
Abstract
β-Cells represent the functional unit of pancreatic islets and they are responsible for glucose homeostasis regulation. β-Cells possess the ability to modify insulin secretion according to the organism-specific needs. Thus, during physiological changes such as pregnancy or obesity, glycemia is increased concomitantly with the ability of β-cells to secrete insulin. However, when demand for insulin chronically increases, a steady stimulation of β-cells eventually may lead to death. In spite of the conducted efforts in order to elucidate the glucotoxicity mechanisms acting on β-cells, they remain largely unknown. Hyperglycemia promotes several metabolic alterations such as glucolipotoxicity, mitochondrial alterations, oxidative stress, endoplasmic reticulum stress, amyloid polypeptide accumulation, and proinflammatory cytokines accumulation. The latter is commonly engaged during apoptosis triggering in β-cells. In recent years, p53 has been also proposed as a major trigger of apoptosis in β-cells during hyperglycemia conditions. Because insulin-producing cells are cultured using high glucose levels, the presence of p53 in mitochondria induce apoptosis. The insight on the mechanisms triggering cell death in pancreatic β-cells will support the proposal of alternatives for prevention and/or cell protection also contributing to treatment of diabetic patients.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Chapter Objectives-
To briefly describe the embryonic development of pancreatic β
-
To analyze the universal literature on the mechanisms that underlie the loss of pancreatic β-cell mass
-
To provide information on the regulation of p53 by hyperglycemia and its participation in the induction of pancreatic β-cell apoptosis
Introduction
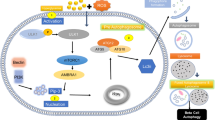
Insulin produced and secreted by β-cells is responsible for blood glucose level regulation. The major stimulus for insulin secretion is glucose itself. When the latter is taken by β-cells in a process mediated by the glucose transporter 2 (GLUT 2), it enters the glycolytic pathway, the Krebs cycle, and oxidative phosphorylation, and it promotes the increase of the ATP/ADP ratio. Subsequently, this leads to the closure of the ATP-dependent potassium channels, to membrane depolarization, and to Ca2+ influx through the voltage-dependent Ca2+ channels. An increase of cytosolic Ca2+ is the signal that triggers glucose-stimulated insulin secretion (GSIS). Alterations of insulin secretion and glycemia increases lead to the settlement of type 2 diabetes (T2D). Additionally, this disease depends on external factors such as diet, body weight, and genetic background that may delay or enhance all clinical sings of the disease. It is known that, in modern society, an increased carbohydrate intake and the lack of exercising lead to the development of obesity. This condition increases insulin demand in order to maintain normal glycemia, thus the ability of β-cells in order to fulfill the insulin requirements is critical to preserve glucose homeostasis. The initial response toward an increased insulin demand is adaptive hyperplasia and an increased synthesis of the hormone. However, if insulin resistance persists during extended time periods, β-cells become exhausted and their mass decreases due to an increase of the apoptotic rate and consequently hyperglycemia appears. This latter condition activates several metabolic pathways impairing β-cells, such as glucolipotoxicity, mitochondrial alterations, reactive oxygen species (ROS) as well as oxidative stress and endoplasmic reticulum stress (ERS), proinflammatory cytokines, deposition of amyloid polypeptide, and p53 translocation to mitochondria (Fig. 12.1). Recently, it has been proposed that p53 protein is a major apoptosis trigger in β-cells during hyperglycemia conditions [1, 2]. These events impair β-cells by hampering their proliferative ability and also by decreasing insulin expression and secretion and promoting their death. Several of these alterations, either separate or combined, may be observed in T2D models, thus it is likely that β-cell loss in humans is linked to the activation of the aforementioned mechanisms and not to just one of them [3]. The contribution from each one of such mechanisms to the decrease of pancreatic β-cell amounts will be reviewed in this chapter.

Metabolic pathways by hyperglycemia-induced β-cell death. ER endoplasmic reticulum
The Origin of β-Cells
Embryonic Development and Differentiation
During embryonic development or even during the first postnatal days, β-cell may be generated by stem or progenitor cells within the pancreatic ducts, bone marrow, or even pancreatic islets. Islet genesis initiates on the week 12 of gestation in humans. In weeks 13–16, the early endocrine precursor cells on the duct’s wall form cell aggregates and generate an early islet. In weeks 17–20, the connection between islets and duct is lost and the islets are properly formed. At this stage, they contain only pancreatic polypeptide, somatostatin and glucagon derived from immature precursor cells. In weeks 21–26, β-cells are located at the islet center [4].
Studies conducted on rodents have allowed the identification of the molecular mechanisms regulating pancreatic β-cell establishment and differentiation. In mouse, it begins on the E8.5 day of development, and it depends on several factors secreted by neighboring early gut cells and mainly from vessels. During pancreas development several transcription factors intervene, such as the pancreatic transcription factor (Ptf1a) and the pancreatic and duodenal homeobox (Pdx1). Other factors such as SRY (sex-determining region Y)-box 9 (SOX9), forkhead box (FOX) A1/2, hepatocyte nuclear factor (HNF) 1β, and GATA4/6 have a critical participation during the establishment of the pancreatic progenitor cell pool. The development of endocrine progenitors requires Notch activation, whereas the formation of β-cells depends on Nkx 6.1, NeuroD1, the regulatory factor x (Rfx) 6, islet 1(Isl), Nkx2.2, and Pax4. The first hormone-producing cells are detected on E9.5 and their numbers increase by E13.5. At that time, the expression of β-cell-specific genes may be observed, such as GLUT 2 [5, 6].
Postnatal Development of the Pancreas
After birth, when feeding initiates, pancreatic mass increases because of the rapid expansion of the exocrine tissue. During the first 2 years of life, β-cell population keeps a 3–4% replication rate, and the pancreas reaches its maximum volume by 5–10 years old due to a decreased replication rate (0.05–0.1%) [7]. By the third decade of life, pancreatic mass is stabilized and constant until approximately 60 years of age. From that moment on, pancreas size begins to decrease [8] because of an increase of cell proliferation inhibitors such as p16, p26, p27, and cyclin D3, whereas Pdx1 decreases. The latter is needed for β-cell differentiation [4]. In rodents, the β-cell population is established during the first 4 weeks of life. In rats, between postnatal days 3 and 24, a proliferation rate of 3% occurs, and during this period apoptosis also contributes to β-cell population curtailing. However, this stage is highly influenced by the prevailing nutritional environment [9].
In adults, during physiological conditions such as pregnancy or during adaptation toward weight increase, it is possible to observe an increase of β-cell replication rates [10]. It has been demonstrated that β-cell replication may be induced in rodents by providing a high-fat diet or a chronic glucose infusion [11]. Hyperglycemia also stimulates cell proliferation by activating glycolysis and by shortening the quiescence period of the cell cycle and also by promoting the G1-S transition through the activation of the ChREBP (carbohydrate response element binding protein) transcription factor [3]. This confirms β-cell adaptive ability when facing a metabolic demand.
β-Cell Dysfunction and the Loss of Pancreatic Β-Cell Mass
Chronic exposition of β-cells to high glucose levels impairs their functions, and it may induce dedifferentiation and even death and a decreased pancreatic β-cell mass (β-cell failure). The onset of β-cell progressive deterioration and loss of function occurs at an earlier stage, before TD2 symptoms even appear, which are evident due to decreased insulin synthesis and secretion [12]. Postmortem studies on human pancreas from patients with a clinical history of fasting glucose alterations demonstrated approximately a 50% decrease of β-cell mass caused by apoptosis [13]. Similarly, an increased glucose-stimulated insulin secretion was observed due to the remaining 50% [14]. However, the decreased β-cell mass is not only induced by an enhanced apoptosis as alterations of cell proliferation rates have also been documented. Additionally, during hyperglycemia conditions other studies have reported that β-cell undergo dedifferentiation or regression processes characterized by a decreased expression of the specific genes for these cells [15].
β-Cell Apoptosis
Apoptosis is a physiological mechanism for cell suppression that enables the elimination of some cells without affecting neighboring cells and without releasing the cell contents, unlike necrosis that is concomitant with an inflammatory reaction (Table 12.1). Apoptosis may be triggered through the activation of two major pathways: intrinsic or extrinsic. The latter is activated by death ligands that bind to cell surface receptors, thus transmitting death signals. Fas and TNFRI are the best described death receptors. They possess a cysteine-rich extracellular domain and a cytoplasmic death domain. Fas ligand (FasL) binds to one of three Fas molecules, and it promotes receptor oligomerization and FADD (Fas-associated protein with death domain or Mort-1) interaction with the receptor’s death domain. Subsequently, FADD binds to procaspase-8 leading to its activation. In turn, caspase-8 leads to the activation of other caspases, such as caspase-9. These are located within cytoplasm as inactive proenzymes, and they may be activated by other caspases, death receptors, or cytochrome c [16] through its interaction with the apoptosis protease-activating factor 1 (Apaf-1) and procaspase-9, thus leading to apoptosome formation and the triggering of the caspase-activating cascade.
