
Abstract
Pancreatic β-cell death in type 2 diabetes has been related to p53 subcellular localisation and phosphorylation. However, the mechanisms by which p53 is phosphorylated and its activation in response to oxidative stress remain poorly understood. Therefore, the aim of this study was to investigate mitochondrial p53 phosphorylation, its subcellular localisation and its relationship with apoptotic induction in RINm5F cells cultured under high glucose conditions. Our results show that p53 phosphorylation in the mitochondrial fraction was greater at ser392 than at ser15. This increased phosphorylation correlated with an increase in reactive oxygen species, a decrease in the Bcl-2/Bax ratio, a release of cytochrome c and an increase in the rate of apoptosis. We also observed a decline in ERK 1/2 phosphorylation over time, which is an indicator of cell proliferation. To identify the kinase responsible for phosphorylating p53, p38 mitogen-activated protein kinase (MAPK) activation was analysed. We found that high glucose induced an increase in p38 MAPK phosphorylation in the mitochondria after 24–72 h. Moreover, the phosphorylation of p53 (ser392) by p38 MAPK in mitochondria was confirmed by colocalisation studies with confocal microscopy. The addition of a specific p38 MAPK inhibitor (SB203580) to the culture medium during high glucose treatment blocked p53 mobilisation to the mitochondria and phosphorylation; thus, the release of cytochrome c and the apoptosis rate in RINm5F cells decreased. These results suggest that mitochondrial p53 phosphorylation by p38 MAPK plays an important role in RINm5F cell death under high glucose conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dysfunctional and decreasing numbers of pancreatic β-cells are determining factors in the development of type 2 diabetes. A 60 % reduction in β-cell mass has been reported to occur prior to any clinical manifestations of the disease [1]. β-cell death is likely a consequence of intracellular changes caused by chronic hyperglycaemia, specifically the increase in mitochondrial oxidative stress [2, 3] and the decreased expression of reactive oxygen species (ROS)-scavenging enzymes [4]. Apoptotic cell death has also been reported in human pancreatic islets and insulin-producing cell lines cultured in the presence of high glucose (30 mM) and mitochondrial respiration inhibitors [5, 6].
Oxidative stress generated as a consequence of chronic hyperglycaemia activates multiple signalling pathways, which in turn initiate phosphorylation cascades involving kinases such as mitogen-activated protein kinase (MAPK) family [extracellular signal-regulated kinase (ERK), c-jun N-terminal kinase (JNK) and p38] [3, 7, 8]. These pathways regulate the ROS response and determine cell fate: proliferation, differentiation, stress adaptation or apoptosis [9]. Recent studies have demonstrated that the expression and phosphorylation of ERK, JNK and p38 MAPK exacerbate endothelial dysfunction in advanced stage of diabetes [10]. Additionally, the activation of MAPKs is critical for cardiovascular dysfunction in diabetes [3, 11] as well as in high glucose-induced cellular hypertrophy [12]. In particular, p38 MAPK activation is a fundamental mechanism for β-cell apoptosis induced by free fatty acids [13]. Other high glucose-activated kinases include DNA-dependent protein kinase and ataxia telangiectasia mutated (ATM) protein kinase [14, 15]. An important protein that is susceptible to phosphorylation by any of these kinases is p53, which plays an important role in β-cell apoptosis [16].
Protein p53 is a major regulator of cell proliferation and apoptosis. Under physiological conditions, p53 is maintained under strict control through binding to Mdm2 (murine double minute 2), a ubiquitin ligase that facilitates the proteasomal degradation of p53. Under stress conditions such as chronic hyperglycaemia, p53 is rapidly stabilised and activated through post-transcriptional modifications [17–20], which appear to influence its participation in β-cell apoptosis [13, 16]. In particular, phosphorylation of the amino and carboxyl terminal regions plays a key role in defining p53 function. Another important feature of p53 functional regulation is its subcellular localisation. The p53 protein has been shown to mobilise to the mitochondria in response to stress [21]. Previous results from our group revealed an increase ROS, p53 mobilisation to the mitochondria, altered membrane potential, cytochrome c release, and apoptosis when the pancreatic cell line RINm5F was cultured in 30 mM glucose [16].
Despite existing studies related to the mechanisms of p53 phosphorylation and its activation in response to oxidative stress, these phenomena remain poorly understood in hyperglycaemia-induced β-cell apoptosis. In this work, we propose that the mitochondrial localisation of p53 is a consequence of its activation and stabilisation via p38 MAPK. The aim of this study was to investigate mitochondrial p53 and p38 MAPK phosphorylation, the subcellular localisation of p53 and its association with the apoptotic index in RINm5F cells cultured under high concentrations of glucose.
Materials and methods
Reagents
RPMI-1640 medium, Hank’s balanced salt solution, trypsin, EDTA, glucose, HEPES, KCl, EGTA, DTT, PMSF, NaF, NaVO4, sucrose and mannitol were obtained from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Foetal bovine serum (FBS), TRIzol, gentamicin and l-glutamine were purchased from Invitrogen Corp. (Carlsbad, CA, USA). Dichlorofluorescein diacetate (DCFH-DA), anti-mouse IgG-Alexa 594, anti-rabbit IgG-Alexa 594 and MitoTracker Green were obtained from Molecular Probes (Eugene, OR, USA). Annexin V-FITC and propidium iodide were purchased from Roche Applied Science (México). Anti-p53 pAb 240, anti-phospho-ser15, anti-phospho-p53-ser392, anti-p38 MAPK, anti-phospho-p38 MAPK, anti-ERK 1/2, anti-phospho-ERK 1/2, anti-Bcl-2, anti-Bax, anti-Cox IV, anti-actin and anti-histone H1 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). An ECL kit was obtained from GE Healthcare (Piscataway, NJ, USA). Sequiblot-PVDF membranes, Precision Plus protein standards and DC protein assay kit were purchase from Bio-Rad Laboratories (Hercules, CA, USA). A First Strand cDNA Synthesis Kit was obtained from Fermentas (MD, USA). A Light Cycler Fast Start DNA Master PLUS SYBR Green I kit was purchased from Applied Biosystems (Foster City, CA, USA). All other analytical grade chemicals were obtained from Merck (Darmstadt, Germany).
