and trade drugs: application in the design of natural product-based combinatorial libraries. J Comb Chem 3:284
Boldi AM (2004) Libraries from natural product-like scaffolds. Curr Opin Chem Biol 8:281
Feher M, Schmidt JM (2002) Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J Chem Inf Model 43:218
Valli M, Dos Santos RN, Figueira LD, Nakajima CH, Castro-Gamboa I, Andricopulo AD, Bolzani VS (2013) Development of a natural products database from the biodiversity of Brazil. J Nat Prod 76:439
Ntie-Kang F, Zofou D, Babiaka SB, Meudom R, Scharfe M, Lifongo LL, Mbah JA, Mbaze LM, Sippl W, Efange SMN (2013) AfroDb: a select highly potent and diverse natural product library from African medicinal plants. PLoS One 8:e78085
Dictionary of marine natural products. http://dmnp.chemnetbase.com
Kong D-X, Jiang Y-Y, Zhang H-Y (2010) Marine natural products as sources of novel scaffolds: achievement and concern. Drug Discov Today 15:884
Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Mainl T, Ohl P, Thiel K, Wiswedel B (2008) KNIME: The Konstanz information miner. Data Anal Mach Learn Appl:319
Henkel T, Brunne RM, Müller H, Reichel F (1999) Statistical investigation into the structural complementarity of natural products and synthetic compounds. Angew Chem Int Ed 38:643
Miyao T, Reker D, Schneider P, Funatsu K, Schneider G (2015) Chemography of natural product space. Planta Med 81:429
Oprea TI, Gottfries J (2001) Chemography: the art of navigating in chemical space. J Comb Chem 3:157
Reutlinger M, Schneider G (2012) Nonlinear dimensionality reduction and mapping of compound libraries for drug discovery. J Mol Graphics Model 34:108
Bajorath J, Peltason L, Wawer M, Guha R, Lajiness MS, Van Drie JH (2009) Navigating structure-activity landscapes. Drug Discov Today 14:698
Quinn RJ, Carroll AR, Pham NB, Baron P, Palframan ME, Suraweera L, Pierens GK, Muresan S (2008) Developing a drug-like natural product library. J Nat Prod 71:464
Charifson PS, Walters WP (2000) Filtering databases and chemical libraries. Mol Divers 5:185
Crane EA, Gademann K (2016) Capturing biological activity in natural product fragments by chemical synthesis. Angew Chem Int Ed 55:3882
Sadowski J, Gasteiger J (1993) From atoms and bonds to three-dimensional atomic coordinates: automatic model builders. Chem Rev 93:2567
Maeda MH (2015) Current challenges in development of a database of three-dimensional chemical structures. Front Bioeng Biotechnol 3:66
Schwab CH (2010) Conformations and 3D pharmacophore searching. Drug Discov Today Technol 7:e245
Albadry MA, Elokely KM, Wang B, Bowling JJ, Abdelwahab MF, Hossein MH, Doerksen RJ, Hamann MT (2013) Computationally assisted assignment of kahalalide Y configuration using an NMR-constrained conformational search. J Nat Prod 76:178
Rupp M, Bauer MR, Wilcken R, Lange A, Reutlinger M, Boeckler FM, Schneider G (2014) Machine learning estimates of natural product conformational energies. PLoS Comput Biol 10:e1003400
Rollinger JM, Schuster D, Danzl B, Schwaiger S, Markt P, Schmidtke M, Gertsch J, Raduner S, Wolberg G, Langer T, Stuppner H (2009) In silico target fishing for rationalized ligand discovery exemplified on constituents of Ruta graveolens. Planta Med 75:195
Atanasov AG, Wang JN, Gu SP, Bu J, Kramer M, Baumgartner L, Fakhrudin N, Ladurner A, Malainer C, Vuorinen A, Noha SM, Schwaiger S, Rollinger JM, Schuster D, Stuppner H, Dirsch VM, Heiss E (2013) Honokiol: a non-adipogenic PPARγ agonist from Nature. Biochim Biophys Acta, Gen Subj 1830:4813