The extrinsic pathway initiates with the release of several proapoptotic factors, such as cytochrome c, from the mitochondrial intermembrane space toward cytosol. As mentioned above, this event leads to caspase activation through the formation of the apoptosome, the activation of caspase-9, and the subsequent activation of executing caspases 3, 6, and 7 and with these the apoptosis proteolytic cascade. Mitochondria also release the apoptosis-induced factor (AIF), a flavoprotein that translocate to the nucleus where it triggers chromatin condensation and DNA fragmentation. Mitochondrial membrane potential (ΔΨm) alterations are also observed as well as respiratory chain uncoupling, all of them identified as early dysfunctions leading to cell death. Ceramides, oxidative agents, and pathologic increases of cytosolic Ca2+ may also induce the disruption of mitochondrial external membrane. Other proteins involved in permeabilization the mitochondrial membrane are those belonging to the Bcl-2 family. All of its members possess one to four preserved residues, known as the Bcl-2 homology domains (BHI to BH4). The apoptosis inhibitors Bcl-2 and BcI-xl possess BH1 and BH2, whereas apoptosis inducers such as Bax, Bak, and Bok display BH1, BH2, and BH3. There are some other proteins that only possess BH3 also known as death proteins. The Bcl-2 family members either promote or inhibit apoptosis in response to different stimuli such as lack of growth factors, Apaf-1 sequestration, and oxidative stress, among others [17].
At physiologic level, apoptosis is crucial for pancreas remodeling in newborns [18]. In adults, β-cell mass may increase and subsequently return to their normal size during some physiologic situations such as pregnancy and depending on the organism requirements. In other conditions as obesity and insulin resistance, it has been proposed that β-cell hyperplasia may be reverted by body weight decreases and the increase of the apoptosis rate [19].
Whereas glucose-mediated stimulation is essential for physiologic maintenance of β-cells, chronic hyperglycemia induces severe damage to these cells, and it creates a vicious cycle contributing to the progressive loss of functional β-cell mass. Chronic hyperglycemia decreases β-cell sensibility toward glucose and induces exhaustion and toxicity. Desensitizing is a reversible protective mechanism facing a steady demand for insulin. When glycemia levels are restored and stimulation ceases, β-cells may regain their sensibility toward glucose [20]. Conversely, if hyperglycemia persists, β-cells become exhausted, thus implicating the loss of insulin granules and that of two transcriptional factors regulating insulin expression: the musculoaponeurotic fibrosarcoma protein A (MafA) and PDX-1. It is important to mention that these effects may also be restored when glycemia decreases [21, 22]. However, if insulin resistance is persistent and glycemia increases, an overstimulation of β-cells occurs, thereby altering their functions and mainly the mechanisms for insulin synthesis and secretion [1, 19, 23,24,25]. If hyperglycemia is not adequately controlled, persistent stimulation of β-cells may eventually lead to degranulation, exhaustion, and apoptosis. In vitro studies have shown that culturing insulin-producing cells (RINm5F) in the presence of high glucose levels, an increased ROS production is observed and it triggers apoptosis [1]. In these conditions, Bax oligomerization also increases along with cytochrome c release and caspase-3 activation [19], but the precise mechanisms involved in β-cell apoptosis have not been completely elucidated.
Glucolipotoxicity
However, free fatty acids (FFA) in physiologic levels contribute to preserve glucose-stimulated insulin secretion (GSIS), exposition toward high FFA levels for prolonged periods, along with hyperglycemia, affects the expression of the Ins gene and induces insulin resistance and also pancreatic β-cell dysfunction. FFA contribute to apoptosis triggering in β-cells through activation of protein kinase C (PKC, apoptosis mediator), increases in ceramide synthesis and Bcl-2 inhibition [26], as well as increased levels of the type 2 uncoupling protein (UCP2) [27], thus decreasing ATP production [28] and glucose-stimulated insulin secretion, besides contributing to both oxidative stress and endoplasmic reticulum stress [29, 30]. It has been observed that unsaturated fatty acids (e.g., palmitic acid) are more toxic when compared to their monounsaturated counterparts (e.g., palmitoleic acid), as the latter may even exhibit a protective effect because they are rapidly esterified in order to form triacylglycerols [31]. However, it is currently accepted that FFA-induced damage depends on concentration, exposure time, and blood glucose levels [30, 32]. When these factors converge, fatty acids and glucose compete for metabolism through the glycolytic pathway. During hyperglycemia, oxidative phosphorylation becomes saturated with glycolytic products, thus promoting the formation of malonyl-CoA that inhibits the β-oxidation of fatty acids. This effect forces β-cells to divert fatty acids to other metabolic pathways in order to metabolize them; thereby it increases the production of esterified fatty acids such as ceramides [33]. Accumulation of the latter occurs from sphingomyelin cleavage and/or de novo ceramide synthesis by condensing serine and non-oxidized palmitoyl-CoA through the activity of serine C-palmitoyltransferase (SPT) located in mitochondria and endoplasmic reticulum. Ceramides affect mitochondrial membrane potential and permeability and they represent a ROS production mechanism. They also enable the release of apoptosis induction factors such as cytochrome c and procaspases, thus leading to β-cell death [34]. Some experiments have demonstrated apoptosis induction mediated by ceramides after inhibiting their synthesis. In such conditions fatty acid-induced apoptosis also decreases [33]. Additionally, ceramides induce the activation of the NF-κB transcription factor that increases inducible nitric oxide synthase (iNOS) and nitric oxide (NO) production. The interaction between NO and superoxide anion (O2) produces peroxynitrite, thus inducing DNA damage and the activation of poly (ADP-ribose) polymerase (PARP), a NAD+-dependent enzyme [34]. Therefore, its over-activation decreases both the NAD+ pool and glycolytic rate and electron transport and ATP synthesis. Besides negatively affecting insulin secretion, this situation may lead to pancreatic β-cell death [35].
Oxidative Stress
Whereas low ROS levels exert a beneficial effect on β-cells [9], their overproduction causes oxidative damage to proteins, lipids, and nucleic acids, and it induces oxidative stress. In hyperglycemia conditions, ROS are mainly generated by glucose auto-oxidation and also through an increased electron flow in mitochondrial respiratory chain [36]. However, in recent years it has been demonstrated an important participation of the NADH oxidase complex [1], as their components have been identified in rat pancreatic β-cells [37]. An increased mitochondrial pathway initially accelerates NADH production. The latter participates in the mitochondrial respiratory chain, and it represents the first step for O°−2 production that also generates other radicals such as hydrogen peroxide (H2O2) and the hydroxyl radical (°OH), one of the most potent oxidants. An increased O°−2inhibits glyceraldehyde 3-phosphate dehydrogenase (GAPDH) inducing accumulation of glyceraldehyde 3-phosphate (G3P) within the glycolytic pathway. This leads to the activation of others ROS-producing pathways and to oxidative stress [38, 39] as: advanced glycation end products (AGE), as their precursor (methylglyoxal) is generated from G3P. It also leads to PKC activation as glycerol (its activator) is also produced from G3P [40] (Fig. 12.2).

Hyperglycemia induces ROS production and oxidative stress by increasing glycolysis and oxidative phosphorylation pathways. This situation induces biomolecule oxidation and β-cell exhaustion and death. ROS reactive oxygen species, O-GlcNAcylation, AGE advanced glycation end products
The alterations induced by oxidative stress range from synthesis modifications and insulin secretion, endoplasmic reticulum stress, and activation of the apoptotic intrinsic pathway [7]. ROS lead to activation mechanisms that reinforce β-cell death and their decreased cell mass. They also induce mitochondrial membrane potential alterations, thus modifying permeability and the release of proapoptotic proteins (cytochrome c, apoptosis-inducing factor, among others) and activating the apoptotic proteolytic cascade [1]. β-Cell susceptibility toward the damage induced by free radicals and their low abundance of antioxidant mechanisms have been previously demonstrated [39]. Therefore, it is considered that oxidative stress is greatly responsible for pancreatic β-cell death after being exposed to hyperglycemia. The ROS-induced damage on β-cells has been quantified by the presence of 8-hydroxy-2′-deoxyguanosine (8-OHdG) on subjects affected by T2D, in animal models [30], and in vitro on insulin-secreting cells [1, 41]. It has been demonstrated that H2O2 addition to rat and mouse β-cells cultures alters mitochondrial membrane potential; it decreases ATP levels as well as glucose-stimulated insulin secretion (GSIS) [42]. This effect is abolished when the expression of antioxidant enzymes is increased. GSIS decrease induced by ROS has been linked to GAPDH and glycolysis, and, as previously mentioned, it consequently decreases ATP levels.
In addition to direct damage induced on biomolecules, oxidative stress favors the activation of other pathways such as O-glycosylation, poly(ADP-ribosylation), and O-linked β -N-acetylglucosaminilation (OGlcNAcylation). All of them may modify or inhibit the function of proteins. Approximately 2–3% of glucose entering β-cells is diverted to the hexosamine biosynthesis pathway in order to synthesize uridine diphosphate N-acetylglucosamine (UDP-GlcNAc); thus O-GlcNAcylation regulates protein function depending on glucose availability. Among the proteins prone to modification by OGT are Pdx1, FoxO1, NeuroD1, IRS2, Akt, and p53; thus it regulates glucotoxicity and β-cell apoptosis [9].