Cell culture
Rat RINm5F (insulin-producing; A.T.C.C) cells were cultured in RPMI-1640 medium with 11 mM glucose (low glucose, LG) supplemented with 10 % FBS (v/v), 1 mM sodium pyruvate, 23.8 mM NaHCO3, 2 mM glutamine and 20 μg/ml gentamicin at 37 °C in a 5 % CO2 environment. The cells were harvested and subcultured for 24, 48 and 72 h in RPMI-1640 with 30 mM glucose (HG). Passage 8 was used until 75 % confluency was achieved [16].
RNA extraction, cDNA synthesis and real-time PCR (RT-PCR) analysis
Total RNA was purified from RINm5F cells (107 cells) using TRIzol following the manufacturer’s instructions. cDNA was synthesised from 5 μg of RNA using a first strand cDNA synthesis kit; the material obtained was used for RT-PCR. The following p53 specific primers were used: 5′-GGACGACAGGCAGACTTTTC-3′ (Tm = 59.85) and 3′-TTTTATGGCGGGACGTAGAC-5′ (Tm = 59.96). RT-PCR was performed in a Light Cycler 2.0 (Roche Molecular Biochemicals, Mannheim Germany) in conjunction with a Light Cycler Fast Start DNA Master PLUS SYBR Green I Kit. The data were normalised to GAPDH. After amplification, melting curves for each PCR product were obtained and used to determine their specificity.
Apoptosis analysis
Flow cytometry
Cells were harvested in 2 mM PBS-EDTA, washed in 0.1 % BSA-PBS (w/v) and centrifuged at 200×g for 5 min. Next, 100 μl of staining solution [20 μl Annexin V-FITC (10 μg/ml) and 20 μl propidium iodide (PI, 50 μg/ml) in 1 ml HEPES buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 5 mM CaCl2)] was added to the pellet followed by incubation in the dark for 15 min. The samples were analysed by flow cytometry (BD FACSAria; Becton–Dickinson System & Co., San Jose, CA, USA) at an excitation wavelength of 488 nm. The emitted light was split and collected at 530 nm for FITC and 585 nm for PI. For each run, 20,000 cells were required. The data were analysed using FACSDIVA™ software (Becton–Dickinson).
DNA fragmentation
After treatment, the cells were harvested with PBS-EDTA and washed twice with PBS. The cell pellet was lysed in 10 μl of buffer (20 mM EDTA, 50 mM Tris–HCl, pH 7.5, 1 % Igepal v/v) per 106, stirred for 10 s and immediately centrifuged for 5 min at 9,200×g and 4 °C. The supernatant was recovered, the pellet was resuspended with lysis buffer and centrifuged, and the supernatants were unified. Subsequently, 1 % SDS was added to the supernatant, followed by incubation for 2 h with RNase (final concentration: 5 μg/μl) at 56 °C, followed by digestion with proteinase K (final concentration: 2 μg/μl) for 2 h at 37 °C. DNA was precipitated with 0.5 volumes of 10 M ammonium acetate and 2.5 volumes of absolute ethanol at −20 °C overnight. The DNA was visualised on 1.3 % agarose gels stained with ethidium bromide [22].
Measurement of reactive oxygen species (ROS)
Following high glucose exposure, RINm5F cells were incubated in the presence of 10 μM 2′,7′- dichlorofluorescein diacetate (DCFH-DA) for 30 min at 37 °C. After incubation, the RINm5F cells were washed with HBSS, trypsinised and resuspended in 1 % FBS-PBS (pH 7.4). The DCF (dichlorofluorescein) fluorescence intensity was measured with a flow cytometer at excitation/emission wavelengths of 488/530 nm, analysing 20,000 cells in each run.
Subcellular fractionation
After treatment, the cells were harvested in PBS (2 mM EDTA, pH 7.4), centrifuged at 250×g for 5 min at 4 °C and washed with PBS. 500 μl of cold buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.1 mM PMSF, 50 mM NaF, 100 mM NaVO4) were added; after 5 min at 4 °C, 20 μl of 10 % Igepal v/v were added and samples were centrifuged at 16,500×g for 1 min. Both the pellet and the supernatant were recovered. The nuclear integrity and purity were verified by staining with 0.1 % toluidine blue. The nuclear fraction was obtained following a previously described method [23]. Mitochondrial and cytosolic fractions: 200 μl of 1 M mannitol and 50 μl of 1.25 M sucrose were added to the supernatants obtained in the previous centrifugation, and the samples were centrifuged at 10,000×g for 20 min at 4 °C. 40 μl of buffer A were added to the resulting pellet (mitochondrial fraction), and the supernatant (cytosolic fraction) was centrifuged at 15,000×g for 10 min at 4 °C; the resulting supernatant was the cytosolic fraction. Cytosolic fractions were analysed for glutamate dehydrogenase (GDH, mitochondrial marker) activity [24] as a purity control. The protein concentration was determined using DC protein assay kit, and the remaining lysates were stored at −70 °C until use.