Sauer WHB, Schwarz MK (2003) Molecular shape diversity of combinatorial libraries: a prerequisite for broad bioactivity. J Chem Inf Model 43:987
Olmedo DA, González-Medina M, Gupta MP, Medina-Franco JL (2017) Cheminformatic characterization of natural products from Panama. Mol Divers 21:779
Ertl P, Schuffenhauer A (2008) Cheminformatics analysis of natural products: lessons from Nature inspiring the design of new drugs. Prog Drug Res 66:217
Lankadurai BP, Nagato EG, Simpson MJ (2013) Environmental metabolomics: an emerging approach to study organism responses to environmental stressors. Environ Rev 21:180
Baker M (2011) Metabolomics: from small molecules to big ideas. Nat Methods 8:117
Ertl P, Schuffenhauer A (2009) Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform 1:8
Grisoni F, Merk D, Consonni V, Hiss JA, Tagliabue SG, Todeschini R, Schneider G (2018) Scaffold hopping from natural products to synthetic mimetics by holistic molecular similarity. Commun Chem 1:44
Wetzel S, Wilk W, Chammaa S, Sperl B, Roth AG, Yektaoglu A, Renner S, Berg T, Arenz C, Giannis A, Oprea TI, Rauh D, Kaiser M, Waldmann H (2010) A scaffold-tree-merging strategy for prospective bioactivity annotation of γ-pyrones. Angew Chem Int Ed 49:3666
Böhm HJ, Flohr A, Stahl M (2004) Scaffold hopping. Drug Discov Today Technol 1:217
Schröder P, Förster T, Kleine S, Becker C, Richters A, Ziegler S, Rauh D, Kumar K, Waldmann H (2015) Neuritogenic militarinone-inspired 4-hydroxypyridones target the stress pathway kinase MAP4K4. Angew Chem Int Ed 54:12398
Rodrigues T (2017) Harnessing the potential of natural products in drug discovery from a cheminformatics vantage point. Org Biomol Chem 15:9275
Merk D, Grisoni F, Friedrich L, Schneider G (2018) Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun Chem 1:68
Hopkins AL (2009) Drug discovery: predicting promiscuity. Nature 462:167
Hopkins AL, Groom CR, Alex A (2004) Ligand efficiency: a useful metric for lead selection. Drug Discov Today 9:430
Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, Whaley R, Glennon RA, Hert J, Thomas KLH, Edwards DD, Shoichet BK, Roth BL (2009) Predicting new molecular targets for known drugs. Nature 462:175
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53:2719
Reker D, Bernardes GJL, Rodrigues T (2019) Computational advances in combating colloidal aggregation in drug discovery. Nat Chem 11:402
Aldrich C, Bertozzi C, Georg GI, Kiessling L, Lindsley C, Liotta D, Merz KM Jr, Schepartz A, Wang S (2017) The ecstasy and agony of assay interference compounds. ACS Cent Sci 3:143
Bisson J, McAlpine JB, Friesen JB, Chen S-N, Graham J, Pauli GF (2016) Can invalid bioactives undermine natural product-based drug discovery? J Med Chem 59:1671
Huth JR, Mendoza R, Olejniczak ET, Johnson RW, Cothron DA, Liu Y, Lerner CG, Chen J, Hajduk PJ (2005) ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J Am Chem Soc 127:217
Baell JB (2016) Feeling Nature’s PAINS: natural products, natural product drugs, and pan assay interference compounds (PAINS). J Nat Prod 79:616
Duan D, Doak AK, Nedyalkova L, Shoichet BK (2015) Colloidal aggregation and the in vitro activity of traditional Chinese medicines. ACS Chem Biol 10:978
Röhrig UF, Majjigapu SR, Vogel P, Zoete V, Michielin O (2015) Challenges in the discovery of indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors. J Med Chem 58:9421
Glaser J, Holzgrabe U (2016) Focus on PAINS: false friends in the quest for selective anti-protozoal lead structures from Nature? Med Chem Commun 7:214