Mitochondrial Alterations
Mitochondrial physiology plays a very important role on the regulation of the insulin secretion mechanism. When exposed to hyperglycemia, β-cell mitochondria exhibit important functional and morphological changes that affect the ATP/ADP ratio required for insulin release. ROS increased due to hyperglycemia modifies mitochondrial permeability and induces apoptosis. ROS oxidizes cardiolipin, a phospholipid responsible to preserve mitochondrial architecture and membrane potential maintenance, and it also provides support for proteins involved in mitochondrial bioenergetics. Through hydrophobic and electrostatic interactions, cardiolipin keeps cytochrome c attached to the inner membrane. During early apoptosis, ROS oxidize cardiolipin, and they disrupt its interaction with cytochrome c that becomes detached from inner membrane, and it is released to cytoplasm [43], where it participates for apoptosome formation in order to activate proapoptotic caspases (intrinsic pathway). Cardiolipin is also the target of the proapoptotic protein t-Bid that is activated by caspase-8 (extrinsic pathway). tBid promotes pore formation on the external membrane mediated by Bax and Bak [44]. Thus, cardiolipin is a central regulator to achieve the activation of both apoptotic pathways.
Mitochondrial integrity and abundance are also regulated by fission and fusion mechanisms. These processes, although opposite, are coordinated in order to preserve mitochondrial morphology, size, and abundance. Mitochondrial fusion is regulated by mitofusins 1 and 2 (Mfn1 and Mfn2) as well as by the Opa1 mitochondrial dynamin like GTPase (Opa1). Mitochondrial fusion allows the exchange and merging of the organelle’s content, including membranes, genetic material, and other metabolites. It also contributes for mitochondrial function preservation in metabolic stress conditions and during glucolipotoxicity [45]. Conversely, mitochondrial fission is mediated by several proteins such as the mitochondrial fission 1 protein (Fis1), the mitochondrial fission factor (Mff) on the external membrane, and the GTPase dynamin-related protein 1 (Drp1). Fission is essential in order to segregate damaged or dysfunctional mitochondria [46]. Alterations of mitochondrial dynamics balance mediated by the loss or gain of proteins regulating fusion or fission events impact on mitochondria structure (fragmentation) and their function as well as glucose-dependent insulin secretion [45].
In response to glucose, cell energy and mitochondrial membrane function are also regulated by uncoupling proteins (UCPs) located at their external membrane. UCPs are mitochondrial transporters on the inner membrane that regulate the coupling status of the respiratory chain as well as ATP synthesis. Thus, they keep the necessary ATP/ADP needed for glucose-stimulated insulin secretion. They also contribute to the mitochondrial antioxidant defense by inducing physiologic uncoupling that accelerates metabolism and decreases ROS and oxidative stress [27]. Five uncoupling proteins have been identified in humans as important regulators of corporal weight gain, the energy balance, and T2D. The most important are UCP-2 and UCP-3 because of their participation for mitochondrial membrane potential maintenance and ATP production. UCP-2 protects β-cells against oxidative stress. INS1 cells cultured in presence of H2O2 increase UCP-2 expression as well as survival rate; they also decrease ROS and caspase activation. However, hyperglycemia and hyperlipidemia increase UCP2 activation; consequently they reduce ATP synthesis and insulin secretion. UCP-2 also promotes mitochondrial membrane potential alterations along with the consequent release of proapoptotic factors and β-cell dysfunction that may lead to apoptosis [47].
Endoplasmic Reticulum Stress
Oxidative stress and endoplasmic reticulum stress (ERS) are interlinked regarding β-cell dysfunction because of their direct effects on insulin biosynthesis and secretion [48]. The endoplasmic reticulum (ER) ensures the appropriate folding and processing of proteins that will be secreted, among them insulin, as well as the degradation of misfolded proteins or those exhibiting alterations. Thus, the organelle’s overload leads to misfolded protein accumulation and ERS. The latter triggers the unfolded protein response (UPR) in order to restore ER homeostasis and to decrease protein synthesis. It also increases the expression of genes involved in protein folding and ER-linked protein degradation. UPR is mediated by proteins bound to the ER membrane: PERK (protein kinase-like ER kinase), IRE1α (inositol-requiring enzyme 1α), and ATF6 (activating transcription factor 6) [49]. When stress occurs, PERK autophosphorylation induces eif2α (eukaryotic translation initiation factor 2α) phosphorylation, a factor that inhibits protein synthesis, whereas it promotes ATF-4 transcription. The latter positively regulates the expression of ERS target genes such as the C/EBP homologous protein (CHOP) and the downstream growth arrest and DNA damage-inducible protein (GADD34). These two proteins activate protein phosphatase-1 (PP-1) that in turn dephosphorylates eif2α, thereby restoring transcription. Acute exposition of β-cells to high glucose levels induces an intermediate UPR signaling characterized by IRE1α phosphorylation and activation as well as glucose-stimulated insulin secretion. However, excessive UPR stimulation induces β-cell death and diabetes. In patients displaying insulin resistance and in islets isolated from ob/ob mice, it has been demonstrated that a constant and steady demand for insulin represent ER constant stimulation and it eventually leads to stress [49]. Additionally, the increase of FFA also induces ER stress as it affects protein processing and trafficking, Ca2+ regulation, and oxidative stress in mouse insulin-producing cells (INS1) and in human cell lines [50]. Palmitate activates the UPR response through phosphorylation of IRE1 and PERK as well as β-cell apoptosis mediated by caspase-12 and caspase-3 activation [51].
Obesity and Inflammation
Currently, obesity stands out as a risk factor to develop T2D. Nevertheless, if obesity actually causes diabetes, most obese individuals sooner or later would develop hyperglycemia and T2D. In spite of this, approximately 20% of all obese individuals are diabetic [52]. This suggests that obesity and resistance toward insulin are factors that increase the risk to develop diabetes, but they are not inductors. Thus, it has been proposed that, in obese individuals, hyperglycemia may be more related to β-cell impaired function and decreased mass [53] and/or their inability to adapt themselves toward the new metabolic demand [54]. Even though some of these studies have demonstrated a decreased β-cell mass in obese humans affected by T2D (postmortem donors) and in those who displayed alterations of fasting glucose levels by 65% and 40%, respectively [10], in obese individuals not affected by T2D, β-cell mass and insulin secretion are increased by 50% in order to cope with resistance to insulin [55]. In obese rodents a physiologic adaptive expansion of β-cells is observed due to increased generation, decreased death, and β-cell hypertrophy [56]. Through this adaptation, β-cells preserve normal glycemia until they become exhausted and eventually die, thus leading to T2D development. Cell expansion is a complex process involving the activation of several pathways that converge to regulate proliferation, survival, cell size, and insulin secretion. Apparently, proliferation and hypertrophy are most important during the β-cell expansion phase, whereas apoptosis may participate in the final phases, during β-cell failure caused by hyperglycemia. Some evidence shows that, in animals displaying resistance toward insulin, the IGF1/PI3k/Akt/mTOR pathway participates during β-cell adaptation induced by a high-fat diet. mTOR (mammalian target of rapamycin) mediates protein synthesis in response to nutrients and growth factors, and it stimulates the phosphorylation of some components of the protein synthesis machinery such as p70S6K (ribosomal S6 kinase protein) and 4E-BP (IF4E binding protein). Akt also participates in cell cycle regulation by inducing phosphorylation and degradation of the cyclin-dependent kinases inhibitors such as p21 and p27. Apparently, the increased β-cell mass in obese individuals may be a reversible event, similarly to pregnancy. Some studies have reported that insulin secretion decreases concomitantly with weight loss or caloric restriction, whereas sensibility toward insulin is regained as well as β-cell function. Furthermore, caloric restriction enhances mitochondrial biogenesis and respiratory efficiency, and it decreases ROS production and promotes metabolic homeostasis [57].
Additionally, because of FFA increase, obesity predetermines a chronic inflammation state in adipose tissue that is characterized by increased proinflammatory adipokines and cytokines. These attract B cells, T cells, and macrophages toward the pancreas and adipose tissue where they secrete even more proinflammatory cytokines and chemokines, thereby contributing to inflammatory reaction and to autoimmune elimination of β-cells. The presence of reactive T cells in islets is observed in patients affected by T2D exhibiting severe β-cells lesions and low insulin secretion [58]. Obesity implies an increased amount of adipocytes and also of their fat content, a vascularization decrease, hypoxia, and cell necrosis. Signal molecules derived from cell elimination may bind to Toll-like receptors (TLRs) and to nucleotide-binding oligomerization domains (NOD) in order to induce a local or generalized immune response. The latter consists on the assembly of cytosolic protein complexes comprised by bound nucleotides, leucine-rich repeats sequences (NLRs) and caspase-1. Once active, they initiate IL-1β production [59]. Hyperglycemia increases the production of the NLRP3 inflammasome, whereas FFA activate TLR2 and TLR4, thus promoting macrophage recruitment and β-cell stress [58].
