
Abstract
Members of the genus Rhodococcus have metabolic versatility and unique adaptation capacities to fluctuating environmental conditions, enabling the colonization of a wide variety of environments; they also play an important role in nutrient cycling and have potential applications in bioremediation, biotransformations and biocatalysis. Rhodococcus spp. are mainly distributed in soil, water and marine sediments, although some of them are also pathogens for humans, animals and plants. Consistent with the wide catabolic diversity, Rhodococcus spp. possess large and complex genomes (up to 10.1 Mbp), which contain a multiplicity of catabolic genes, high genetic redundancy of biosynthetic pathways and large catabolic plasmids, the latter encoding peculiar metabolic and physiological traits. Recently, the progress in sequencing technology led to a dramatic increase in the number of sequenced Rhodococcus genomes, which have been investigated through diverse bioinformatic approaches. In particular, whole-genome comparative and genome-based functional studies were associated to omic technologies for the study of the global Rhodococcus cell response with the aim of providing insight into the genetic basis of specific catabolic capacities and phenotypic traits. Lastly, genome-based advances in Rhodococcus engineering led to the first design of molecular toolkits for tunable and targeted genome editing. Besides this, genome-based metabolic models were developed to make metabolic predictions of the Rhodococcus cell response to specific growth conditions. Both the synthetic and system approaches offered new opportunities for genome-scale rational design of Rhodococcus cell for environmental and industrial applications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The Rhodococcus genus comprises of Gram-positive, nonmotile, nonsporulating, aerobic bacteria, with a high G+C content and a mycolic acid-containing cell wall. Members of Rhodoccocus genus are genetically and physiologically diverse bacteria, widely distributed in soil, water and marine sediments; some Rhodococcus spp. are also pathogens for plants (R. fascians), animals and humans (R. equi). Members of Rhodococcus have also been recovered from extreme environments such as the deep sea, oil-contaminated soils and freeze-thaw tundra on glacial margins (Sheng et al. 2011; Shevtsov et al. 2013; Konishi et al. 2014).
Rhodococcus genus is featured by a broad metabolic versatility and environmental persistence supporting its clinical, industrial and environmental significance. In particular, Rhodococcus strains have peculiar degradative capacities towards a variety of organic compounds, including toxic and recalcitrant molecules like chlorinated hydrocarbons (Grӧning et al. 2014; Cappelletti et al. 2017), herbicides (Fang et al. 2016), 4,4′-dithiodibutyric acid (DTDB) (Khairy et al. 2016) and dibenzothiophene (DBT) (Tao et al. 2011) as well as the ability to resist to various stress conditions (desiccation, radiation, heavy metals) (LeBlanc et al. 2008; Taketani et al. 2013; Cappelletti et al. 2016). They are also able to mediate a broad range of biotransformation, including enantioselective syntheses, and to produce biosurfactants, which facilitate the cell contact with hydrophobic substrates (Martìnkovà et al. 2009). Due to their metabolic flexibility and their tolerance to various stresses, they play an important role in nutrient cycling and have potential applications in bioremediation, biotransformations and biocatalysis.
Because of their biotechnological applications, the massive application of high-throughput genomic technologies has dramatically increased the number of sequenced Rhodococcus genomes, and a great effort has been directed towards the computational analysis of genomic data. Since the first complete genome (of R. jostii RHA1) published in 2006, an exponentially increasing number of genomes of Rhodococcus strains have been sequenced due to the progress of the sequencing technologies and to the reduction of the DNA sequencing costs. In July 2018, 218 complete genomes of Rhodococcus are available in NCBI (www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=1827), most of them being available as draft genomes. Collectively, the analysis of genome sequences has provided insights into the genetic basis for diversity, plasticity and adaptation characteristic of Rhodococcus spp.
2 The Rhodococcus Genome: General Features and Structure
Rhodococcus genomes show high GC amount (61–71%) and range in size from around 4 Mb to over 10 Mb. In particular, the genome size seems to be at least partially dependent on the Rhodococcus strain lifestyle and niche complexity; indeed, the pathogenic R. equi has a substantially smaller genome than the soil-associated versatile biodegrader R. jostii RHA1 (9.7 Mb) and the other two environmental rhodococci, Rhodococcus erythropolis PR4 (6.9 Mb) and Rhodococcus opacus B4 (8.2 Mb) (Letek et al. 2010). Indeed, obligate pathogens live inside a host, reducing the need to adapt to sudden environmental changes and presence of competitors, which may contribute to genome minimization (Moran 2002).
Among Rhodococcus strains of the same species, the amount of GC in the genome was quite constant, showing the highest values (>70% GC-content) in R. aetherivorans and R. ruber species (Table 1). On the other hand, a remarkable variability was observed in the rhodococcal genome structure and plasmid presence.
Chromosomal and plasmid DNA of Rhodococcus strains is either linear or circular. Both R. jostii RHA1 and R. opacus B4 have linear chromosomes, while R. erythropolis PR4 and R. equi chromosomes are circular (Table 1). Interestingly, chromosome topology does not seem to correlate with taxonomy or phylogenetic relationship, as R. equi and R. erythropolis belong to different subclades, and the latter is considered the prototype of the “erythropolis subclade”, which also includes R. opacus (Letek et al. 2010). Further, not all actinomycetales genera present linear chromosomes. Streptomycetes typically have linear chromosomes of very large size (>8.5 Mb), so linearization appears to have occurred independently in different actinobacterial lineages during evolution, apparently in association with increasing genome size (Bentley et al. 2002).