Dahlin JL, Nelson KM, Strasser JM, Barsyte-Lovejoy D, Szewczyk MM, Organ S, Cuellar M, Singh G, Shrimp JH, Nguyen N, Meier JL, Arrowsmith CH, Brown PJ, Baell JB, Walters MA (2017) Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors. Nat Commun 8:1527
Matlock MK, Hughes TB, Dahlin JL, Swamidass SJ (2018) Modeling small-molecule reactivity identifies promiscuous bioactive compounds. J Chem Inf Model 58:1483
Ekins S, Freundlich JS (2011) Validating new tuberculosis computational models with public whole cell screening aerobic activity datasets. Pharm Res 28:1859
Erlanson D (2013) Fragmenting natural products – sometimes PAINfully. http://practicalfragments.blogspot.com/2013/02/fragmenting-natural-products-sometimes.html. Accessed 18 Jun 2018
Irwin JJ, Duan D, Torosyan H, Doak AK, Ziebart KT, Sterling T, Tumanian G, Shoichet BK (2015) An aggregation advisor for ligand discovery. J Med Chem 58:7076
Pohjala L, Tammela P, Pohjala L, Tammela P (2012) Aggregating behavior of phenolic compounds – a source of false bioassay results? Molecules 17:10774
Rao H, Li Z, Li X, Ma X, Ung C, Li H, Liu X, Chen Y (2010) Identification of small molecule aggregators from large compound libraries by support vector machines. J Comput Chem 31:752
Feng BY, Shelat A, Dorman TN, Guy RK, Shoichet BK (2005) High-throughput assays for promiscuous inhibitors. Nat Chem Biol 1:146
Weaver S, Gleeson MP (2008) The importance of the domain of applicability in QSAR modeling. J Mol Graphics Model 26:1315
Dragos H, Gilles M, Alexandre V (2009) Predicting the predictability: a unified approach to the applicability domain problem of QSAR models. J Chem Inf Model 49:1762
Ingólfsson HI, Thakur P, Herold KF, Hobart EA, Ramsey NB, Periole X, de Jong DH, Zwama M, Yilmaz D, Hall K, Maretzky T, Hemmings HC, Blobel C, Marrink SJ, Koçer A (2014) Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem Biol 9:1788
Cristani M, D’Arrigo M, Mandalari G, Castelli F, Grazia Sarpietro M, Micieli D, Venuti V, Bisignano G, Saija A, Trombetta D (2007) Interaction of four monoterpenes contained in essential oils with model membranes: implications for their antibacterial activity. J Agric Food Chem 55:6300
Ossman T, Fabre G, Trouillas P (2016) Interaction of wine anthocyanin derivatives with lipid bilayer membranes. Comput Theor Chem 1077:80
Pillong M, Hiss JA, Schneider P, Lin B, Blatter M, Müller AT, Bachler S, Neuhaus CS, Dittrich PS, Altmann KH, Wessler S, Schneider G (2017) Rational design of membrane-pore-forming peptides. Small 13:1701316
Lyu Y, Xiang N, Mondal J, Zhu X, Narsimhan G (2018) Characterization of interactions between curcumin and different types of lipid bilayers by molecular dynamics simulation. J Phys Chem B122:2341
Capuzzi SJ, Muratov EN, Tropsha A (2017) Phantom PAINS: problems with the utility of alerts for pan-assay interference compounds. J Chem Inf Model 57:417
Jasial S, Hu Y, Bajorath J (2017) How frequently are pan-assay interference compounds active? Large-scale analysis of screening data reveals diverse activity profiles, low global hit frequency, and many consistently inactive compounds. J Med Chem 60:3879
Vidler LR, Watson IA, Margolis BJ, Cummins DJ, Brunavs M (2018) Investigating the behavior of published PAINS alerts using a pharmaceutical company data set. ACS Med Chem Lett 9:792
Owen SC, Doak AK, Wassam P, Shoichet MS, Shoichet BK (2012) Colloidal aggregation affects the efficacy of anticancer drugs in cell culture. ACS Chem Biol 7:1429
Baell JB, Nissink JWM (2018) Seven year itch: pan-assay interference compounds (PAINS) in 2017 – utility and limitations. ACS Chem Biol 13:36
Lagunin A, Filimonov D, Poroikov V (2010) Multi-targeted natural products evaluation based on biological activity prediction with PASS. Curr Pharm Des 16:1703