Plenty of evidence exists on the importance of proinflammatory cytokines (IL-1β, TNFα, and interferon-γ (IFNγ)) to activate signaling cascades in β-cells, such as NF-κB, the mitogen-activated protein kinase (MAPK), and the Janus kinase/signal transducer and activator of transcription (JAK/STAT). β-Cell elimination by the proinflammatory cytokines IL-1β, TNFα, and IFNγ begins by their binding to specific receptor in β-cells and to the endoplasmic reticulum [59]. The consequences of increased ROS were previously mentioned. Conversely, in the presence of cytokines, the 12/15-lipoxygenase (12/15-LO) induces the cleavage of arachidonic acid to produce highly reactive metabolic such as 12-hydroxyeicosatetraenoic that may induce oxidative stress and mitochondrial dysfunction [30]. It has been also demonstrated that thioredoxin-interacting protein (TXNIP) interacts with NLRP3 and contributes to IL-1β production induced by hyperglycemia [60]. The latter contributes to β-cell dysfunction and apoptosis in T2D. TNFα negatively regulates the insulin receptor substrate-2 (IRS-2) in β-cells by inducing its phosphorylation and modifying insulin signaling. In obesity leptin secretion by adipose tissue also predominates. Leptin inhibits glucose-stimulated insulin secretion in β-cell lines and normal mice [61], and it also contributes to intolerance toward glucose in diabetes.
β-Islet Amyloid Polypeptide (IAPP)
Amylin is synthesized by pancreatic islets and it is secreted along with insulin. Amylin is comprised by 37 amino acids, although it may produce polypeptides and be accumulated in islets in response to stress. IAPP effects initiate after binding to its receptors. Only three of them are known. They contain the calcitonin receptor in their inner structure and one of the following three receptor activity-modifying proteins: RAMP1, RAMP2, or RAMP3. The accumulation of intracellular amylin has been linked to both oxidative and endoplasmic reticulum stresses. β-Amyloid plates are a common feature in patients affected by T2D. During hyperglycemia/hyperlipidemia, IAPP synthesis also increases in β-cells along with proinsulin, and they reach enough levels in order to allow for oligomer formation [18]. They also stimulate IL-1β, islet inflammation, and β-cell apoptosis. IAPP soluble peptides have been detected, they represent early intermediates for fibril formation and they are also responsible for cell death. In peripheral tissues, IAPP modifies glucose metabolism [62], it suppresses glucose uptake induced by insulin in muscle cells [63] and digestive secretion (gastric acid, pancreatic enzymes), and it delays gastric emptying. IAP administration to rats decreases food intake [64], but when an IAPP antagonist is provided, food intake increases as well as body weight [65].
β-Cell Apoptosis and p53
β-Cell apoptotic death after being exposed to high glucose levels has been associated with p53 protein translocation toward mitochondria [1]. Furthermore, β-cell population recovery and the rescue from the diabetic phenotype was demonstrated in p53-knockout mice, thus highlighting the importance of this protein for diabetes establishment [2].
p53 protein is a transcription factor engaged in DNA damage monitoring. Depending on the severity of the damage, p53 triggers apoptosis or arrests the cell cycle until the DNA repairing mechanisms are activated. p53 activation occurs in response to several types of stress, mainly those damaging DNA, and this leads to its stabilization and accumulation in cells submitted to stress conditions. p53 is also involved in apoptosis triggering by interacting and forming complexes with Bcl-2 and Bcl-XL through its DNA-binding domain, thus allowing Bax-Bax oligomerization and the release of cytochrome c [66].
Apoptosis onset induced by p53 at mitochondria level is associated with oxidative stress. In insulin-producing cells (RINm5F) cultured on glucose 30 mM, p53 translocation to mitochondria, cytochrome c release, and apoptosis were induced because of oxidative stress [1]. Taking this into account, it was proposed that glucose increase modifies intracellular p53 distribution and it promotes its mitochondrial localization besides inducing p53 phosphorylation, impairing its degradation, and increasing its biologic activity [67]. The presence of p53 in mitochondria is correlated with a decreased Bcl-2/Bax ratio, a decreased mitochondrial membrane potential [1], p53 activation, and increases of p21, Bax, and apoptosis [68]. This emphasizes p53 participation during the decrease of β-cell mass induced by hyperglycemia.
Hyperglycemia regulates p53 stability and function by inducing posttranslational modifications such as phosphorylation, poly(ADP-ribosylation), and N-acetylglucosamination [69].
p53 Phosphorylation
Hyperglycemia promotes p53 mitochondrial localization and its phosphorylation at serine 392 (homologous to Ser289 in mouse). This correlates with a Bcl-2 decrease, Bax increase, and β-cell apoptosis. The inhibition of the p38 MAPK hampered p53 phosphorylation, and it curtailed β-cell apoptosis induced by hyperglycemia, thereby suggesting its participation during the decrease of pancreatic β-cell mass. As in mitochondria p53 is engaged in complex formation with other antiapoptotic and/or proapoptotic proteins and as it triggers the mitochondrial permeation process, it is likely that its phosphorylation is a requirement that may enable its interaction with such proteins and to induce cell death. Additionally this process stimulates the interaction between p53 and the p300/CBP and P/CAF coactivators that promote its acetylation, thereby inhibiting its ubiquitination and degradation [70]. These results indicate the importance of p53 phosphorylation as one of the factors contributing to β-cell elimination as consequence of hyperglycemia through mitochondria (Fig. 12.3). Hyperglycemia also leads to ATM activation in cytosol, which in turn phosphorylates p53 at serine 15, thus avoiding its recognition by Mdm2, its ubiquitination, and nuclear degradation and also contributing to apoptosis triggering in response to hyperglycemia in β-cells [71].

Hyperglycemia promotes p53 translocation to mitochondria and its phosphorylation. There, p53 contributes to mitochondrial permeability, cytochrome c release, and β-cell apoptosis. P: phosphorylation, ROS: reactive oxygen species, Cyt c: cytochrome c
p53 O-N-Acetylglucosamination
Hyperglycemia promotes O-GlcNAcylation of several proteins, including p53. This consists on the addition of an N-acetylglucosamine moiety in serine or threonine residues. O-GlcNAcylation is analogue to phosphorylation, and it regulates the stability, activity, or subcellular localization of target proteins. This modification depends on glucose availability, and it represents a cellular regulation mechanism according to nutritional environment. In a glucose-rich environment, the O-N-acetylglucosamination of p53 has been observed and it is liked to its stability and it prevents its degradation. O-GlcNAc in p53 at Ser149 enhances its stability by interfering with phosphorylation at Thr155, by overcoming its interaction with Mdm2 and its ubiquitination and its subsequent proteolysis. All of this results in higher p53 stability [72]. Thus, it has been proposed that O-GlcNAc stabilizes p53 and may represent a signal for its translocation to mitochondria [71], where it may contribute to the release of proapoptotic factors [73].
p53 Poly(ADP-Ribosylation)
Another protein that becomes active when DNA suffers damage is poly (ADP-ribose) polymerase (PARP). This enzyme catalyzes the transfer of ADP-ribose units from NAD+, an essential cofactor for ATP synthesis and redox potential balance, to the carboxylic residues glutamic and aspartic acids of several nuclear proteins. Poly(ADP-ribosylation) is important for DNA replication and repair, transcription, inflammatory response, and cell death mainly caused by genotoxic agents, infection, and stress. PARP fragmentation is concomitant with β-cell apoptosis triggering induced by hyperglycemia. These results are in agreement with previous reports showing that hyperglycemia triggers apoptosis in β-cells and pancreatic cell lines such as RINm5F. Poly(ADP-ribosylation) of p53 in presence of high glucose levels is an early response that may contribute for protein stabilization and probably to its translocation to mitochondria as well as to the increased apoptotic rate of RINm5F cells caused by high glucose levels [73].
p53 Regulation by Mdm2
Cell survival depends in great extent on the balance between synthesis and degradation of p53. Among the mechanisms regulating p53, the expression and activation of Mdm2 (murine double minute 2) is of great importance. Mdm2 is an E3 ubiquitin ligase that binds p53 and it transfers ubiquitin to the latter residues, thereby enabling its recognition by proteasomes for targeted degradation. Depending on the amount of ubiquitin residues attached to p53, this protein will be degraded or exported to the cytosol. Mono- and/or poly-ubiquitination of p52 is defined by the concentration and activation status of Mdm2.
As previously mentioned, the interaction between p53 and Mdm2 depends on intracellular environment impacting on those posttranslational modifications displayed by these proteins. In the case of Mdm2, ubiquitination, sumoylation, and phosphorylation disrupt the formation of the p53-Mdm2 diene, and they stabilize p53 levels. It has been demonstrated that Mdm2 phosphorylation on Ser395 mediated by ATM decrease its ability to target p53 for degradation. Previously it was demonstrated that p53 phosphorylation by Akt participates in its regulation as it attenuated transactivation and increased p53 ubiquitination [74]. High glucose concentration decreases the expression of Mdm2 mRNA and its protein levels on both nucleus and cytosol [75]. Mdm2 expression is regulated by p53. Although we previously demonstrated its stabilization in presence of high glucose levels, this protein is not targeted toward nucleus, but it translocates to other organelles such as mitochondria; thus it cannot stimulate Mdm2 expression. Additionally, DNA fragmentation induced by hyperglycemia also may affect the expression of Mdm2 mRNA [1].