The co-existence of large linear and circular plasmids of different sizes within the same cell is also a characteristic of several actinomycetes genera; however, linear plasmids are mainly described in Streptomyces and Rhodococcus strains. The linearity of DNA molecules in Rhodococcus strains has been mainly associated to the presence telomere-like structures comprising of terminal inverted repeats (TIRs) and proteins bound to their 5′ ends (Kikuchi et al. 1985; Dib et al. 2015). This structure, named as invertron, has been detected in both chromosomes and plasmids of Rhodococcus and has been extensively studied in Streptomyces strains (Ventura et al. 2007). Most commonly, both ends of linear plasmids are the same or featured by only few mismatches, giving recognizable terminal inverted repeats (TIRs). For instance, plasmid pHG207 of R. opacus MR22 carries an imperfect terminal inverted repeat (TIR) of 583/560 bp (Kalkus et al. 1993). The RHA1 replicons have been described as typical actinomycete invertrons, containing two sets of inverted repeats flanking the GCTXCGC central motif with covalently associated proteins (Warren et al. 2004). The chromosomal inverted repeats (of around 10 Kp in RHA1) are much longer than those of the plasmids (e.g. the pRHL1 telomeres are 500 bp). The RHA1 telomeric inverted repeats are similar to those of Streptomycetes, particularly over the first 300 bp. The replication of linear Streptomyces chromosomes and plasmids is initiated from a fairly centrally located replication origin rich in DnaA box sequences and proceeds bidirectionally towards the telomeres. The telomeres themselves are replicated by a mechanism that includes priming from the terminal proteins covalently bound to the 5′ ends. It has been proposed that linear plasmids evolved from bacteriophages and that linear chromosomes originated from single-crossover recombination between an initially circular chromosome and a linear plasmid (McLeod et al. 2006; Ventura et al. 2007).
2.1 Genomic and Metabolic Traits of Reference Rhodococcus Strains
The analysis of single bacterial genomes and its annotation confer a wide range of knowledge in relation to the general genome features and chromosomal structure but also in relation to identification of specific genes in a genome and the detection of regions containing functionally connected genes. Although most of them are still in a draft version, the sequencing of individual Rhodococcus genomes has often provided important inputs for wet-lab experiments aimed at investigating the relevant metabolic capacities shown by the isolates.
Table 1 reports the genome features of representative Rhodococcus strains belonging to different species featured by specific metabolic and physiological traits.
Members of Rhodococcus jostii, R. opacus and R. wratislaviensis species possess genomes of very large size, ranging from 8.5 to over 10 Mb (Table 1). R. jostii RHA1 is considered the model of the genus as its genome was the first completely sequenced from a Rhodococcus strain. R. jostii RHA1 was isolated from lindane-contaminated soil (McLeod et al. 2006) and was widely investigated for the strong ability to degrade a wide variety of aromatic compounds and polychlorinated biphenyls (PCBs), for lipid accumulation and for stress condition resistance (Gonçalves et al. 2006; LeBlanc et al. 2008; Patrauchan et al. 2012; Costa et al. 2015).
R. opacus R7 and R. wratislaviensis NBRC 100605 have the largest Rhodococcus genomes described up to date (10.1 and 10.4 Mbp, respectively) and two of the largest bacterial genome reported in the literature. While NBRC 100605 metabolic features have been scarcely investigated (Guo and Wu 2017), R. opacus R7 has been thoroughly studied considering both the broad degradative abilities towards aromatic and aliphatic hydrocarbons and the associated genome features (Di Gennaro et al. 2001, 2010; Zampolli et al. 2014; Orro et al. 2015; Di Canito et al. 2018). Based on the terminal sequence signatures defining the possible linearity of replicons, R7 possesses one linear chromosome and additional five linear replicons, ranging in size from 20 Kb to 430 Kb (Tables 1 and 2) (Di Gennaro et al. 2014). Other Rhodococcus opacus strains described from a genomic point of view have broad organic compound degradative abilities and capacity to produce and accumulate lipids for possible biodiesel application. In particular, R. opacus PD630 is the most studied bacterium for its ability to produce and accumulate lipids (mostly triacylglycerols, TAGs) using different carbon sources (Alvarez et al. 1996; Castro et al. 2016). Indeed, it was shown to accumulate significant amounts of lipids, namely, 76 and 87%, of its cellular dry weight, when grown on gluconate and olive oil, respectively (Alvarez et al. 1996; Voss and Steinbuchel 2001). R. opacus B4 was the first R. opacus with a completely sequenced genome; it was described to be able to transform and degrade several types of hydrocarbons, to stabilize water-oil phases and to accumulate TAG (Na et al. 2005; Honda et al. 2008; Sameshima et al. 2008). The analysis of R. opacus B4 genome revealed an 8.8-Mb-long genome composed by one linear chromosome, two linear plasmids of pROB series of size range of 2–4 Kb and three circular plasmids of pKNR series of size range of 110–550 Kb (Table 2).
Members of R. erythropolis are largely distributed in the environment and exhibit a remarkable metabolic versatility related to their capacity to degrade complex compounds, such as quorum-sensing signals N-acylhomoserine lactones, phenols, sterols and fuel derivatives (Table 1). Moreover, many R. erythropolis strains have been isolated from petroleum-contaminated sites and have been described for their ability to degrade various hydrocarbons using multiple catabolic pathways (de Carvalho and da Fonseca 2005). All the R. erythropolis strains whose complete genome has been sequenced showed the presence of plasmids ranging in length from 3 Kb to 261 Kb (Table 2).
The genomes of R. aetherivorans and R. ruber species showed the highest amount of GC, being around 70% in all the strains described up to date. Members of these two species have been described for different biosynthetic and biodegradative capacities. R. aetherivorans BCP1 (formerly Rhodococcus sp. BCP1) is able to degrade a wide range of alkanes, naphthenic acids and chlorinated alkanes (Frascari et al. 2006; Cappelletti et al. 2011, 2015; Presentato et al. 2018a; Ciavarelli et al. 2012) and to produce metal-based nanostructures (Presentato et al. 2016, 2018b, c). BCP1 has a genome of 6.2 Mbp composed by one chromosome and two plasmids (of 100–120 Kb size) (Tables 1 and 2). R. aetherivorans IcdP1 was isolated from an abandoned coking plant; it was proven to be able to degrade numerous high-molecular-weight polycyclic aromatic hydrocarbons and organochlorine pesticides (Qu et al. 2015). Although their genome data are not available, R. aetherivorans IAR1 was described to accumulate polyhydroxybutyrate PHB, while R. aetherivorans IG24 was employed by Merck Corporation for the production of the anti-HIV drug Crixivan™ (Treadway et al. 1999; Hori et al. 2009).
Rhodococcus pathogenic strains belong to R. equi and R. fascians species, which cause disease to animals and plants, respectively. In line with their lifestyle inside a host, their genomes are generally smaller (<6 Mb) compared to the environmental Rhodococcus strains (Table 1). Recently, numerous genomes of both these two species have been sequenced to conduct phylogenomic analyses and to investigate on the evolutionary mechanisms of virulence traits (Letek et al. 2010; Creason et al. 2014a, b). In particular, virulence genes have been associated to plasmids. Several R. equi strains have been described to possess circular plasmids, whereas R. fascians members are featured by linear plasmids. The presence of linear plasmids in R. fascians has been correlated with phytopathogenicity, although it was described not to be strictly necessary (Creason et al. 2014a).