Sá MS, de Menezes MN, Krettli AU, Ribeiro IM, Tomassini TC, Ribeiro dos Santos R, de Azevedo WF, Soares MB (2011) Antimalarial activity of physalins B, D, F, and G. J Nat Prod 74:2269
Johnson MA, Maggiora GM (1990) Concepts and applications of molecular similarity. Wiley, New York
Rogers D, Hahn M (2010) Extended-connectivity fingerprints. J Chem Inf Model 50:742
MACCS Structural Keys (2005) – MDL Information Systems Inc
Nickel J, Gohlke B-O, Erehman J, Banerjee P, Rong WW, Goeade A, Dunkel M, Preissner R (2014) SuperPred: update on drug classification and target prediction. Nucleic Acids Res 42:W26
Antolín AA, Jalencas X, Yélamos J, Mestres J (2012) Identification of Pim kinases as novel targets for PJ34 with confounding effects in PARP biology. ACS Chem Biol 7:1962
Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguez RM, Huang X-P, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FRC, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL (2012) Automated design of ligands to polypharmacological profiles. Nature 492:215
Reutlinger M, Rodrigues T, Schneider P, Schneider G (2014) Combining on-chip synthesis of a focused combinatorial library with computational target prediction reveals imidazopyridine GPCR ligands. Angew Chem Int Ed 53:582
Schuster D, Spetea M, Music M, Rief S, Fink M, Kirchmair J, Schütz J, Wolber G, Langer T, Stuppner H, Schmidhammer H, Rollinger JM (2010) Morphinans and isoquinolines: Acetylcholinesterase inhibition, pharmacophore modeling, and interaction with opioid receptors. Bioorg Med Chem Lett 18:5071
Fakhrudin N, Ladurner A, Atanasov AG, Heiss EH, Baungartner L, Markt P, Schuster D, Ellmerer EP, Wolber G, Rollinger JM, Stuppner H, Dirsch VM (2010) Computer-aided discovery, validation, and mechanistic characterization of novel neolignan activators of peroxisome proliferator-activated receptor. Mol Pharmacol 77:559
Schneider G, Böhm H-J (2002) Virtual screening and fast automated docking methods. Drug Discov Today 7:64
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3:935
Lyne PD (2002) Structure-based virtual screening: an overview. Drug Discov Today 7:1047
Do Q-T, Lamy C, Renimel I, Sauvan N, Andre P, Himbert F, Morin-Allroy L, Bernard P (2007) Reverse pharmacognosy: identifying biological properties for plants by means of their molecule constituents: application to meranzin. Planta Med 73:1235
Bajorath J (2002) Affinity fingerprints – leading the way? Drug Discov Today 7:1035
Wassermann AM, Lounkine E, Urban L, Whitebread S, Chen S, Hughes K, Guo H, Kutlina E, Fekete A, Klumpp M, Glick M (2014) A screening pattern recognition method finds new and divergent targets for drugs and natural products. ACS Chem Biol 9:1622
Reutlinger M, Koch CP, Reker D, Schneider TN, Rodrigues T, Schneider G (2013) Chemically advanced template search (CATS) for scaffold-hopping and prospective target prediction for “orphan” molecules. Mol Inf 32:133
Schneider G, Neidhart W, Giller T, Schmid G (1999) “Scaffold-hopping” by topological pharmacophore search: a contribution to virtual screening. Angew Chem Int Ed 38:2894
Kohonen T (1990) The self-organizing map. Proc IEEE 78:1464
Schneider P, Tanrikulu Y, Schneider G (2009) Self-organizing maps in drug discovery: compound library design, scaffold-hopping, repurposing. Curr Med Chem 16:258
Digles D, Ecker GF (2011) Self-organizing maps for in silico screening and data visualization. Mol Inf 30:838