Western blot analysis
Forty microgram samples of total protein from different subcellular fractions were subjected to 10 % SDS-PAGE and electroblotted onto PVDF membranes overnight (40 V, 4 °C). The samples were incubated for 2.5 h with the appropriate primary antibody (1:500 dilution; anti-p53 pAb 240, anti-phospho-p53-ser15, anti-phospho-p53-ser392, anti-p38 MAPK, anti-phospho-p38 MAPK-Tyr182, anti-ERK 1/2, anti-phospho-ERK 1/2, anti-Bcl-2, anti-Bax, anti-cytochrome c, anti-Cox IV (cytochrome oxidase), anti-actin or anti-histone H1). Next, the samples were incubated with HRP-conjugated secondary antibodies (1:2,000), and an enhanced chemiluminescence kit (ECL) was used to develop the luminescent signal. Immunoreactive bands were quantified by densitometric scanning, and the density of the individual bands was calculated using a Kodak 1500 Imaging System and Kodak molecular imaging software.
Immunoprecipitation
The appropriate antibodies (anti-p53 or anti-Bax in a 5 μl volume) were added to 200 μg of total protein and incubated for 1 h at 4 °C under orbital agitation. Thereafter, 20 μl of Protein G PLUS-Agarose (Santa Cruz Biotechnology) was added, followed by overnight incubation under orbital agitation. The immunoprecipitates were collected by centrifugation, boiled in Laemmli sample buffer, subjected to 10 % SDS-PAGE and electroblotted onto PVDF membranes. After blocking, the p38 MAPK antibody or anti-phosphoserine antibody (1:500) in 1 % BSA-TBS was added, followed by overnight incubation at 4 °C. Detection was performed as previously described.
Colocalisation of phospho-p53-ser392 and phospho-p38 MAPK
RINm5F cells were cultured in 8-well Lab-Tek Chamber Slides with covers (Nalgene Nunc International) in RPMI 1640-10 % FBS (v/v) with 11 or 30 mM glucose. After 72 h, the cells were incubated in culture medium with 200 nM MitoTracker Green for 30 min; the dye was then removed, and the cells were washed and fixed in 4 % formaldehyde in culture medium for 30 min at 37 °C. After rinsing with PBS-Tween 20®, the cells were permeabilised with 0.3 % Triton™ X-100 in PBS- for 15 min at room temperature. After blocking, anti-phospho-p53-ser392 or anti-phospho-p38 MAPK antibody (1:10 dilution) was added, and the samples were incubated at 4 °C overnight. The next day, anti-mouse IgG-Alexa 594 or anti-rabbit IgG-Alexa 594 (1:50 dilution) was added, followed by incubation for 4 h. The cells were then washed, the nuclei were stained with DRA-Q7 (Biostatus), and the samples were mounted with VECTASHIELD®. Microscopic analysis was performed with a confocal microscope (Carl Zeiss, Axiovert), and capture and intensity analysis was performed with the program Zen 2009 (Carl Zeiss, Goettingen, Germany). For imaging, 488- and 543-nm laser and short-pass filters (BP 505-530) were used for MitoFluor Green, a short-pass filter (BP 565-585) was used for phospho-p53-ser392 and phospho-p38 MAPK, and a long-pass filter (LP 650) was used for DRA-Q7. All images were captured at 40×.
Statistical analysis
The results are expressed as the mean ± SD of at least three independent experiments. The data were subjected to variance analysis followed by Tukey′s test for multiple comparisons. Differences with P values <0.05 were considered to be significant. The overlap coefficient in confocal micrographs was determined by the degree of colocalisation between channel 1 and channel 2 shown in quadrant 3. Each fluorescent contains 256 intensity levels (8 bits) and is represented by a two-dimensional graph or a dot plot of 256 × 256 levels of intensity with the criterion of 50 × 50 background intensity levels, as indicated by the program. Statistical analysis was performed using Pearson′s colocalisation coefficient in quadrant 3 with Zen software (Carl Zeiss).
Results
Reactive oxygen species levels, changes in the Bcl-2/BAX ratio and ERK 1/2 phosphorylation induced by high glucose treatment increase the apoptosis rate in RINm5F cells
Previously, we demonstrated that high glucose concentrations (30 mM) in RINm5F cells increase the apoptotic index after 48 h of culture [16]. Therefore, in the present study, we sought to determine the effect of high glucose on cell viability after a longer time period (72 h). Figure 1a illustrates the mitochondrial Bcl-2/Bax ratio, which was quantified as an indicator of the cellular tendency to undergo apoptotic cell death. Our results indicate that as time passes, the ratio tends to favour the pro-apoptotic protein as opposed to the anti-apoptotic protein. This result correlates with the observed ROS increase after a longer period of exposure to high glucose, as measured by flow cytometry using DCF (Fig. 1b). The percentage of apoptotic cells was also determined using annexin V-FITC, and the results indicate that although the previous three parameters suggested a propensity to undergo apoptosis at 24 h, actual cell death was only observed at 48 and 72 h (37 and 44 %, respectively, P < 0.01) (Fig. 1c). Finally, because a reduction in β-cell mass has been reported to occur as a very early event in diabetic pathology, ERK 1/2 activation was determined as an indicator of cell proliferation and β-cell mass. Figure 1d shows that high glucose decreased ERK 1/2 activation by approximately 20 % after 24 h, by 40 % at 48 and by 47 % at 72 h.