Compared to the other species, only few genomic studies are available for R. qingshengii, R. pyridinivorans and R. rhodochrous. Nevertheless, members of these species have been reported to possess peculiar metabolic activities such as degradative capacity towards rubber, fungicide and other hydrocarbon-related complex molecules. In relation to this, additional genome sequencing efforts promise to expand the knowledge on the genetic and metabolic diversity of Rhodococcus bacterial strains (Table 1) (Xu et al. 2007; Dueholm et al. 2014; Watcharakul et al. 2016).
3 Comparative Genomics of Rhodococcus
Comparative genomics allow providing genomic basis for the differences observed among microorganisms in terms of phenotypes, metabolic capacities and cellular response to specific environmental conditions. Recent studies have described genome-wide comparative analyses of Rhodococcus strains with the aim of defining the phylogeny and evolutionary relationship, determining the Rhodococcus genus core genome, analysing their catabolic potentials and stress response (Orro et al. 2015; Cappelletti et al. 2016). Genome comparative analysis is performed through pairwise or multiple (>3 genomes) whole-genome alignments. Among the applicable programs, Artemis Comparison Tool (ACT), available within the Integrated Microbial Genomes (IMG) system, was used for genome comparison of strain R. opacus M213 with different Rhodococcus strains (Pathak et al. 2016). Mauve (http://darlinglab.org/mauve/mauve.html) performs multiple sequence alignment and has been used for comparative genomic analyses of different Rhodococcus strains, e.g. R. opacus R7 and R. aetherivorans BCP1 (Orro et al. 2015), R. opacus M213 (Pathak et al. 2016) and Rhodococcus fascians strains (Creason et al. 2014b) (Fig. 1).

Whole-genome sequence alignment with program Mauve diagram. The alignment of three pairs of genomes is shown: R. jostii RHA1 and R. opacus R7 (panel A), R. jostii RHA1 and R. aetherivorans BCP1 (panel B), R. opacus R7 and R. aetherivorans BCP1 (panel C). The coloured region represents a block of sequences that is collinear to a corresponding block of sequences in the other genome sequence. [Modified from Orro et al. (2015)]
Comparative genome analysis allows to detect genomic regions that are conserved or syntenic (Pathak et al. 2016), representing conserved genome sections that have not undergone internal rearrangements and inversions. This conservation typically reflects phylogenetic correlation and represents the indication of functional evolutionary constraints. In comparative studies concerning Rhodococcus, the conservation of collinear blocks in different R. fascians strains was shown to be in accordance with their phylogenetic relationship (Creason et al. 2014b; Pathak et al. 2016). The 16S rRNA-based affiliation of Rhodococcus sp. M213 to opacus species was challenged by the higher M213 syntenic affiliation with R. jostii RHA1 compared to R. opacus strains B4 and PD630 (Pathak et al. 2016).
Another useful way to compare whole genomes is by computing a value that summarizes their similarity or distance. The average nucleotide identity (ANI) value calculated from whole-genome alignments was described to provide a tool for taxonomy and evolutionary studies and species definition (Creason et al. 2014b; Anastasi et al. 2016). Whole-genome alignment analysis of three Rhodococcus equi strains allowed to detect single nucleotide polymorphisms (SNPs) in specific coding regions that were suggested to possibly reflect differences in the lifestyle of these three isolates (Sangal et al. 2014).
In addition to gene/protein sequence-based alignment, gene content comparison can be done in terms of function using, for instance, the concept of functional categories (available in RAST subsystem) or COG (cluster of orthologous groups of proteins). Typically, statistical tools analyse gene functions (or annotations) as categorized by COGs and determine whether certain functions or functional categories are statistically enriched in certain genomes compared to others. In this respect, the R. opacus M213 genome analysis at functional (COG) level further reinforced the closer relationship of M213 with R. jostii RHA1 and other Rhodococcus strains (e.g. R. wratislaviensis IFP 2016, R. imtechensis RKJ300, Rhodococcus sp. strain DK17) compared to R. opacus strains (Pathak et al. 2016). On the other hand, by comparing the RAST-based functional categories, R. opacus R7 genome resulted to be enriched in genes belonging to all the functional categories, compared to R. aetherivorans BCP1, except for those involved in some central metabolic process (e.g. DNA, iron, potassium and phosphorous metabolism, dormancy and sporulation, motility and chemotaxis) (Orro et al. 2015).
3.1 Phylogenomics
The use of the only 16S rRNA sequence as taxonomic marker was proved to limit the resolution of Rhodococcus phylogenetic studies (Gürtler et al. 2004; Pathak et al. 2016; Creason et al. 2014b) and more in general to lead to artefacts in the bacterial phylogenetic tree construction (Ventura et al. 2007). More recent phylogenetic investigations have been carried out using alternate gene/protein sequences (e.g. functional genes or molecular chaperone-encoding genes) as phylogenetic markers, which often evolve faster than the rRNA operon. This provides a larger phylogenetic resolution that can help to distinguish closely related bacterial species. For this purpose, the alkane monooxygenase (alkB) gene was applicable as phylogenetic marker of Rhodococcus strains (Táncsics et al. 2015).
The availability of genome sequences has provided new opportunities to get information on applicable phylogenetic markers, and new approaches arose to examine rhodococcal phylogeny based upon multiple gene or protein sequences. Among these approaches, the construction of phylogenetic trees based on concatenated sequences for large numbers of proteins (also called multilocus sequence analysis—MLSA) has proven particularly useful for evolutionary relationship studies (Gao and Gupta 2012). Phylogenetic tree based upon multiple conserved (or slow-evolving) genes/proteins was demonstrated to properly assign environmental isolates to existing Rhodococcus species (Orro et al. 2015) and to resolve evolutionary relationships of Rhodococcus strains belonging to the same species (Creason et al. 2014b; Letek et al. 2010). MLSA mainly employed sequence concatenamers of housekeeping genes including 16S rRNA, recA, gyrB, rpoB, rpoC and secY (Orro et al. 2015; Kwasiborski et al. 2015). Orro et al. (2015) assigned the species to two Rhodococcus strains on the basis of MLSA with four of these marker genes (Fig. 2). MLSA provided a framework for defining Rhodococcus genus within the Actinobacteria phylum using seven marker genes (ftsY, infB, rpoB, rsmA, secY, tsaD and ychF) (Creason et al. 2014b). In line with the whole-genome based studies by Letek et al. (2010), this study placed the R. equi species within the Rhodococcus genus, which was in contrast with other inconclusive phylogenetic studies based on 16S rDNA sequences (Goodfellow et al. 1998). More genome sequences and informative marker sequences might help in improving MLSA-mediated taxonomy and discern the finer details within the suborder. In this respect, available genome sequences were used to discover novel molecular characteristics that could be used for evolutionary and systematic studies of Rhodococcus genus. In this regard, novel specific conserved signature indels (CSIs) and conserved signature proteins (CSPs) have been proposed as possible molecular markers on the basis of their conservation among different Rhodococcus species (Gao and Gupta 2012).