Reker D, Seet M, Pillong M, Koch CP, Schneider P, Witschel MC, Rottmann M, Freymond C, Brun R, Schweizer B, Illarionov B, Bacher A, Fischer M, Diederich F, Schneider G (2014) Deorphaning pyrrolopyrazines as potent multi-target antimalarial agents. Angew Chem Int Ed 53:7079
Schneider G, Reker D, Chen T, Hauenstein K, Schneider P, Altmann K-H (2016) Deorphaning the macromolecular targets of the natural anticancer compound doliculide. Angew Chem Int Ed 55:12408
Rodrigues T, Werner M, Roth J, da Cruz EHG, Marques MC, Akkapeddi P, Lobo SA, Koeberle A, Corzana F, da Silva Junior EN, Werz O, Bernardes HJL (2018) Machine intelligence decrypts β-lapachone as an allosteric 5-lipoxygenase inhibitor. Chem Sci 9:6899
Schneider P, Schneider G (2017) A computational method for unveiling the target promiscuity of pharmacologically active compounds. Angew Chem Int Ed 56:11520
Joseph JA, Fisher DR, Cheng V, Rimando AM, Shukitt-Hale B (2008) Cellular and behavioral effects of stilbene resveratrol analogues: implications for reducing the deleterious effects of aging. J Agric Food Chem 56:10544
Rollinger JM, Steindl TM, Schuster D, Kirchmair J, Anrain K, Ellmerer EP, Langer T, Stuppner H, Wutzler P, Schmidtke M (2008) Structure-based virtual screening for the discovery of natural inhibitors for human rhinovirus coat protein. J Med Chem 51:842
Brand S, Roy S, Schröder P, Rathmer B, Roos J, Kapoor S, Patil S, Pommeremnke C, Maier T, Janning P, Eberth S, Steinhilber D, Schade D, Schneider G, Kumar K, Ziegler S, Waldmann H (2018) Combined proteomic and in silico target identification reveal a role for 5-lipoxygenase in developmental signaling pathways. Cell Chem Biol 25:1095
Schmeller T, Sauerwein M, Sporer F, Wink M, Müller WE (1994) Binding of quinolizidine alkaloids to nicotinic and muscarinic acetylcholine receptors. J Nat Prod 57:1316
Foley DJ, Craven PGE, Collins PM, Doveston RG, Aimon A, Talon R, Churcher I, von Delft F, Marsden SP, Nelson A (2017) Synthesis and demonstration of the biological relevance of sp3-rich scaffolds distantly related to natural product frameworks. Chem Eur J 23:15227
Engels MFM, Thielemans T, Verbinnen D, Tollaenere JP, Verbeek R (2000) CerBeruS: a system supporting the sequential screening process. J Chem Inf Comput Sci 40:241
Renner S, Van Otterlo WAL, Dominguez Seoane M, Möcklinghoff S, Steinhilber D, Brunsveld L, Rauh D, Waldmann H (2009) Bioactivity-guided mapping and navigation of chemical space. Nat Chem Biol 5:585
Koch MA, Wittenberg L-O, Basu S, Jeyrai DA, Gouroulidou E, Reinecke K, Odermatt A, Waldmann H (2004) Compound library development guided by protein structure similarity clustering and natural product structure. Proc Natl Acad Sci U S A 101:16721
Schmid F, Jessen HJ, Burch P, Gademann K (2013) Truncated militarinone fragments identified by total chemical synthesis induce neurite outgrowth. Med Chem Commun 4:135
Jessen HJ, Schumacher A, Shaw T, Pfaltz A, Gademann K (2011) A unified approach for the stereoselective total synthesis of pyridone alkaloids and their neuritogenic activity. Angew Chem Int Ed 50:4222
Bon RS, Waldmann H (2010) Bioactivity-guided navigation of chemical space. Acc Chem Res 43:1103
Gunasekera SP, McCarthy PJ, Kelly-Borges M, Lobkovsky E, Clardy J (1996) Dysidiolide: a novel protein phosphatase inhibitor from the Caribbean sponge Dysidea etheria de Laubenfels. J Am Chem Soc 118:8759
Pascolutti M, Quinn RJ (2014) Natural products as lead structures: chemical transformations to create lead-like libraries. Drug Discov Today 19:215
Friedrich L, Rodrigues T, Neuhaus CS, Schneider P, Schneider G (2016) From complex natural products to simple synthetic mimetics by computational de novo design. Angew Chem Int Ed 55:6789