ROS production, apoptosis, and proliferation markers induced by high glucose. a Representative western blot and densitometric analysis of mitochondrial Bcl-2 and Bax and the ratio of Bcl-2/Bax. Protein expression levels were normalised to the Cox IV expression level; results are shown in the lower panel (*, ** and ***, P < 0.001). b Intracellular ROS production was measured by flow cytometry with DCF staining. The histogram is representative of three independent experiments. c Percentages of apoptotic cells were measured with Annexin V-FITC and propidium iodide (*P < 0.001 vs. low glucose). d Western and densitometric analyses of basal and phospho-activated levels of ERK 1/2 in the cytosol. Expression levels were normalised to actin; results are shown in the lower panel (* and **, P < 0.05). HG high glucose, 30 mM; LG low glucose, 11 mM for 72 h. The values are reported as the mean ± SD of five independent experiments
High glucose concentrations modify the subcellular distribution of p53
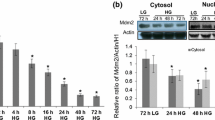
To ensure the enrichment of the mitochondrial fraction, GDH activity was measured in the mitochondrial and cytosolic fractions of cells with low and high glucose. GDH activity was lower in the cytosolic fraction (0.83 ± 0.25 and 0.96 ± 0.28 U/mg protein) for HG and LG, respectively) compared with the mitochondrial fraction (17.81 ± 1.95 and 18.3 ± 0.103 U/mg protein, respectively). Furthermore, the purity of the nuclei was verified by staining with toluidine blue before lysis of the nuclear fraction. High glucose did not alter p53 mRNA expression in RINm5F cells at any of the studied times (Fig. 2a); rather, it favoured a p53 subcellular redistribution. As shown in Fig. 2b, p53 is primarily located in the cytosol and nucleus in control cells; however, after 24 h of incubation in the presence of 30 mM glucose, a fraction of p53 (0.37 ± 0.03) was mobilised from the cytosol and nucleus into the mitochondria. A similar behaviour was observed after 48 h and 72 h, where the majority of p53 was found in the mitochondria (0.97 ± 0.097) (Fig. 2b).

p53 mRNA expression and intracellular distribution. a p53 mRNA levels was determined by RT-PCR, and GADH was used to normalise expression levels. b Representative western blot and densitometric analyses. p53 protein expression levels were normalised to actin (cytosol), H1 (nucleus) or Cox IV (mitochondria) expression levels; results are shown in the lower panel. Cytosol *, **, P < 0.001. Nucleus *, **, §, †, P < 0.05. Mitochondria *, **, §, †, P < 0.001. C cytosol; M mitochondria; N nucleus; T total; H1 histone 1; Cox IV cytochrome oxidase; HG high glucose, 30 mM; LG low glucose, 11 mM for 72 h. The values are reported as the mean ± SD of five independent experiments
High glucose induces p53 and p38 MAPK phosphorylation in mitochondria
In RINm5F cells cultured in low glucose (11 mM, LG) for up to 72 h, p53 was unphosphorylated (in both the cytosol and mitochondria). High glucose induced p53-ser392 phosphorylation in the mitochondria starting at 24 h, and this effect was maintained at 72 h. Under these conditions, p53-ser15 phosphorylation was also observed, but it was markedly lower (approximately fourfold) than ser392 phosphorylation. Because p38 MAPK is known to be activated in response to oxidative stress [25], the activation of this kinase was examined in the mitochondrial and cytosolic fractions. The results showed that the protein kinase p38 MAPK was found only in the cytosol in its inactive form in RINm5F cells cultured in LG, but in cells cultured in high glucose, p38 MAPK was found both in the cytosol and in the mitochondria. However, the phosphorylated form was exclusively found in the mitochondrial fraction (Fig. 3a).

p53 and p38 MAPK phosphorylation in the absence or presence of the MAPK inhibitor SB203580. a Western and densitometric analyses of basal and mitochondrial p53 phosphorylation at ser15 and ser392, p38 MAPK and P-p38 MAPK. b Immunoprecipitation with anti-p53 and immunoblotting with anti-p38 MAPK in RINm5F cells cultured in high glucose with SB203580. c Western and densitometric analyses of mitochondrial phospho-p53-ser392 with SB203580. d Western blot and densitometric analyses of basal p53 in RINm5F cells cultured in high glucose with SB203580. e Immunoprecipitation with anti-Bax and immunoblotting with anti-phosphoserines. Cox IV and actin were used as loading controls to normalise protein expression levels. C cytosol; M mitochondria; HG high glucose, 30 mM; LG low glucose 11 mM for 72 h. The values are reported as the mean ± SD of five independent experiments (*, **, P < 0.05)
p38 MAPK co-precipitates with p53 under high glucose conditions
To investigate the interaction between p38 MAPK and p53, RINm5F cells were cultured with high glucose and at the end of the treatment, immunoprecipitation was performed with an antibody against p53, and western blotting was performed using an antibody against p38 MAPK. The results showed the formation of a complex between p38 MAPK and p53 in cells treated with high glucose. When the pyridinyl imidazole SB203580 (10 μM), a potent inhibitor of p38 MAPK, was added to the culture, the interaction between p53 and p38 MAPK was abrogated (Fig. 3b).
SB203580 inhibits p53-ser392 phosphorylation and regulates the intracellular localisation of p53
To further investigate whether the apoptosis and p53-ser392 phosphorylation observed in RINm5F cells cultured in high glucose were due to p38 MAPK activation, we cultured cells in 30 mM glucose with the p38 MAPK inhibitor SB203580 and examined p53 phosphorylation. The inhibition of p38 MAPK completely abolished p53-ser392 phosphorylation in the mitochondria (Fig. 3c). These results indicate that p53-ser392 phosphorylation regulates the subcellular distribution of p53 as well as its degradation because the addition of SB203580 decreased p53 levels in a time-dependent manner after treatment (Fig. 3d). Thus, it is likely that preventing the phosphorylation of p53 promotes its recognition by molecules that lead to its degradation.