Formation of the p53-Mdm2 complex increased in presence of high glucose, although its ubiquitination was not observed. The latter demonstrates that glucose increased levels induce Mdm2 activation in cytosol and its interaction with p53 is also promoted, whereas its ubiquitination is inhibited [71]. It is known that the E3 ubiquitin ligase activity of Mdm2 depends on other domains comprising this protein. The latter activity is located within the RING finger domain at the C-terminal. This region also contains the substrate lysine acceptor and its main function is to label p53 for degradation. The central acid domain of Mdm2 binds to the RING finger domain and it stimulates catalytic activity, thereby promoting ubiquitin release from the E3 enzyme. The interaction between the acid domain and the RING finger domain depends on its phosphorylation by ATM [76]. An increase of ATM-mediated phosphorylation has been also observed in hyperglycemia. Thus, it is not excluded that stress and the phosphorylation cascade induced by high glucose levels may phosphorylate some residues on the RING finger domain and/or on nearby regions and on the acid central domain in Mdm2 leading to the inhibition of p53 poly-ubiquitination and degradation.
Conversely, p53 ubiquitination also depends on ATP levels. In hyperglycemia conditions, ATP decreases due to increased ROS and mitochondrial uncoupling. Therefore, if ATP decreased levels occur, ubiquitin ligases are unable to condensate the glycine residues on their C-terminal region with the lysine residues on p53. Thus, it is necessary to analyze these factors in hyperglycemia conditions in order to know about its participation for p53 ubiquitination in RINm5F cells.
Alterations in the Cell Cycle
Cell Proliferation Rate
In obese subjects, β-cell failure to compensate for insulin resistance has been related to either an inappropriate cell mass expansion or the inability of preexisting cells to respond toward glucose. This may be generated from defects of insulin signaling or the absence of insulin receptors and IGF-I in these cells [62]. For instance, knockout mice for these receptors exhibit a decreased cell mass and develop diabetes from an early age. It has been pointed out that progression through the cell cycle is also altered. It has been observed that cyclin inhibitor p27kip1 is progressively accumulated inside β-cell nucleus in mice lacking the insulin receptor substrate-2 (IRS-2) and also in db/db mice. p27kip1 accumulation in oxidative stress and hyperglycemia conditions may be another pathway by which ROS decrease β-cell mass as the deletion of its gene hampers hyperglycemia effects and induce β-cell proliferation [77]. Another important regulator of β-cell replication is the cell cycle inhibitor p21, which is expressed at high levels in adult β-cells, and it has been linked to proliferation decrease during senescence. It is known that β-cell replication potential is lost with age and it is correlated with the loss of expression of genes such as EZH2 (enhancer of zeste homologue 2), a histone methyl transferase that represses the transcription of cell cycle inhibitors when histone 3 is trimethylated in its lysine 27 residue (H3K27me3). EZH2 decreases H3K27me3 and increases p16 and p19 expression and it inhibits β-cell proliferation [78].
β-Cell Dedifferentiation
During recent years it has been observed that pancreatic β-cells undergo a dedifferentiation process when metabolic demand increases and also during hyperglycemia and inflammation. In mouse, hyperglycemia modifies the expression of transcription factors and insulin secretion. For instance, the loss of FOXO1 leads to β-cell dedifferentiation, decreases insulin content, and reverts its phenotype toward progenitor-like cells, characterized by the expression of Ngn3. Although changes in transcriptional factors have been observed on humans and primates affected by diet-induced prediabetes, a dedifferentiation process has not been demonstrated. β-cell dedifferentiation induced by hyperglycemia is reverted when blood glucose values are restored. Insulin immunostaining loss correlates with an increased glucagon staining in several diabetic mouse models. In one of these, it was observed that small β-cells begin to express glucagon, although it is not known if these cells will transform into α-cells or if they represent an intermediate cell type expressing glucagon [54, 79]. The Pax4 gene also participates in β-cell dedifferentiation. The latter is an embryonic development regulator of pancreatic islets, and its presence on adult β-cells from animals confers protection against stress-induced apoptosis, and it stimulates cell proliferation. However, the sustained Pax4 expression promotes β-cell dedifferentiation and hyperglycemia. Pax4 overexpression is concomitant with Ngn3 expression. This suggests that an acute Pax4 increase protects cells, but its steady or chronic expression induces β-cell dedifferentiation into progenitor cells apparently as a protective mechanism against deleterious environmental effects [80].
Conclusions
Obesity, insulin resistance, and glucose intolerance affect pancreatic β-cell functional status. Particularly, generation of new cells (neogenesis, replication) is decreased, whereas the apoptotic death rate increases. Among the mechanisms leading to β-cell alterations, it has been observed the participation of several proteins synthesized by the cell itself, such as amyloid polypeptide and the type 2 uncoupling protein, the molecules expressed and released by adipose tissue (free fatty acids and cytokines), the presence of high glucose levels, and the reactive oxygen species produced by glucolipotoxicity. It was recently proposed that high ROS appearing levels during hyperglycemia promote phosphorylation, poly(ADP-ribosylation), and/or O-GlcNAcylation, and they may interfere with p53 degradation by inhibiting the Mdm2 E2 ubiquitin ligase. Therefore, p53 degradation is avoided, and its recruitment to mitochondria and the apoptotic mechanisms are promoted along with β-cell dysfunction.
Loss of β-cell mass may reach a critical point in which the deleterious effects mediated by the aforementioned molecules might not be reverted, thus decreasing insulin production and release and contributing to diabetes development. Several treatments exist in order to attenuate β-cell deterioration. Changes of dietary routine and increased physical activity are among them. The objective is to promote weight loss and specially to decrease abdominal fat and insulin resistance. Thus, eventually β-cell mass may be regained.
Concluding Remarks
-
Activating hyperglycemia metabolic pathways induces β-cells apoptosis.
-
Free fatty acids are more toxic when hyperglycemia is present.
-
Oxidative and endoplasmic reticulum stress, mitochondrial alterations, inflammatory cytokines, islet amyloid polypeptide, together or separate, can decrease pancreatic β-cell mass.
-
Hyperglycemia induces posttranslational changes in p53 that inhibits its degradation and promotes mitochondrial location.
-
Many factors trigger the death of pancreatic β-cells and decrease β-cell mass. However, most appear to have their origin in hyperglycemia, so control of glucose levels is of great importance in preserving the mass and function of pancreatic β-cells.
Multiple-Choice Questions
-
1.
At what stage of embryonic life is β-cell mass set?
-
(a)
Before birth
-
(b)
At 2 years old
-
(c)
While breastfeeding
-
(d)
Between 5 and 10 years old
-
(e)
At 5 years old
-
(a)
-
2.
A decrease of β-cell mass was observed in persons without diabetes but with:
-
(a)
Chronic hyperglycemia
-
(b)
Metabolic syndrome
-
(c)
Impaired glucose tolerance
-
(d)
Obesity
-
(e)
Type 2 diabetes
-
(a)
-
3.
All are apoptotic cellular death characteristics, except:
-
(a)
DNA oligonucleosomal fragmentation
-
(b)
Phosphatidylserine exposition
-
(c)
Death cell phagocytosis
-
(d)
Intracellular content release
-
(e)
Formation of apoptotic bodies
-
(a)
-
4.
Increased production of reactive oxygen species in hyperglycemic conditions is due to
-
(a)
Microsomes
-
(b)
Oxidative phosphorylation
-
(c)
Macrophages
-
(d)
Endoplasmic reticulum
-
(e)
NADPH oxidase
-
(a)
-
5.
Endoplasmic reticulum stress is characterized by:
-
(a)
DNA oligonucleosomal fragmentation
-
(b)
Increased insulin demand
-
(c)
Unfolded protein response
-
(d)
Oxidative stress
-
(e)
Changes in mitochondrial permeability
-
(a)
-
6.
In hyperglycemic conditions, p53-induced apoptotic β-cell pathway:
-
(a)
Releases intracellular content
-
(b)
Changes mitochondrial permeability
-
(c)
Activates death receptors
-
(d)
Expresses proapoptotic proteins
-
(e)
Inhibits cell cycles
-
(a)
-
7.
Hyperglycemia induces the activation of metabolic pathways related to β-cell death as:
-
(a)
An increase in reactive oxygen species
-
(b)
Accumulation of amyloid polypeptide
-
(c)
Stress of the endoplasmic reticulum
-
(d)
A hexosamine pathway
-
(e)
All of the above
-
(a)
-
8.
What mitochondrial alterations contribute to the dysfunction of β-cells in diabetes?
-
(a)
Fission and fusion events
-
(b)
Loss of glucose sensitivity
-
(c)
Changes in ATP/ADP rate
-
(d)
Increased proinflammatory cytokines
-
(e)
Modification of NAD/NADH+ rate
-
(a)
-
9.
Chronic hyperglycemia affects the survival of β-cells by:
-
(a)
Decreasing beta-cell mass in diabetes by apoptosis
-
(b)
Causing β-cell hyperplasia and exhaustion
-
(c)
Causing cell dedifferentiation
-
(d)
Creating a loss of glucose sensitivity
-
(e)
All of the above
-
(a)
Correct Answers
-
1.
(d) Between 5 and 10 years old
At this stage of development, the rate of cell replication is reduced and the pancreas reaches its full size
-
2.
(c) Impaired glucose tolerance
Postmortem studies in humans showed a decrease in pancreatic cell mass of up to 50% in people with impaired fasting glucose
-
3.