Rupp M, Schröter T, Steri R, Zettl H, Proschak E, Hansen K, Rau O, Schwarz O, Müller-Kuhrt L, Schubert-Zsilavecz M, Müller K-R, Schneider G (2010) From machine learning to natural product derivatives selectively activating transcription factor PPAR. ChemMedChem 5:191
Schneider P, Schneider G (2016) De novo design at the edge of chaos. J Med Chem 59:4077
Schneider G (2013) De novo design–hop(p)ing against hope. Drug Discov Today Technol 10:e453
Rodrigues T, Roudnicky F, Koch CP, Kudoh T, Reker D, Detmar M, Schneider G (2013) De novo design and optimization of aurora A kinase inhibitors. Chem Sci 4:1229
Perna AM, Rodrigues T, Schmidt TP, Böhm M, Stutz K, Reker D, Pfeiffer B, Altmnn K-H, Backert S, Wessler S, Schneider G (2015) Fragment-based de novo design reveals a small-molecule inhibitor of Helicobacter pylori HtrA. Angew Chem 127:10382
Rodrigues T, Kudoh T, Roudnicky F, Lim YF, Lin Y-C, Koch CP, Seno M, Detmar M, Schnedier G (2013) Steering target selectivity and potency by fragment-based de novo drug design. Angew Chem Int Ed 52:10006
Schneider G, Hartenfeller M, Reutlinger M, Tanrikulu Y, Proschak E, Schneider P (2009) Voyages to the (un)known: adaptive design of bioactive compounds. Trends Biotechnol 27:18
Spänkuch B, Keppner S, Lange L, Rodrigues T, Zettl H, Koch CP, Reutlinger M, Hartenfeller M, Schneider P, Schneider G (2013) Drugs by numbers: reaction-driven de novo design of potent and selective anticancer leads. Angew Chem Int Ed 52:4676
Rodrigues T, Reker D, Welin M, Caldera M, Brunner C, Gabernet G, Schneider P, Walse B, Schneider G (2015) De novo fragment design for drug discovery and chemical biology. Angew Chem Int Ed 54:15079
Gupta A, Müller AT, Huisman BJH, Fuchs JA, Schneider P, Schneider G (2018) Generative recurrent networks for de novo drug design. Mol Inf 37:1700111
Segler MHS, Kogej T, Tyrchan C, Waller MP (2018) Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci 4:120
Blaschke T, Olivecrona M, Engkvist O, Bajorath J, Chen H (2018) Application of generative autoencoder in de novo molecular design. Mol Inf 37:1700123
Renner S, Schneider G (2006) Scaffold-hopping potential of ligand-based similarity concepts. ChemMedChem 1:181
Newman DJ, Cragg GM (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75:311
Jayaseelan KV, Moreno P, Truszkowski A, Ertl P, Steinbeck C (2012) Natural product-likeness score revisited: an open-source, open-data implementation. BMC Bioinformatics 13:106
Murray CW, Rees DC (2009) The rise of fragment-based drug discovery. Nat Chem 1:187
Acknowledgments
Daniel Reker is a Swiss National Science Foundation Fellow (Grant P2EZP3_168827 and P300P2_177833). His work is partly supported by the MIT-IBM Watson AI Lab and the MIT SenseTime Alliance. Daniel Reker is grateful to his Ph.D. advisor Prof. Gisbert Schneider and his postdoctoral advisors Prof. Robert S. Langer and Prof. Giovanni Traverso for invaluable guidance and mentorship.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Reker, D. (2019). Cheminformatic Analysis of Natural Product Fragments. In: Kinghorn, A., Falk, H., Gibbons, S., Kobayashi, J., Asakawa, Y., Liu, JK. (eds) Progress in the Chemistry of Organic Natural Products 110. Progress in the Chemistry of Organic Natural Products, vol 110. Springer, Cham. https://doi.org/10.1007/978-3-030-14632-0_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-14632-0_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14631-3
Online ISBN: 978-3-030-14632-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)