A p38 MAPK inhibitor (SB203580) inhibits Bax phosphorylation
Previous studies have shown that p38 MAPK regulates both Bax translocation to mitochondria and the release of cytochrome c [26, 27]. Likewise, Bax has been shown to be phosphorylated by p38 MAPK [28]. As expected in RINm5F cells, Bax phosphorylation increased in a time-dependent manner, and when the SB203580 inhibitor was added, Bax phosphorylation decreased significantly, as was observed in the immunoprecipitation experiments (Fig 3e).
Phosphorylated p53-ser392 and p38 MAPK colocalise with mitochondria in RINm5F cells cultured in high glucose
The phosphorylation of p53 and that of p38 MAPK were separately analysed by confocal microscopy after staining the mitochondria with MitoFluor Green. The results are shown in Fig. 4. Phosphorylated p53-ser392 in cells harvested after 72 h of treatment with high glucose colocalised with the mitochondrial fraction (Fig. 4a). The same behaviour was observed for phosphorylated p38 MAPK (Fig. 4b). The average coefficients of colocalisation (overlap coefficients) were 0.79 ± 0.042 and 0.82 ± 0.013 for phospho-p53-ser392 and phospho-p38 MAPK with HG, respectively. For the cells treated with LG, the coefficients of colocalisation were 0.48 ± 0.02 for phospho-p53-ser392 and 0.52 ± 0.03 for phospho-p38 MAPK, show statistically significant differences (P < 0.001) compared with cells exposed to high glucose (Fig. 4c).

The colocalisation coefficients of P-p53ser392 and P-p38 MAPK in RINm5F cells cultured in high glucose (30 mM, 72 h). Mitochondria were labelled with MitoFluor Green (green). a Phosphorylation of p53 ser392 (red, quadrant 1). b Phosphorylation of p38 MAPK (red, quadrant 2). Colocalisation with the mitochondria is shown in yellow (quadrant 3). The dot plot shows a representative image of the overlap coefficient, and the white lines delimit the quadrants based on a background value of 50 × 50 intensity levels. c The colocalisation of phosphorylated p53-ser392 and p38 MAPK with mitochondria was analysed by Pearson′s colocalisation coefficient and is shown in the lower histogram. The image is representative of 5–6 independent experiments, and the values are reported as the mean ± S.D. (*P < 0.0001 vs. 72 h LG)
SB203580 inhibits RINm5F cell apoptosis induced by high glucose
When RINm5F cells were cultured in high glucose with SB203580, a significant decrease in two important indicators of apoptosis, caspase 3 activation (Fig. 5a) and oligonucleosomal DNA fragmentation (Fig. 5b), was observed. These results agree with previous studies showing that high glucose concentrations alter mitochondrial permeability and induce the release of cytochrome c and the incidence of apoptosis in RINm5F cells [16]. In this study, we also observed that these two effects are diminished by the addition of SB203580 to RINm5F cells cultured in high glucose (Fig. 5c and 5d).

SB203580 reduces apoptosis of RINm5F cells induced by high glucose. a Analysis of oligonucleosomal DNA fragmentation. b Western and densitometric analyses of caspase 3. Western and densitometric analyses of cytochrome c release into the cytosol without (c) and with (d) the p38 MAPK inhibitor. The densitometric analyses of three independent western blots are shown in the lower panels, and the protein expression levels were normalised to actin or Cox IV expression levels (*, **, §, P < 0.001 without SB203580 and P < 0.05 with SB203580)
Discussion
Hyperglycaemia has been associated with oxidative stress production and cell death. Therefore, to analyse whether the glucose concentration and mitochondrial stress are determinant factors able to modify p53 expression, cellular distribution and phosphorylation, pancreatic RINm5F cells were cultured using two different glucose concentrations. The results showed that the high glucose treatment increased the ROS levels and the apoptosis rate in a time-dependent manner. p53 localisation to the mitochondria and its phosphorylation therein were also promoted, and this event was abrogated by the inhibition of p38 MAPK.
In our model, an increased rate of apoptosis under high glucose conditions was observed at 48 h and was still maintained at 72 h. This response is likely related to differences in the cell’s sensitivity to glucose. Nevertheless, our study was performed using a cell line that was not synchronised, therefore each cell in the population could have been in a different stage of the cell cycle, a condition that might be able to decide the cellular response to different stressors. It has been suggested that β-cell subpopulations prevent or reverse apoptotic pathway activation [29]. This outcome occurs in addition to the protective effect attributed to glucose for β-cell survival, in which the synthesis of anti-apoptotic proteins is stimulated in a time- and glucose concentration-dependent manner [30].
Along with the increase rate of apoptosis and in accordance with previous reports [31, 32], we observed a significant increase in the percentage of cells producing ROS, coinciding with a significantly decrease mitochondrial Bcl-2/Bax ratio. Our results are consistent with previous studies demonstrating that high glucose induces apoptosis in β-cells by decreasing Bcl-2 expression while increasing Bax expression in association with cytochrome c release and caspase 3 activation [32, 33]. Furthermore, high glucose levels diminished ERK 1/2 phosphorylation, which could affect β-cell survival. It has been demonstrate that ERK 1/2 phosphorylation modulates genes associated with cell proliferation, differentiation, migration and death [34]. In human ductal pancreatic cells, ERK 1/2 phosphorylation increases cell survival by promoting the activity of anti-apoptotic proteins [35], whereas ERK 1/2 in β-cells stimulates insulin synthesis and secretion in response to glucose and inhibits apoptosis [36].