Multilocus sequence analysis-based tree of 28 Rhodococcus strains. Phylogenetic tree of 28 Rhodococcus strains based on MLSA using the sequence of the four marker genes, 16S rRNA, secY, rpoC and rpsA. [Modified from Orro et al. (2015)]
In addition to phylogenomic analysis based on the concatenation of different taxonomic markers, Rhodococcus genome-based trees have been constructed on the basis of average sequence similarity. The genome similarity is at the basis of the wet-lab DNA-DNA hybridization (DDH) technique, which represents the gold standard for bacterial taxonomy classification (Richter and Rosselló-Móra 2009). In the era of genomics, different attempts have been made to replace the traditional wet-lab DDH with in silico genome-to-genome comparison (Goris et al. 2007; Richter and Rosselló-Móra 2009). Among these, the average nucleotide identity (ANI) estimates the average nucleotide identity between two genomic datasets, and an ANI value in the range of 94–96% has been proposed to be the criterion for bacterial species delineation (corresponding to a wet-lab DDH value of 70%), while ANI values of 70–75% identified members of the same genus (Konstantinidis and Tiedje 2005; Goris et al. 2007; Richter and Rosselló-Móra 2009). Creason et al. (2014b) conducted phylogeny study of Rhodococcus genus by using the average nucleotide identity (ANI) approach and found that the genus Rhodococcus can be represented by as many as 20 distinct species (Fig. 3).

Average nucleotide identity (ANI) dendrogram for 59 isolates of Rhodococcus. Complete genome sequences for 59 isolates of Rhodococcus were used to generate an ANI matrix. The matrix was used to calculate an ANI divergence dendrogram. Branches are coloured based on the ANI threshold values. [Modified from Creason et al. (2014b)]
Using 27 de novo sequenced R. equi genomes, the ANI value was 99.13%, well above the consensus 95–96% threshold for prokaryotic species definition (Anastasi et al. 2016). This result highlighted the high genetic homogeneity of R. equi group and was found to correspond to a 16S rRNA sequence identity of 100%.
3.2 Core and Accessory Genome
Whole-genome comparative analysis can be used to obtain information on the pan genome, core genome and accessory genome of a group of bacterial strains belonging to a single species or different species belonging a single genus. The pan genome comprises all the genes present in a given bacterial group (the collection of all genetic material). The core genome is defined as the set of all genes shared by the genomes as orthologs and therefore considered as conserved by all the bacterial strains under analysis (Ventura et al. 2007). The genes included within the core genome are supposed to have strong functional significance for cell physiology and replication. Moreover, as the core genome can be considered to define the members of the bacterial group under analysis, the core genes are suitable as molecular targets to infer phylogeny (Ventura et al. 2007). On the other hand, the accessory or variable genome is represented by the genes, which are confined to a single member of the bacterial group and are therefore specific for a single strain (Ali 2013). The accessory genome therefore represents those gene families that can be associated with phenotypic traits that differentiate each member within a given bacterial population under analysis.
Comparative genomic studies on Rhodococcus strains have assessed the core genome shared by either strains belonging to the same species or strains belonging to different species. The resulting core-genome size and identity reported in each work was dependent on the Rhodococcus strains under analysis. Within the Rhodococcus genome set composed by R. opacus R7, R. aetherivorans BCP1, R. jostii RHA1, R. opacus PD630, R. opacus B4 and R. pyridinivorans SB3094, the core genome corresponded to around 50% of all the identified CDSs in each strain (Orro et al. 2015) (Fig. 4a). This indicated a large genetic variability among these Rhodococcus strains, some of them belonging to different species. The portion of core genes increased by considering Rhodococcus strains belonging to the same species, such as R. opacus R7, B4 and PD630 (Fig. 4a) (Orro et al. 2015), R. erythropolis (Kwasiborski et al. 2015) and R. equi (Anastasi et al. 2016). In particular, the genome of R. erythropolis R138 was compared to the genomes of other ten R. erythropolis strains, either available in the database or newly sequenced. The core-genome genes represented up to 87% of all the CDSs identified in R. erythropolis R138. A large core genome was also identified in a comparative study with 29 R. equi strains, which showed to have ~80% of shared genes in their genome (Anastasi et al. 2016). Some of these comparative studies also found that the core genes were mainly localized on the chromosome, whereas the unique genes (accessory genome) were harboured by the endogenous plasmids in each strain (Orro et al. 2015; Kwasiborski et al. 2015). For instance, considering R. erythropolis strains, up to 97% of the shared sequences were located on the circular chromosome of R138 strain (Kwasiborski et al. 2015). These results are general indications of a core set of functions localized on the Rhodococcus chromosome, whereas plasmids have variable genetic contents. The variable accessory genome underlies the peculiar capacities of each Rhodococcus strain and contributes functions for niche adaptation (Kwasiborski et al. 2015). In line with this, considering R. equi strains, the core genome included traits necessary for bacterial physiology, virulence and niche adaptation, including tolerance to desiccation and oxidative stress (Anastasi et al. 2016). Similarly, R. erythropolis core genome was highly represented by gene categorized within the primary metabolism of amino acids, carbohydrates, cofactors and proteins (Kwasiborski et al. 2015). Interestingly, genes encoding enzymes involved in the degradation of complex molecules (i.e. N-acylhomoserine lactones, phenol, catechol and sterol derivatives) were also shared by the 11 R. erythropolis under analysis by Kwasiborski et al. (2015). On the other hand, functions encoded by unique genes (accessory genome) were associated to degradative enzymes in R. opacus M213 (Pathak et al. 2016), R. aetherivorans BCP1 and R. opacus R7 (Orro et al. 2015). In particular, R. aetherivorans BCP1 possessed unique genes involved in short-chain n-alkanes degradation which were not present in the other Rhodococcus strains under analysis (Orro et al. 2015) Conversely, R. opacus R7 genome had a high number of unique genes involved in aromatic degradation metabolism. These genetic findings generally correlated with the different types of contaminants persisting in each Rhodococcus strain isolation source. (Di Gennaro et al. 2001; Frascari et al. 2006; Orro et al. 2015).