(d) Intracellular content release
An important characteristic of apoptosis is the formation of apoptotic bodies, which consists of the invagination of the plasma membrane that surrounds the subcellular remains and prevents the release of intracellular material
-
4.
(b) Oxidative phosphorylation
Mitochondria are the main sources of endogenous ROS in hyperglycemic conditions. Of the oxygen consumed by mitochondria, ∼1–5% is converted to ROS as by-products of the flow of electrons in the respiratory chain
-
5.
(c) Unfolded protein response
Constant requirements of insulin during glucose intolerance and insulin resistance lead to alterations in the processing of proteins in the RE. This situation stimulates an ER response known as unfolded protein response and ER stress
-
6.
(b) Changes mitochondrial permeability
P53 in the mitochondria releases Bax from Bcl-2, allowing Bax oligomerization and pore formation to release proapoptotic factors as cyt c
-
7.
(e) All of the above
During hyperglycemia, the activation of all these metabolic pathways was observed, which concluded with the activation of the intrinsic pathway of apoptosis
-
8.
(a) Fission and fusion events
Hyperglycemia alters the expression of the proteins that regulate mitochondrial fusion/fission events, which modifies the ΔΨm and allows the output of proapoptotic factors
-
9.
(e) All of the above
Chronic hyperglycemia decreases pancreatic β-cell mass by activating apoptosis and inhibiting the cell cycle, in addition to inducing the dedifferentiation of β-cells and the sensitivity to glucose is lost
Abbreviations
- °OH:
-
Hydroxyl radical
- 8-OHdG:
-
8-hydroxy-2′-deoxyguanosine
- AGE:
-
Advanced glycation end products
- AIF:
-
Apoptosis-inducing factor
- Apaf-1:
-
Apoptotic protease-activating factor 1
- ATF6:
-
Activating transcription factor 6
- ATM:
-
ATM serine/threonine kinase protein
- Bak:
-
Bcl-2 homologous antagonist killer
- Bax:
-
Bcl-2-associated X protein
- Bcl-2:
-
B-cell lymphoma 2
- Bcl-xl:
-
B-cell lymphoma-extra large
- BH (1–4):
-
Bcl-2 homology 1–4 domains
- Bok:
-
Bcl-2 related ovarian killer
- Caspase:
-
Cysteine-aspartic proteases, cysteine aspartases
- CHOP:
-
C/EBP homologous protein
- ChREBP:
-
Carbohydrate response element binding protein
- Drp1:
-
Dynamin-related protein 1
- eif2α:
-
Eukaryotic translation initiation factor 2α
- ER:
-
Endoplasmic reticulum
- ERS:
-
Endoplasmic reticulum stress
- EZH2:
-
Enhancer of zeste homologue 2
- FADD:
-
Fas-associated death domain
- Fas:
-
Death receptor
- FFA:
-
Free fatty acids
- Fis1:
-
Mitochondrial fission 1 protein
- FOXA1/2:
-
Forkhead box A1/2
- G3P:
-
Glyceraldehyde 3-phosphate
- GADD34:
-
Downstream growth arrest and DNA damage-inducible protein
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- GATA4/6:
-
GATA-binding protein 4/6
- GLUT:
-
Glucose transporter
- GSIS:
-
Glucose-stimulated insulin secretion
- H3K27me3:
-
Histone H3 tri methyl K27
- HNF1β:
-
Hepatocyte nuclear factor 1β
- IAPP:
-
Islet amyloid polypeptide
- IFNγ:
-
Interferon gamma
- IGF1:
-
Insulin-like growth factor 1
- IL-1β:
-
Interleukin 1 beta
- iNOS:
-
Inducible nitric oxide synthases
- Ins :
-
Insulin gene
- INS1:
-
Insulin-secreting beta cell-derived line
- IRE1α:
-
Inositol-requiring enzyme 1α
- IRS-2:
-
Insulin receptor substrate-2
- Isl:
-
Islet
- MafA:
-
Musculoaponeurotic fibrosarcoma protein A
- Mdm2:
-
Murine double minute 2
- Mff:
-
Mitochondrial fission factor
- Mfn:
-
Mitofusin
- Mouse db/db:
-
Model of obesity, diabetes, and dyslipidemia with a mutation in leptin receptor
- mTOR:
-
Mammalian target of rapamycin
- NAD+:
-
Nicotinamide adenine dinucleotide
- NADH:
-
Nicotinamide adenine dinucleotide reduced
- NADPH oxi:
-
Nicotinamide adenine dinucleotide phosphate-oxidase
- NeuroD1:
-
Neurogenic differentiation 1
- NF-κB:
-
Nuclear factor kappa B
- Nkx:
-
Homeobox protein
- NLRP3:
-
NACHT, LRR, and PYD domains-containing protein 3
- NLRs:
-
Nucleotide oligomerization domain (NOD)-like receptors
- NO:
-
Nitric oxide
- NOD:
-
Nucleotide oligomerization domain
- Notch:
-
Transcription factor
- O.-2:
-
Superoxide anion
- O-GlcNAc:
-
O-linked β-N-acetylglucosamine
- Opa1:
-
Opa1 mitochondrial dynamin like GTPase
- P/CAF:
-
P300/CBP-associated factor
- p16:
-
Cyclin-dependent kinase inhibitor 2A, multiple tumor suppressor 1
- p21:
-
Cyclin-dependent kinase inhibitor 1 or CDK-interacting protein 1
- p27:
-
Cyclin-dependent kinase inhibitor 1B
- p300/CBP:
-
E1A binding protein p300/CREB-binding protein
- p38 MAPK:
-
P38 mitogen-activated protein kinases
- p53:
-
Tumor protein p53
- PARP:
-
Poly (ADP-ribose) polymerase
- Pax4:
-
Transcription factors paired box gene 4
- Pdx1:
-
Pancreatic and duodenal homeobox 1
- PERK:
-
Protein kinase-like ER kinase
- PI3k:
-
Phosphatidylinositol-3-kinase
- PKC:
-
Protein kinase C
- PP-1:
-
Protein phosphatase-1
- Ptf1α:
-
Pancreas transcription factor 1α
- RAMP1:
-
Receptor activity-modifying protein 1
- Rfx 6:
-
Regulatory factor x 6
- RING finger:
-
Really Interesting New Gene
- RINm5F:
-
Rat insulinoma cells
- ROS:
-
Reactive oxygen species
- Sox9 SRY:
-
Sex-determining region Y-box 9
- SPT:
-
Serine C-palmitoyltransferase
- T2D:
-
Type 2 diabetes
- TLRs:
-
Toll-like receptors
- TNFRI:
-
Tumor necrosis factor receptor type I
- TNFα:
-
Tumor necrosis factor alpha
- TXNIP:
-
Thioredoxin-interacting protein
- UCP2:
-
Uncoupling protein 2
- UDP-GlcNAc:
-
Uridine diphosphate N-acetylglucosamine
- UPR:
-
Unfolded protein response
- ΔΨm:
-
Mitochondrial membrane potential
References
Ortega-Camarillo C, Guzmán-Grenfell AM, García-Macedo R, Rosales-Torres AM, Ávalos-Rodríguez A, Duran-Reyes G, et al. Hyperglycemia induces apoptosis and p53 mobilization to mitochondria in RINm5F cells. Mol Cell Biochem. 2006;281:163–70.
Hinault C, Kawamori D, Liew CW, Maier B, Hu J, Keller SR, et al. D40 isoform of p53 controls β-cell proliferation and glucose homeostasis in mice. Diabetes. 2011;60:1210–22.
Jurczyk A, Bortell R, Alonso LC. Human β-cell regeneration: progress, hurdles, and controversy. Curr Opin Endocrinol Diabetes Obes. 2014;21(2):102–8.
Yagihashi S, Inaba W, Mizukami H. Dynamic pathology of islet endocrine cells in type 2 diabetes: β-cell growth, death, regeneration and their clinical implications. J Clin Invest. 2016;7:155–65.
Oliver-Krasinski JM, Stoffers DA. On the origin of the β cell. Genes Dev. 2008;22(15):1998–2021.
Gittes GK, Rutter WJ. Onset of cell-specific gene expression in the developing mouse pancreas. Proc Natl Acad Sci U S A. 1992;89:1128–32.
Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. β-Cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008;57(6):1584–94.
Mizukami H, Takahashi K, Inaba W, Osonoi S, Kamata K, Tsuboi K, et al. Age-associated changes of islet endocrine cells and the effects of body mass index in Japanese. J Diabetes Investig. 2014;5(1):38–47.
Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Méneur C, Bernal-Mizrachi E. Natural history of β-cell adaptation and failure in type 2 diabetes. Mol Asp Med. 2015;42:19–41. https://doi.org/10.1016/j.mam.2014.12.002.
Butler AE, Janson J, Bonner-Weir S. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10.
Khadra A, Schnell S. Development, growth and maintenance of β-cell mass: models are also part of the story. Mol Asp Med. 2015;42:78–90.
Ward W, Bolgiano D, McKnight B, Halter J, Porte DJ. Diminished β cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–28.
Meier JJ. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia. 2008;51:703–13.
Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 2003;144(12):5159–65.