We previously reported that the localisation of p53 in the mitochondria correlates with increased apoptosis in RINm5F cells cultured in high glucose for 48 h [16]. The present study shows that the p53 levels in mitochondria with HG were higher at 72 h. Interestingly, high glucose treatment of the RINm5F cells did not modify p53 mRNA expression, suggesting that p53 mobilisation and not de novo synthesis might be the trigger that modifies mitochondrial permeability and initiates apoptosis. These results are similar to observations in other cell types in which DNA damage, oncogene expression or ROS induce p53 mobilisation to the mitochondria as an event prior to or independent of p53-mRNA transcription [21]. Moreover, the level of p53 depends on a balance between its synthesis and degradation, and any increase in p53 levels may have different consequences for the cell. Because high glucose did not alter p53 transcription, the mobility and increased levels of p53 in the mitochondria might be explained by its stabilisation, which may depend on post-translational modifications such as phosphorylation, which not only prevents p53 degradation but also regulates its biological activity. N-terminal p53 phosphorylation in the cytosol has been linked to Bax transcription [37, 38], and ser389 phosphorylation at the C-terminus has been related to tetramer formation, DNA binding, apoptosis and tumour suppression [20, 39]. To examine the role of p53 phosphorylation in the apoptosis induction during high glucose treatment, we studied two key phosphorylation sites (ser15 and ser392) that regulate p53 activation and are involved in cell fate. Our results showed an increase in ser392 phosphorylation with respect to ser15, emphasising the relevance of ser392 in the glucose-mediated induction of apoptosis via the mitochondrial pathway. Thus, a contribution from this study is that ser392-p53 phosphorylation in the mitochondria promoted by high glucose is related to apoptosis induction. Although the purpose and physiological relevance of p53 phosphorylation at ser15 and ser392 in the mitochondria with high glucose remains unclear, this mechanism is likely a requirement that allows p53 interaction with pro- and/or anti-apoptotic proteins. The present study did not focus on the interaction of p53 with other proteins; however, we previously demonstrated that p53 mobilization to the mitochondria is associated with alterations in mitochondrial permeability that are concomitant with cytochrome c release in RINm5F cells grown in high glucose [16]. It might be interesting to further explore p53 interaction with several pro- and anti-apoptotic proteins and their relationship with cytochrome c release in this model. In RBK-2H3 cells (mastocytes) treated with eugenol (an antiallergen), p53 phosphorylation at ser15 in the mitochondria permits its interaction with Bcl-2 and Bcl-xL and induces changes in the mitochondrial membrane potential (Δψm) and cytochrome c release [40]. In contrast, there is evidence suggesting that ser15 phosphorylation in the mitochondria stimulates ATP production and Δψm, which act as pro-survival signals [41]. The phosphorylation of p53 at ser392 in mitochondria after high glucose treatment has not been reported to date in β-cells. However, prior studies reported that the phosphorylation of this serine residue by p38 MAPK was associated with Bax activation and apoptosis in myocytes cultured in high glucose [42, 43]. Although these studies were conducted with total cell extracts, the observed Bax activation was likely due to p53 mobilisation to the mitochondria, yet the possible interaction of p53 with Bcl-2, Bcl-XL or Bak, or its participation in mitochondrial permeability modifications cannot be excluded.
Because p53 has multiple phosphorylation sites and multiple kinases, including those activated by oxidative stress, such as p38 MAPK [3, 14], regulate this process, we analysed p38 MAPK activation and its participation in p53 phosphorylation in RINm5F cells cultured in high glucose. Our results show a significant increase in p38 MAPK phosphorylated in the high glucose-treated RINm5F mitochondrial fraction; which coincided with the immunoprecipitation of the p53-p38 MAPK complex, p53 phosphorylation and an increased apoptosis rate. These results agree with previous studies that have shown the involvement of p38 MAPK and p53 in β-cells (NIT-1) [13, 44] and neuroblastoma cells [45] apoptosis induced by oxidative stress. In this study, p38 MAPK phosphorylation was found exclusively in the mitochondria under conditions of high glucose, and this result was likely due to the increased ROS observed under these conditions, as previously demonstrated [45, 46].
To determine whether phosphorylated p53-ser392 and p38 MAPK co-localise in mitochondria, in situ immunofluorescence experiments were performed after high glucose treatment (72 h), with the confocal analysis showing a positive colocalisation of phosphorylated p53 and p38 MAPK within the mitochondria. Furthermore, experiments utilising the inhibitor SB203580 demonstrated that p38 MAPK plays an important role in the mitochondrial localisation of p53 and its phosphorylation induced by high glucose. p38 MAPK inactivation by SB203580 was verified by measuring Bax phosphorylation, an alternative target of p38 MAPK [27, 28], and our results showed that Bax phosphorylation was also inhibited.
Interestingly, SB203580 not only inhibited p53 and Bax phosphorylation but also decrease cytochrome c release, caspase 3 activation and DNA fragmentation. These results suggest that p53 phosphorylation at ser392 is related to mitochondrial membrane permeability and high glucose-induced apoptosis. Our results also suggests that p53 phosphorylation at ser392 may prevent p53 degradation, and act as a signal for its translocation to the mitochondria to initiate the apoptotic cascade. In other cells experiencing hyperglycaemia or oxidative or UV stress, p38 MAPK regulates apoptosis by phosphorylating p53 at ser392 [20, 43, 47] and ser15 [48]. Within this context, p38 MAPK also participates in Bax activation, Δψm collapse and apoptosis [27]. Other important functions of p38 MAPK include its significant contribution to insulin secretion and β-cells survival [49, 50]. Furthermore, it has been demonstrated that the p38 MAPK/p53 signalling pathway is activated in H2O2-induced tumour cell apoptosis [51]. Lastly, based on our results, we propose that β-cell apoptosis induced by high glucose requires p38 MAPK activation, which leads to the mobilisation of p53 to the mitochondria and its phosphorylation. However, to confirm a direct causality, experiments with a molecular knockout or small interfering RNA against p53 or p38 MAPK in addition to an animal model of experimental diabetes are recommended.