Venn diagram displaying the core-genome and the unique genes (accessory genome) specific to each strain. The number of genes shared by all strains included within each genome set (i.e. the core genome) is in the centre. Numbers in nonoverlapping portions of each oval show the number of genes unique to each strain. The total number of protein coding genes found in each genome is listed below the strain name. Panel (A), core-genome and unique genes resulting from the comparison of the genomes from R. opacus R7, R. aetherivorans BCP1, R. jostii RHA1, R. opacus PD630, R. opacus B4, R. pyridinivorans SB4094; panel (B), core-genome and unique genes resulting from the comparison of the genomes from R. opacus R7, R. opacus PD630 and R. opacus B4
3.3 Functional Genomics
Genome sequence-based analysis relies on homology-based gene annotation and protein domain identification to annotate open reading frames (ORFs) and to perform functional predictions. Bioinformatic tools have been applied for the functional analysis of Rhodococcus genomes to determine the genetic basis possibly involved in relevant metabolic activities and phenotypic traits, e.g. degradation abilities of specific xenobiotics, plant growth-promoting effect and stress tolerance (Francis et al. 2016; Cappelletti et al. 2016; Pathak et al. 2016). For instance, genome-based analyses assessed the occurrence and distribution of key genes and pathways involved in the synthesis and accumulation of triacylglycerols (TAG), wax esters, polyhydroxyalkanoates, glycogen and polyphosphate (Hernández et al. 2008; Villalba et al. 2013). In line with the extensive experimental work demonstrating the ability of Rhodococcus strains to synthesize and accumulate neutral lipids, numerous genes/enzymes predicted to be involved in TAG biosynthesis and degradation and fatty acid β-oxidation were identified in the genomes of R. jostii RHA1, R. opacus PD630, R. opacus B4, R. erythropolis PR4, R. equi 103S and R. fascians F7 (Villalba et al. 2013). The comparative genome analysis of psychrophilic Rhodococcus sp. JG3 and other mesophilic rhodococci allowed to detect the cold adaptive traits which confer JG3 with the ability to survive the extremely arid, cold and oligotrophic conditions of permafrost (Goordial et al. 2016). A bioinformatic analysis of the genomes of 20 Rhodococcus strains allowed identifying numerous biosynthetic gene clusters encoding pathways involved in the possible production of novel secondary metabolites (Ceniceros et al. 2017).
Several works have assessed a large range of metabolic abilities of strains using Phenotype Microarray (PM) technologies. PM results were interpreted using genomic analysis to predict the genes involved in central metabolic pathways, xenobiotic degradation metabolisms and stress response (Letek et al. 2010; Holder et al. 2011; Orro et al. 2015; Cappelletti et al. 2016).
The large-scale functional analysis of Rhodococcus genomes has been further integrated with the results obtained through omic technologies. In particular, high-throughput experiments were conducted to obtain global datasets on the expression of genes (transcriptomic analyses through RNA-seq or microarray) and on the protein profiles induced under specific growth conditions or after specific cell treatments. Transcriptomic studies were performed to define the genes involved in the degradation of aromatic compounds and steroids (chlorate and cholesterol) (Gonçalves et al. 2006; van der Geize et al. 2007; Swain et al. 2012), of PCBs and diesel oil (Puglisi et al. 2010, Laczi et al. 2015) and of synthetic polymers (Gravouil et al. 2017) but also in lipid metabolism and accumulation (Chen et al. 2014; Amara et al. 2016) and isoprene metabolism (Crombie et al. 2015). Proteome studies were combined with genome analysis for the study of the degradation of xenobiotic 4,4-dithiodibutyric acid (DTDB) by R. erythropolis MI2 (Khairy et al. 2016) and the metabolism of short-chain alkanes in R. aetherivorans BCP1 (Cappelletti et al. 2015) and to study the global response to stress conditions such as desiccation and carbon starvation in R. jostii RHA1 (LeBlanc et al. 2008; Patrauchan et al. 2012). In many of these studies, the involvement of multiple homologous genes was identified in specific metabolic pathways, providing clues to the redundancy of the different catabolic pathways in Rhodococcus (Gonçalves et al. 2006; Swain et al. 2012). Further, some omic studies gave indications on the involvement of possible regulators in the specific catabolic pathways under analysis (Crombie et al. 2015). In R. ruber C208, transcriptomic results indicated the involvement of alkane degradation pathway and β-oxidation of fatty acids as the main catabolic route for polyethylene degradation; further, it also indicated metabolic limiting steps which could represent molecular target to optimize the biodegradation process (Gravouil et al. 2017). In addition to catabolic genes/proteins, many genes/proteins involved in stress response (e.g. chaperone-like proteins, superoxide dismutase, catalase/peroxidase) were reported to be induced during the degradation of organic compounds (Tomás-Gallardo et al. 2006; Puglisi et al. 2010; Khairy et al. 2016). The microarray analysis of R. aetherivorans I24 cells exposed to PCBs reported the transcriptional response to be primarily directed towards reducing oxidative stress rather than catabolism (Puglisi et al. 2010).
4 The Genomic Basis of Metabolic Versatility in Rhodococcus
The extraordinary metabolic versatility of Rhodococcus strains is reflected in their genomic attributes, like (1) large genome sizes (up to 10 Mbp) encoding numerous catabolic pathways for a variety of chemical compounds; (2) a significant degree of gene redundancy that ensures functional robustness and free-to-evolve genetic reservoirs; and (3) the occurrence of circular and linear (mega)plasmids that generally evolve more rapidly than the chromosome and consist of an additional pool of DNA that can evolve and can be easily transferred (Redenbach and Altenbuchner 2002; Van der Geize and Dijkhuize 2004). In this context, the broad Rhodococcus adaptability is related to their genome “flexibility” which refers to the genomic rearrangements mainly occurring on the large linear plasmids and due to still mostly unknown molecular mechanisms promoting the frequent non-homologous illegitimate recombination. This last aspect has been discussed in several review articles by Larkin and collaborators (Larkin et al. 1998, 2010; Kulakov and Larkin 2002). Additionally, recent works have underlined how the equipment of genes for the transport of many different substrates (de Carvalho et al. 2014) and the numerous genes encoding oxygenases, typically catalysing the hydroxylation and cleavage of organic compounds, are at the basis of the wide metabolic capacities of Rhodococcus strains. Only a few portion of the oxygenase genes have been acquired through horizontal gene transfer events, while most of them were chromosomally located, suggesting their fundamental role in Rhodococcus physiology (Mcleod et al. 2006).