Kjørholt C, Åkerfeldt MC, Biden TJ, Laybutt DR. Chronic hyperglycemia, independent of plasma lipid levels, is sufficient for the loss of β-cell differentiation and secretory function in the db/db mouse model of diabetes. Diabetes. 2005;54(9):2755–63.
Thornberry AN, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6.
Schwartzman RA, Cidlowski JA. Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocr Rev. 1993;14(2):133–51.
Höppener JW, Ahrén B, Lips CJ. Islet amyloid and type 2 diabetes mellitus. N Engl J Med. 2000;343(9):411–9.
Tomita T. Apoptosis in pancreatic β-islet cells in type 2 diabetes. Bosn J Basic Med Sci. 2016;16(3):162–79. https://doi.org/10.17305/bjbms.2016.919.
Kilpatrick ED, Robertson RP. Differentiation between glucose-induced desensitization of insulin secretion and β-cell exhaustion in the HIT-T15 cell line. Diabetes. 1998;47(4):606–11.
Robertoson RP. β-cell deterioration during diabetes: what’s in the gun? Trends Endocrinol Metab. 2009;20(8):388–93.
Kajimoto Y, Watada H, Matsuoka T-a, Kaneto H, Fujitani Y, Miyazaki J-i, et al. Suppression of transcription factor PDX-1/IPF1/STF-1/IDX-1 causes no decrease in insulin mRNA in MIN6 cells. J Clin Invest. 1997;100(7):1840–6.
Federici M, Hribal M, Perego L, Ranalli M, Caradonna Z, Perego C, et al. High glucose causes apoptosis in cultured human pancreatic islets of Langerhans. A potential role for regulation of specific Bcl family genes toward an apoptotic cell death program. Diabetes. 2001;50:1290–300.
Donath MY, Gross DJ, Cerasi E, Kaiser N. Hyperglycemia-induced -cell apoptosis in pancreatic islets of Psammomys obesus during development of diabetes. Diabetes. 1999;48:738–44.
Kim W-H, Lee JW, Suh YH, Hong SH, Choi JS, Lim JH, et al. Exposure to chronic high glucose induces β-cell apoptosis through decreased interaction of glucokinase with mitochondria. Diabetes. 2005;54:2602–11.
Piro S, Anello M, Pietro CD, Lizzio MN, Patanè G, Rabuazzo AM, et al. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism. 2002;51(10):1340–7.
Chan CB, Saleh MC, Koshkin V, Wheeler MB. Uncoupling protein 2 and islet function. Diabetes. 2004;53:S136–S42.
Joseph JW, Koshkin V, Saleh MC, Sivitz WI, Zhang C-Y, Lowell BB, et al. Free fatty acid-induced -cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004;279(49):51049–56.
Laybutt DR, Preston AM, Åkerfeldt MC, Kench JG, Busch AK, Biankin AV, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50(4):752–63.
Ogihara T, Mirmira RG. An islet in distress: β cell failure in type 2 diabetes. J Diabetes Investig. 2010;1(4):123–33.
Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem. 2003;278:30015–21.
Stein DT, Esser V, Stevenson BE, Lane KE, Whiteside JH, Daniels MB, et al. Essentiality of circulating fatty acids for glucose-stimulated insulin secretion in the fasted rat. J Clin Invest. 1996;97(12):2728–35.
Véret J, Coant N, Berdyshev EV, Skobeleva A, Therville N, Bailbé D, et al. Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 β-cells. Biochem J. 2011;438:177–89.
Galadari S, Rahman A, Pallichankandy S, Galadari A, Thayyullathil F. Role of ceramide in diabetes mellitus: evidence and mechanisms. Lipids Health Dis. 2013;12:98–114.
Unger RH, Zhou Y-T. Lipotoxicity of b-cells in obesity and in other causes of fatty acid spillover. Diabetes. 2001;50(1):S118–S21.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20.
Oliveira HR, Verlengia R, Carvalho CRO, Britto LRG, Curi R, Carpinelli AR. Pancreatic β-cells express phagocyte-like NAD(P)H oxidase. Diabetes. 2003;52(6):1457–63.
Yan L-J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. Journal of Diabetes Research. 2014;2014:137919.
Grankvist K, Marklund SL, Täljedal I-B. CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J. 1981;199:393–8.
Brownlee M. The pathology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–25.
Nishikawa T, Edelstein D, Du JX, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90.
Maechler P, Wollheim CB. Mitochondrial function in normal and diabetic β-cells. Nature. 2001;414:807–12.
Orrenius S, Zhivotovsky B. Cardiolipin oxidation sets cytochrome c free. Nat Chem Biol. 2005;1(4):188–9.
Ma ZA, Zhao Z, Turk J. Mitochondrial dysfunction and β-cell failure in type 2 diabetes mellitus. Exp Diabetes Res. 2012;2012(703538):1–11.
Kaufman BA, Li C, Soleimanpour SA. Mitochondrial regulation of β-cell function: maintaining the momentum for insulin release. Mol Asp Med. 2015;2015(42):91–104.
Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23(15):5409–20.
Maassen JA, Romijn JA, Heine RJ. Fatty acid-induced mitochondrial uncoupling in adipocytes as a key protective factor against insulin resistance and beta cell dysfunction: a new concept in the pathogenesis of obesity-associated type 2 diabetes mellitus. Diabetologia. 2007;50:2036–41.
Hasnain SZ, Prins JB, McGuckin MA. Oxidative and endoplasmic reticulum stress in b-cell dysfunction in diabetes. J Mol Endocrinol. 2016;56:R33–54.
Karunakaran U, Kim H-J, Kim J-Y, Lee I-K. Guards and culprits in the endoplasmic reticulum: glucolipotoxicity and β-cell failure in type II diabetes. Exp Diabetes Res. 2012;2012(Article ID 639762):9 pages https://doi.org/10.1155/2012/639762.
Sharma RB, Alonso LC. Lipotoxicity in the pancreatic beta cell: not just survival and function, but proliferation as well? Curr Diab Rep. 2014;14(6):492–508.
Cui W, Ma J, Wang X, Yang W, Zhang J, Ji Q. Free fatty acid induces endoplasmic reticulum stress and apoptosis of β-cells by Ca2+/Calpain-2 pathways. PLoS One. 2013;8(3):e59921.
Meigs JB, Wilson PWF, Fox CS, Vasan RS, Nathan DM, Sullivan LM, et al. Body mass index, metabolic syndrome, and risk of type 2 diabetes or cardiovascular disease. J Clin Endocrinol Metab. 2006;91(8):2906–12.
Meier JJ, Bonadonna RC. Role of reduced Β-cell function in the pathogenesis of type 2 diabetes. Diabetes Care. 2013;36(2):S13–S9.
Cantley J, Ashcroft FM. Q&A: insulin secretion and type 2 diabetes: why do β-cells fail? BMC Biol. 2015;13:33–40.
Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. 2013;36:111–7.
Hull RL, Kodama K, Utzschneider KM, Carr DB, Prigeon RL, Kahn SE. Dietary-fat induced obesity in mice results in beta cell hyperplasia but not increased insulin release: evidence for specificity of impaired beta cell adaptation. Diabetologia. 2005;48(7):1350–8.
Cheng Z, Almeida FA. Mitochondrial alteration in type 2 diabetes and obesity. Cell Cycle. 2014;13(6):890–7.
Itariu BK, Stulnig TM. Autoimmune aspects of type 2 diabetes mellitus – a mini-review. Gerontology. 2014;60:189–96.
Araki E, Oyadomari S, Mori M. Impact of endoplasmic reticulum stress pathway on pancreatic β-cells and diabetes mellitus. Exp Biol Med. 2003;228:1213–7.
Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta cell apoptosis. Diabetes. 2008;57(4):938–44.
Teff KL, Elliott SS, Tschöp M, Kieffer TJ, Rader D, Heiman M, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab. 2004;89(6):2963–72.
Lowell B, Shulman I. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–7.
Ritzel RA, Meier JJ, Lin C-Y, Veldhuis JD, Butler PC. Human islet amyloid polypeptide oligomers disrupt cell coupling, induce apoptosis, and impair insulin secretion in isolated human islets. Diabetes. 2007;56:65–71.
Arnelo U, Permert J, Larsson J, Reidelberger RD, Arnelo C, Adrian TE. Chronic low dose islet amyloid polypeptide infusion reduces food intake, but does not influence glucose metabolism, in unrestrained conscious rats: studies using a novel aortic catheterization technique. Endocrinology. 1997;138:4081–5.
Hull RL, Westermark GT, Westermark P, Kahn SE. Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab. 2004;89:629–3643.
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11(3):577–90.
Ortega-Camarillo C, Flores-López LA, Ávalos-Rodríguez A. The role of p53 in pancreatic β-cell apoptosis. Immunoendocrinology. 2015;2:E1075. https://doi.org/10.14800/Ie.1075.
Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–77.
Elkholi R, Chipuk JE. How do I kill thee? Let me count the ways: p53 regulates PARP-1 dependent necrosis. BioEssays. 2014;36(1):46–51.
Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006;13:941–50.
Flores-López LA, Díaz-Flores M, García-Macedo R, Ávalos-Rodríguez A, Vergara-Onofre M, Cruz M, et al. High glucose induces mitochondrial p53 phosphorylation by p38 MAPK in pancreatic RINm5F cells. Mol Biol Rep. 2013;40(8):4947–58.
Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol. 2006;8:1074–83.
Flores-López LA, Cruz-López M, García-Macedo R, Gómez-Olivares JL, Díaz-Flores M, Konigsberg-Fainstein M, et al. Phosphorylation, ON-acetylglucosaminylation and poly-ADP-ribosylation of p53 in RINm5F cells cultured in high glucose. Free Radical Biol Med. 2012;53:S95.
Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277(24):21843–50.
Barzalobre-Gerónimo R, Flores-López LA, Baiza-Gutman LA, Cruz M, García-Macedo R, Ávalos-Rodríguez A, et al. Hyperglycemia promotes p53-Mdm2 interaction but reduces p53 ubiquitination in RINm5f cells. Mol Cell Biochem. 2015;405:257–64. https://doi.org/10.1007/S11010-015-2416-0.
Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem. 2000;275:8945–51.
Zhong L, Georgia S, Tschen S-i, Nakayama K, Nakayama K, Bhushan A. Essential role of Skp2-mediated p27 degradation in growth and adaptive expansion of pancreatic β cells. J Clin Invest. 2007;117(10):2869–76.
Vetere A, Choudhary A, Burns SM, Warner BK. Targeting the pancreatic β-cell to treat diabetes. Nat Rev Drug Discov. 2014;13:278–89.
Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic β-cell dedifferentiation in diabetes and re-differentiation following insulin therapy. Cell Metab. 2014;19(5):872–82.
Lorenzo PI, Fuente-Martín E, Brun T, Cobo-Vuilleumier N, Jimenez-Moreno CM, Gomez IGH, et al. PAX4 defines an expandable β-cell subpopulation in the adult pancreatic islet. Sci Rep. 2015;5:15672.
Suggested/Further Reading
Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–25. The author presents a unified mechanism that links overproduction of superoxide by the mitochondrial electron-transport chain to high glucose-mediated damage and diabetes complications. This paper provides the basis for understanding of the origin of ROS and oxidative stress in diabetes.
Hasnain SZ, et al. Oxidative and endoplasmic reticulum stress in b-cell dysfunction in diabetes. J Mol Endocrinol. 2016;56:R33–54. https://doi.org/10.1530/JME-15-0232. Here, the importance of deleterious effects of oxidative stress and endoplasmic reticulum stress-induced unfolded protein response is evaluated on β-cell insulin synthesis and secretion as well as on inflammatory signaling and apoptosis. Additionally, the authors describe recent findings on how inflammatory cytokines contribute to β-cell dysfunction and protect interleukin 22.
Kaufman B, et al. Mitochondrial regulation of β-cell function: maintaining the momentum for insulin release. Mol Aspects Med. 2015;42:91–104. https://doi.org/10.1016/j.mam.2015.01.004. Pancreatic β-cell function and insulin release is mitochondria dependent. In this work, the authors review mitochondrial metabolism and control of mitochondrial mass as they relate to pancreatic β-cell function.
Ortega-Camarillo C, et al. The role of p53 in pancreatic β-cell apoptosis. Immunoendocrinology. 2015;2:e1075. https://doi.org/10.14800/ie.1075; © 2015. This paper examines p53 mobilization to a mitochondrion and its phosphorylation, as well as the activation of the intrinsic route of β-cell apoptosis by hyperglycemia. They also describe how hyperglycemia affects the p53 degradation pathways.
Sharma RB, Alonso LC. Lipotoxicity in the pancreatic beta cell: not just survival and function, but proliferation as well? Curr Diab Rep. 2014;14(6):492. https://doi.org/10.1007/s11892-014-0492-2. This paper reviews free fatty acids’ (FFAs) positive and negative effects on beta cell survival and insulin secretion. It also examines strong new findings that lipids may also impair compensatory beta cell proliferation.
Strycharz J, et al. Is p53 involved in tissue-specific insulin resistance formation? Oxid Med Cell Longev 2017; Article ID 9270549, 23 p. https://doi.org/10.1155/2017/9270549. The protein p53 is connected with metabolic defects underlying cellular aging, obesity, inflammation and β-cells apoptosis. Additionally, the authors discuss p53 regulation of multiple biochemical processes such as glycolysis, oxidative phosphorylation, lipolysis, lipogenesis, 𝛽-oxidation, gluconeogenesis, and glycogen synthesis.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Glossary
- Adipokines
-
Cytokines (cell signaling proteins) secreted by adipose tissue.
- Amylin
-
A 37-amino acid peptide hormone, discovered in 1987, which is co-located and co-secreted with insulin by the pancreatic beta cells in response to nutrient stimuli.
- Antioxidant
-
Molecule that inhibits the oxidation of other molecules.
- Apoptosis (a-po-toe-sis)
-
Was first used by Kerr, Wyllie, and Currie in 1972 to describe a morphologically distinct form of cell death and energy-dependent biochemical mechanisms.
- Apoptosome
-
Molecular complex of two major components – the adapter protein apoptotic protease-activating factor 1 (Apaf1) and the procaspase-9. These are assembled during apoptosis upon Apaf1 interaction with cytochrome c. Apoptosome assembly triggers effector caspase activation.
- Cardiolipin
-
Phospholipid important of the inner mitochondrial membrane, where it constitutes about 20% of the total lipid composition.
- Caspase (cysteine-aspartic proteases, cysteine aspartases or cysteine-dependent aspartate-directed proteases)
-
Family of protease enzymes playing essential roles in apoptosis and inflammation.
- Ceramides
-
Family of waxy lipid molecules. A ceramide is composed of sphingosine and a fatty acid.
- Cytochrome c
-
Heme protein serving as electron carrier in respiration. Cytochrome c is also an intermediate of apoptosis.
- Cytokines
-
Cell signaling small proteins. Involved in autocrine signaling, paracrine signaling, and endocrine signaling as immunomodulating agents.
- Dedifferentiation process
-
Processes by which cell that were specialized for a specific function lose their specialization.
- Fission
-
Division of mitochondria into new mitochondria.
- Flavoprotein
-
Proteins that contain a nucleic acid derivative of riboflavin: the flavin adenine dinucleotide (FAD) or flavin mononucleotide (FMN).
- Fusion
-
Process mediated by several large GTPases whose combined effects lead to the dynamic mitochondrial networks seen in many cell types.
- Glucolipotoxicity
-
Combined, deleterious effects of elevated glucose and fatty acid levels on pancreatic beta-cell function and survival.
- Hyperlipidemia
-
Elevation of fats or lipids in the blood.
- Hyperplasia
-
Enlargement of an organ or tissue caused by an increase in the cell proliferation rate.
- Inflammasome
-
A multiprotein cytoplasmic complex which activates one or more caspases, leading to the processing and secretion of proinflammatory cytokines – e.g., IL-1 beta, IL-18, and IL-33. Assembly of inflammasomes depends on the NOD-like receptor family members, such as the NALP protein kinase: enzyme catalyzing phosphorylation of an acceptor molecule by ATP.
- Misfolded proteins
-
Are proteins structurally abnormal and thereby disrupt the function of cells, tissues, and organs. Proteins that fail to fold into their normal configuration; in this misfolded state, the proteins can become noxious in some way and can lose their normal function.
- Mitofusins
-
Proteins that participate in mitochondrial fusion.
- Necrosis
-
Morphological changes in cell death caused by enzymatic degradation.
- Neogenesis
-
Generation of new cells.
- Oxidative stress
-
Pathological changes in living organisms in response to excessive levels of intracellular free radicals.
- Proenzyme
-
Precursor of an enzyme, requiring some change (hydrolysis of an inhibiting fragment that masks an active grouping) to render it active form.
- Proteasome
-
An intracellular complex enzymatic that degrades misfolded or damaged proteins (proteolysis), after damaged proteins are tagged by ubiquitin.
- Resistance to insulin
-
Pathological condition in which cells fail to respond normally to the hormone insulin.
- RING finger domain
-
Really Interesting New Gene finger is a proteins domain that plays a key role in the ubiquitination process.
- Stem cells
-
Undifferentiated biological cells that can differentiate into specialized cells and can divide.
- Sumoylation
-
Small Ubiquitin-like Modifier (or SUMO) proteins are a family of small proteins that are covalently attached to and detached from other proteins in cells to modify their function. Posttranslational modification involved in various cellular processes.
- Triacylglycerol
-
Ester of glycerol with three molecules of fatty acid.
- Ubiquitin
-
Small (8.5 kDa) regulatory protein that has been found in almost all tissues (ubiquitously) of eukaryotic organisms and regulated proteolysis.
- Ubiquitin ligase
-
Protein that recruits, recognizes a protein substrate, and catalyzes the transfer of ubiquitin from the E2 enzyme to the protein substrate.
- Uncoupling proteins
-
Proteins that uncouple phosphorylation of ADP from electron transport.
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ortega-Camarillo, C. (2019). Dysfunction and Death of Pancreatic Beta Cells in Type 2 Diabetes. In: Rodriguez-Saldana, J. (eds) The Diabetes Textbook. Springer, Cham. https://doi.org/10.1007/978-3-030-11815-0_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-11815-0_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11814-3
Online ISBN: 978-3-030-11815-0
eBook Packages: MedicineMedicine (R0)