In conclusion, we propose that high glucose induces apoptosis in RINm5F cells via an oxidative stress response involving p53 mobilisation to the mitochondria and its phosphorylation at ser392. These events might be associated with p38 MAPK activation. p53 phosphorylation might also be an important modification that allows its interaction with anti- and/or pro-apoptotic proteins and might also be involved in the regulation of mitochondrial membrane permeability. In addition, we cannot rule out a certain degree of p53 transcriptional activation.
References
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Brownlee M (2005) The pathology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625
Evans JL, Goldfine DI, Maddux AB, Grodsky MG (2002) Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23:599–622
Grankvist K, Marklund SL, Täljedal I-B (1981) CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and glutathione peroxidase in pancreatic islets and other tissues in the mouse. Biochem J 199:393–398
Sakai K, Nakamaru K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M, Araki E (2003) Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochem Biophys Res Commun 300:216–222
Robertson RP, Harmon JS (2007) Pancreatic islet β-cell and oxidative stress: the importance of glutathione peroxidase. FEBS Lett 581:3743–3748
Hou N, Torii S, Saito N, Hosaka M, Takeuchi T (2008) Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology 149(4):1654–1665
Kim WH, Lee JW, Gao B, Jung MH (2005) Synergistic activation of JNK/SAPK induced by TNF-α and IFN-γ: apoptosis of pancreatic β-cells via the p53 and ROS pathway. Cell Signal 17:1516–1532
Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaronson SA (2003) Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol 23(23):8576–8585
Huang A, Yang YM, Yan C, Kaley G, Hintze TH, Sun D (2012) Altered MAPK signaling in progressive deterioration of endothelial function in diabetic mice. Diabetes 61(12):3181–3188
Liu Z, Cao W (2009) p38 mitogen-activated protein kinase: a critical node linking insulin resistance and cardiovascular diseases type 2 diabetes mellitus. Endocr Metab Immune Disord Targets 9(1):38–46
Fujita H, Omori S, Ishikura K, Hida M, Awazu M (2004) ERK and p38 mediate high glucose-induced hypertrophy and TGF-beta expression in renal tubular cells. Am J Physiol Renal Physiol 286(1):F120–F126
Yuan H, Zhang X, Huang X, Lu Y, Tang W, Man Y, Wang S, Xi J, Li J (2010) NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of β-cells via JNK, p38 MAPK and p53. PLoS One 5(12):e15726–e15726
Chen K, Albano A, Ho A, Keaney JF (2003) Activation of p53 by oxidative stress involves platelet-derived growth factor-b receptor-mediated ataxia telangiectasia mutated (ATM) kinase activation. J Biol Chem 278:39527–39533
Bonini P, Cicconi S, Cardinales A, Vitale C, Serafino AL, Ciotti MT, Marlier NJ-L (2004) Oxidative stress induced p53-mediated apoptosis in glia: p53 transcription-independent way to die. J Neurosci Res 75:83–95
Ortega-Camarillo C, Guzmán-Grenfell AM, García-Macedo R, Rosales-Torres AM, Ávalos-Rodríguez A, Duran-Reyes G, Medina-Navarro R, Cruz M, Díaz-Flores M, Kumate J (2006) Hyperglycemia induces apoptosis and p53 mobilization to mitochondria in RINm5F cells. Mol Cell Biochem 281:163–170
Yap DBS, Hsieh J-K, Zhong S, Heath V, Gusterson B, Crook T, Lu X (2004) Ser392 phosphorylation regulates the oncogenic function of mutant p53. Cancer Res 64:4749–4754
Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, Vassilev LT (2004) Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem 279:53015–53022
Shieh S-Y, Ahn J, Tamai K, Taya Y, Prives C (2000) The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 14:289–300
Huang C, Ma W-Y, Maxiner A, Sun Y, Dong Z (1999) p38 kinase mediates UV-induced phosphorylation of p53 protein at serine 389. J Biol Chem 274:12229–12235
Marchenko DN, Zaika A, Moll MU (2000) Death signal-induced localization of p53 protein to mitochondria. J Biol Chem 275:16202–16212
Herrmann M, Lorenz HM, Voll R, Grünke M, Woith W, Kalden JR (1994) A rapid and simple method for the isolation of apoptotic DNA fragments. Nucleic Acid Res 22(24):5506–5507
Schreiber E, Matthias P, Müller MM, Schaffner W (1989) Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acid Res 17:6419
Bergmeyer HU (1963) Methods of enzymatic analysis. Academic Press, New York
Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC (2002) Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem 277(33):30010–30018
Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE (2003) p38 mitogen-activated protein kinase regulates Bax translocation in cyanide-induced apoptosis. Toxicol Sci 75:99–107
Gomez-Lazaro M, Galindo MF, RMM-Fd Mera, Fernandez-Gómez FJ, Concannon CG, Segura MF, Comella JX, Prehn JHM, Jordan J (2007) Reactive oxygen species and p38 mitogen-activated protein kinase activate Bax to induce mitochondrial cytochrome c release and apoptosis in response to malonate. Mol Pharmacol 71(3):736–743