4.1 Catabolic Gene Redundancy
Since the first complete genome sequence obtained from a Rhodococcus strain, the presence of multiple catabolic genes encoding homologous enzymes, called isozymes, attracted research interest. Recently, the analysis of whole-genome sequences of several rhodococci confirmed the redundancy in catabolic pathways initially only hypothesized for this genus (McLeod et al. 2006). Catabolic gene redundancy is often considered at the basis of the Rhodococcus catabolic versatility, functional robustness, adaptation to polluted and extreme environments and high-performing environmental competition (Mcleod et al. 2006; Pérez-Pérez et al. 2009). Several examples of genetic redundancy originated either from gene duplication or horizontal gene transfer events have been described in Rhodococcus strains, the latter being supported by the proximity of transposase and invertase sequences to duplicated gene clusters (Taguchi et al. 2007). In general, the duplicate genes have been described to evolve and to encode isoenzymes possessing similar sequences but different substrate specificities and induction patterns. On the other hand, some duplicates have been described to be, at some extent, functionally redundant (Zhang 2012), catalysing the same metabolic reaction and presenting overlapping substrate range and inducing profiles. This type of duplication has probably occurred in relative recent period of time or has not been subject to strong selective pressure. For instance, in R. jostii RHA1, it has been hypothesized that the multiple homologous genes involved in the central aromatic pathways have received low selective pressure for functional diversification or gene removal (McLeod et al. 2006).
In this framework, the presence of multiple homologous genes strongly contributes to the wide Rhodococcus versatility in terms of catabolic activities including aliphatic and aromatic hydrocarbon catabolism, chlorinated hydrocarbons transformation and steroid degradation. Further, the presence of multiple functional homologs was found in central metabolic pathways (e.g. tricarboxylic acid cycle) (Van der Geize and Dijkhuize 2004).
While the presence of multiple copies of specific genes was not a universal feature of Rhodococcus, genomic redundancy generally characterizes the members of this genus in relation to metabolic and physiological traits useful for the adaptation to the environmental niche from which each Rhodococcus strain was isolated. For instance, the genome analysis of the psychrophilic Rhodococcus sp. JG3 isolated from a permafrost soil with a possible alkane source showed genomic redundancy in relation to genes involved in the adaptation to cold temperatures such as those associated to osmotic stress and to genes encoding alkane 1-monooxygenases (Goordial et al. 2016).
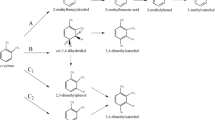
Functional redundancy has referred to the multiple alkane hydroxylase genes found in Rhodococcus strains featured by versatile alkane degradation capacity and typically isolated from petroleum-contaminated (sediment or marine) sites (van Beilen et al. 2002) (Fig. 5a). In particular, alkB gene encodes the membrane alkane hydroxylase AlkB, which catalyses the initial oxidation of alkanes. Four alkB homologous genes have been detected in R. erythropolis NRRL B-16531 and R. qingshengii Q15 (Whyte et al. 2002); three to five alkB genes were found in different R. erythropolis strains from various contaminated soils and in Rhodococcus sp. strain TMP2 (van Beilen et al. 2002; Takei et al. 2008). The multiple alkB genes found in Rhodococcus strains were mainly localized on the chromosome, and some of them showed high sequence divergence and different flanking regions (Whyte et al. 2002). The different AlkBs often catalysed the oxidation of different ranges or classes of alkanes and were differently expressed depending on the substrate and growth condition (Takei et al. 2008; Laczi et al. 2015). Because of this, the presence of multiple AlkB hydroxylases has been associated to broad metabolic capacities of Rhodococcus strains towards medium- and long-chain alkane and also to branched alkanes. In R. aetherivorans BCP1, two homologous gene clusters encoding alkane monooxygenases of SDIMO (soluble di-iron monooxygenases) family were involved in the oxidation of short-chain alkanes (Cappelletti et al. 2013, 2015). This functional redundancy was correlated with the strong capacity of BCP1 strain to utilize gaseous alkanes and to co-metabolize chlorinated alkanes (Frascari et al. 2006; Cappelletti et al. 2012). In some cases, alkane hydroxylation functional redundancy was also associated to the co-existence in a single Rhodococcus strains of genes encoding different alkane hydroxylases, AlkB, SDIMOs and cytochrome P450 belonging to the CYP153 family. For instance, the versatile and efficient degradation of alkanes by R. erythropolis PR4 was associated to the presence of four alkB genes, two CYP153 genes and other genes coding P450 on its genome, which are differently expressed on alkanes and hydrocarbon mixtures (Laczi et al. 2015).

Examples of genetic redundancy in Rhodococcus. Genetic organization of (a) alkB genes in Rhodococcus strain NRRLB-16531, (b) catabolic island including tpa, pat and pad gene that is duplicated in the two linear plasmids in R. jostii RHA1, (c) the three homologous gene clusters (bphA, etb1A and etb2A genes) encoding dioxygenase systems involved in the initial hydroxylation of substituted benzenes in R. jostii RHA1. The plasmidic or chromosomal localization is indicated in correspondence to each gene cluster. [Modified from Whyte et al. (2002), Hara et al. (2007), Gonçalves et al. (2006), respectively]
Catabolic gene redundancy in some Rhodococcus strains has been extensively reported for aromatic compound degradation pathways. In particular, R. jostii RHA1, isolated from γ-hexachlorocyclohexane (lindane)-contaminated site, was described to be featured by high redundancy of catabolic pathways involved in aromatic hydrocarbon catabolism, which are also responsible for the co-metabolic transformation of polychlorinated biphenyls (PCBs). Several homologous genes were predicted to encode enzymes involved in the upper and lower degradation pathways of substituted benzenes, like ethylbenzene (ETB) and biphenyl (BPH). In RHA1, three homologous gene clusters (bphA, etb1A and etb2A genes) were found to encode dioxygenase systems, which are all induced by BPH and ETB by a possible common regulatory system (Fig. 5b). Nevertheless, these dioxygenase systems have shown distinct substrate specificity in terms of both aromatic hydrocarbons and PCB congeners (Iwasaki et al. 2006, 2007; Patrauchan et al. 2008). The gene cluster bphA is localized on the large linear plasmid pRHL1, while etb1A and etb2A are carried on the other linear plasmid pRHL2 and share high sequence similarity. Multiple copies of bph genes (bphC-G) were also predicted to be involved in the benzene ring cleavage downstream of the initial hydroxylation. Among these multiple homologs, only a few numbers of genes were transcriptionally induced during the growth on BPH and ETB, while the most part was expressed at constitutive levels (Gonçalves et al. 2006). In particular, only three out of the eight bphEFG clusters were up-regulated during RHA1 growth on BPH and ETB, independently on their genome localization (Gonçalves et al. 2006; Patrauchan et al. 2008). This indicated that out of the eight bphEFG, five homologous clusters produced paralog enzymes involved in distinct physiological processes (Irvine et al. 2000; Taguchi et al. 2004). Other Rhodococcus strains, isolated from the termite ecosystem typically exposed to plant-derived lignin and aromatics, showed multiple copies of bph gene clusters involved in PCB/biphenyl degradation; many of these were present on linear plasmids and in the proximity of transposase and invertase sequences (Taguchi et al. 2007). In addition to the catabolic enzymes, functional redundancy was found in the regulatory system responsible for the growth of RHA1 on aromatic compounds. In particular, two copies of the gene cluster bphS and bphT encode the two-component systems, BphS1T1 and BphS2T2, which showed high similarity in the amino acid sequence (>92%) and the same substrate spectrum, except for biphenyl (Takeda et al. 2010).
Additional genomic redundancy found in the genome of aromatic Rhodococcus degraders was related to genes involved in the catabolism of phthalate (pad), terephthalate (tpa), catechol (cat), protocatechuate (pca) and benzoate (ben) (Fig. 5c). In particular, two identical copies of a catabolic island including the pad and tpa clusters were found on the linear plasmids of R. jostii RHA1 and Rhodococcus sp. DK17, flanked by transposase-encoding genes (Patrauchan et al. 2005; Choi et al. 2005). The duplicated phthalate-degrading operons resulted to be simultaneously expressed and equally functional during the DK17 growth on phthalate, allowing this strain to achieve the maximal degradation of phthalate (Choi et al. 2007). Genes involved in naphthalene and phthalate were found duplicated on two separate genomic islands in R. opacus M213, and, in Rhodococcus sp. TFB, it was demonstrated that naphthalene degradation probably results from the activities of different isozymes (Tomás-Gallardo et al. 2006). Genetic redundancy in aromatic degradation pathways was observed in R. opacus R7 possibly in relation to both the size of the genome and the degradation abilities towards different aromatic classes (Di Gennaro et al. 2014; Orro et al. 2015) and in R. opacus M213.
The catabolic redundancy was also associated to different substrate specificities and to possible mechanisms of metabolic intermediate detoxification in the steroid metabolism (involving kshAB and kshD genes) by some Rhodococcus strains. Four homologous ksh gene clusters are present in R. jostii RHA1 genome; the cluster 1 was shown to encode enzymes involved in cholesterol catabolism (van der Geize et al. 2007), while the homologous cluster 3 supported the catabolism of cholate (Swain et al. 2012). In R. erythropolis SQ1, ksh cluster 1 was involved in steroid metabolism, while ksh cluster 2 was supposed to have a role in limiting the intracellular accumulation of toxic metabolic intermediates (van der Geize et al. 2008). The genome analysis of R. rhodochrous DSM43269A revealed five kshA homologous genes (kshA1 to kshA5) which were phylogenetically distinct, and each one showed a unique steroid induction pattern and substrate range, ensuring a fine-tuned steroid catabolism (Petrusma et al. 2011).
The redundancy of genes encoding (chloro)phenol hydroxylases supported the ability of R. opacus 1CP to metabolize a large spectrum of phenolic and chloro-phenolic compounds (Grӧning et al. 2014). All the three homologous phenol hydroxylases were able to convert the tested phenolic substrates at significant rates. This was probably due to the broad substrate specificity of these phenol hydroxylases and, therefore, low specialization (Grӧning et al. 2014). Multiple genes (from 3 to 5) encoding phenol hydroxylases (pheA1/pheA2) were identified in other Rhodococcus strains (Grӧning et al. 2014) In particular, in R. jostii RHA1, differences in the transcriptional regulation of two pheA1/pheA2 clusters suggested their activation under different growth conditions (Szőköl et al. 2014).
Interestingly, the functional redundancy found in Rhodococcus was strongly associated to evolutionary mechanisms related to niche adaptation. In this respect, the genome and microarray analysis of R. aetherivorans I24 study of this strain to PCB/biphenyl exposure indicated the involvement of the only core enzymes in the substituted benzene metabolism, while most of the isozymes found in RHA1 were missing (Puglisi et al. 2010). This was associated to the fact that, unlike RHA1, I24 strain was not isolated from a PCB-contaminated site. Therefore, as previously mentioned, despite the gene redundancy is recognized as a trait of Rhodococcus genus, the presence of functional isozymes involved in specific catabolic pathways reflects the selective pressure imposed by the environmental habitat and is also in part associated to the size of the genomes, as in the case of R. jostii and R. opacus strains featured by larger genomes compared to R. aetherivorans and R. ruber (Table 1).
4.2 Plasmids
Plasmids are circular or linear extrachromosomal DNA elements, which are capable of semi-autonomous or self-replication; they do not typically encode essential genes for the host but instead carry genes that may help the organism to adapt to novel environments or nutrient sources (Aminov 2011; Carroll and Wong 2018). In this respect, most of the Rhodococcus genes belonging to the variable accessory genome have plasmidic localization (see Par. 3.2) (Creason et al. 2014a; Orro et al. 2015; Pathak et al. 2016). Plasmids are considered a major driving force in prokaryotic evolution as they can be transferred between cells, as mobile genetic elements, mediating horizontal gene transfer events (Hülter et al. 2017). Accordingly, in bacteria, plasmids are thought to play critical roles in the evolution, propagation and assembly of catabolic pathways, antibiotic and metal resistance mechanisms, antimicrobial biosynthetic pathways and pathogenicity (Meinhardt et al. 1997; Shimizu et al. 2001).