Z-q Yuan, Feldman RI, Sussman GE, Coppola D, Nicosia SV, Cheng JQ (2003) AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1. J Biol Chem 278(26):23432–23440
Pipeleers DG (1992) Heterogeneity in pancreatic beta-cell population. Diabetes 41:777–781
Martens GA, Pipeleers DG (2009) Glucose, regulator of survival and phenotype of pancreatic beta cells. Vitam Horm 80:507–539
Nishikawa T, Edelstein D, Du JX, Yamagishi S, Matsumura T, Kaneda Y, Yorek AM, Beebe D, Oates JP, Hemmes PH, Giardino I, Brownlee M (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Adams JM, Cory S (1998) The bcl-2 protein family: arbiters of cell survival. Science 281:1322–1325
Huang Q, Bu S, Yu Y, Guo Z, Ghatnekar G, Bu M, Yang L, Lu B, Feng Z, Liu S, Wang F (2007) Diazoxide prevents diabetes through inhibiting pancreatic β-cells from apoptosis via Bcl-2/Bax rate and p38-b mitogen-activated protein kinase. Endocrinology 148(1):81–91
Murphy LO, Blenis J (2006) MAPK signal specificity: the right place at the right time. Trends Biochem Sci 31:268–275
Balmanno K, Cook SJ (2009) Tumor cell survival signalling by the ERK 1/2 pathway. Cell Death Differ 16:368–377
Lawrence M, Shao C, Duan L, McGlynn K, Cobb MH (2008) The protein kinases ERK 1/2 and their roles in pancreatic beta cells. Acta Physiol (Oxf) 192:11–17
Ito K, Nakazato T, Yamato K, Miyakawa Y, Yamada T, Hozumi N, Segawa K, Ikeda Y, Kizaki M (2004) Induction of apoptosis in leukemic cells by homovanillic acid derivative, capsaicin, through oxidative stress. Cancer Res 64:1071–1078
Lavin MF, Gueven N (2006) The complexity of p53 stabilization and activation. Cell Death Differ 13:941–950
Hoogervorst EM, Bruins W, Zwart E, CTMv Oostrom, GJvd Aardweg, Beems RB, Jvd Berg, Jacks T, Steeg Hv, Ad Vries (2005) Lack of p53 Ser389 phosphorylation predisposes mice to develop 2-acetylaminofluorene-induced bladder tumors but not ionizing radiation-induced lymphomas. Cancer Res 65:3610–3616
Park BS, Song YS, Yee SB, Lee BG, Seo SY, Park YC, Kim JM, Kim HM, Yoo YH (2005) Phospho-ser 15–p53 translocates into mitochondria and interacts with Bcl-2 and Bcl-xL in eugenol-induced apoptosis. Apoptosis 10(1):193–200
Nantajit D, Fan M, Duru N, Wen Y, Reed JC, Li JJ (2010) Cyclin B1/Cdk1 phosphorylation of mitochondrial p53 induces anti-apoptotic response. PLoS One 5(8):e12341–e12354
Malhotra A, Vashistha H, Yadav VS, Dube MG, Kalra SP, Abdellatif M, Meggs LG (2009) Inhibition of p66ShA redox activity in cardiac muscle cells attenuates hyperglycemia-induced oxidative stress and apoptosis. Am J Physiol Heart Circ Physiol 296(2):H380–H388
Fiordaliso F, Leri A, Cesselli D, Limana F, Safai B, Nadal-Ginard B, Anversa P, Kajstura J (2001) Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes 50:2363–2375
Ravindran J, Gupta N, Agrawal M, Bala Bhaskar AS, Lakshmana Rao PV (2011) Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 16(2):145–161
Cao M, Jiang J, Du Y, Yan P (2012) Mitochondria-targeted antioxidant attenuates high glucose-induced p38 MAPK pathway activation in human neuroblastoma cells. Mol Med Rep 5(4):929–934
Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT (2002) Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol 282:L1324–L1329
Thornton TM, Rincon M (2009) Non-classical p38 MAP kinase functions: cell cycle checkpoints and survival. Int J Biol Sci 5(1):44–52
She QB, Chen N, Dong Z (2000) ERKs and p38 kinase phosphorylate p53 protein at Serine 15 in response to UV radiation. J Biol Chem 275(27):20444–20449
Sumara G, Formentini I, Collins S, Sumara I, Windak R, Bodenmiller B, Ramracheya R, Caille D, Jiang H, Platt KA, Meda P, Aebersold R, Rorsman P, Ricci R (2009) Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis. Cell 136:235–248
Lu X, Liu J, Hou F, Liu Z, Cao X, Seo H, Gao B (2011) Cholesterol induces pancreatic β cell apoptosis through oxidative stress pathway. Cell Stress Chaperones 16:539–548
Filomeni G, Piccirillo S, Rotilio G, Ciriolo MR (2012) p38(MAPK) and ERK1/2 dictate cell death/survival response to different pro-oxidant stimuli via p53 and Nrf2 in neuroblastome cells SH-SY5Y. Biochem Pharmacol 83(10):1349–1357
Acknowledgments
This work was partially funded by the Consejo Nacional de Ciencia y Tecnología, México (Grant 54878/FIS/IMSS/PROT/144), and the Fondo de Investigación en salud, IMSS (Grant FIS/IMSS/PROT/145). We thank the Fundación IMSS A.C. and the Fundación Río Arronte I.A.P. for their support in purchasing laboratory equipment. We would also like to thank the Universidad Autónoma Metropolitana—Izt (postgraduate section), the Consejo Nacional de Ciencia y Tecnología and the Coordinación de Investigación en Salud, IMSS (LA Flores-López, scholarship).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Flores-López, L.A., Díaz-Flores, M., García-Macedo, R. et al. High glucose induces mitochondrial p53 phosphorylation by p38 MAPK in pancreatic RINm5F cells. Mol Biol Rep 40, 4947–4958 (2013). https://doi.org/10.1007/s11033-013-2595-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-013-2595-2