Many Rhodococcus strains harbour plasmids, which can be linear or circular, of small (of few Kbp) or large size (of hundreds of Kbp), with cryptic or catabolic functions. Some Rhodococcus strains have been described to simultaneously possess several plasmids of different types and sizes, e.g. R. erythropolis PR4 contains one linear plasmid, pREL1 (~270 Kb), and two circular plasmids, pREC1 (~100 Kb) and pREC2 (~3.5 Kb) (Sekine et al. 2006); R. opacus B4 possesses two linear plasmids of pROB series (pROB01 of ~560 Kb, pROB01 of ~240 Kb) and three circular plasmids of pKNR series (pKNR of 111 Kb, pKNR01 of 4.4 Kb, pKNR02 of 2.8 Kb) (Na et al. 2005). Many Rhodococcus plasmids, both linear and circular, encode catabolic functions associated to xenobiotic degradation (Shimizu et al. 2001), chemolithoautotrophical growth on gaseous hydrogen and carbon dioxide, hydrogen production (Grzeszik et al. 1997), metal toxicity resistance (Cappelletti et al. 2016) and pathogenicity (Letek et al. 2010). In this respect, deletions in several Rhodococcus plasmids have resulted in the loss of degradative genes and specific growth deficiencies; e.g. deletions on pRHL1 and pRHL2 in R. jostii RHA1 and on pTA422 in R. erythropolis TA421 affected biphenyl degradation (Fukuda et al. 1998; Kosono et al. 1997). In some cases, catabolic plasmids were also found to harbour homologous genes, and their mutation was found to exert moderate or null phenotypic effects (Patrauchan et al. 2005). In this framework, catabolic plasmids are thought to play a key role in Rhodococcus catabolic versatility and efficiency by harbouring unique set of genes involved in specific catabolic pathways but also contributing to Rhodococcus multiple pathways and functional redundancy (Kim et al. 2018). Additionally, Rhodococcus plasmids have been described to have a much higher density of DNA mobilization genes (e.g. insertion sequences, transposase genes), pseudogenes, unique species-specific genes and niche-specific determinants (e.g. genes involved in pathogenesis in R. equi and in peripheral aromatic clusters in R. jostii RHA1 and R. opacus R7) than the corresponding chromosomes. Rhodococcal plasmids are therefore under less stringent selection and are key players in rhodococcal genome plasticity and niche adaptability (Sekine et al. 2006).
Linear plasmids, which are often very large, are widespread and diverse among Rhodococcus strains (Ventura et al. 2007). As they can reach lengths of several hundreds of kilobases, they are frequently referred to as megaplasmids. For instance, pRHL2 from R. jostii RHA1 is 443 kb (Shimizu et al. 2001), pPDG1 from R. opacus R7 is 656 kb (Di Gennaro et al. 2014), and pRHL1 from R. jostii RHA1 is 1.12 Mb (McLeod et al. 2006). The reason of extensive research attention on Rhodococcus large linear plasmids is connected with the fact that they are associated with catabolic genes and that they confer advantageous abilities on their hosts (Meinhardt et al. 1997). For instance, linear plasmids have been described to encode enzymes involved in the catabolism of naphthalene (Kulakov et al. 2005; Orro et al. 2015; Pathak et al. 2016), biphenyl (Taguchi et al. 2004), toluene (Priefert et al. 2004), alkylbenzene (Kim et al. 2002), isopropylbenzene and trichloroethylene (Meinhardt et al. 1997), phthalate (Patrauchan et al. 2005), gaseous n-alkanes (Cappelletti et al. 2015), chloroaromatic compounds (Konig et al. 2004), isoprene (Crombie et al. 2015) and triazine compounds (Dodge et al. 2011) and also in the desulphurization of organosulphur compounds (Denis-Larose et al. 1997), resistance to toxic metals (Meinhardt et al. 1997; Cappelletti et al. 2016), pathogenicity towards bovines (Valero-Rello et al. 2015) and phytopathogenicity (Francis et al. 2012). In many cases, catabolic genes present in linear plasmids contribute to degradation pathways together with genes located on the chromosome (Gonçalves et al. 2006).
As a distinctive feature, linear megaplasmids from Rhodococcus strains are capable of conjugal transfer, being responsible for genetic information sharing through HGT (Meinhardt et al. 1997; Dib et al. 2015). The importance of linear elements is also associated to their higher “flexibility” compared to circular plasmids. In particular, the telomeres are considered to be frequently subject to recombinational events (Volff and Altenbuchner 2000; Chen et al. 2002). In the case of HGT, intermolecular recombination events can take place between plasmids but also between host chromosomes of compatible species. Both the frequent observation of linear plasmids and illegitimate recombination in Rhodococcus have led to the hypothesis of the hyper-recombinational gene storage strategy. This is related to the function of the plasmids as storage of large number of catabolic genes that can represent recombination sources to respond and adapt to novel compounds in the native soil environments.
As for most bacteria, circular plasmids are also very common in Rhodococcus species. Circular plasmids in Rhodococcus have been generally shown to possess smaller size compared to the linear ones (Table 2). However, they often contribute to the catabolic capacities of the Rhodococcus host strain. Several circular plasmids harbour genes encoding part or complete xenobiotic degradation pathways in Rhodococcus strains such as pRTL1 (100 Kb) encoding haloalkane degradation enzymes in R. rhodochrous NCIMB13064 (Kulakova et al. 1995); pREC1 contains a complete set of genes for the β-oxidation of fatty acids (Sekine et al. 2006) and the large circular plasmids pKNR (111 kb) in R. opacus B4 (Honda et al. 2012).
Additionally, many Rhodococcus plasmids are self-transmissible but phenotypically cryptic. The cryptic plasmids described in Rhodococcus strains have principally small size and circular structure, e.g. pREC2 (3.6 Kb) in R. erythropolis PR4, pB264 (4.9 Kb) in Rhodococcus sp. B264-1. In some cases, these plasmids have been isolated to develop vector systems for DNA manipulation and protein expression in Rhodococcus strains. For instance, pKA22 (4.9 Kb) from R. rhodochrous NCIMB13064 (Kulakov et al. 1997), pFAJ2600 (5.9 Kb) from R. erythropolis NI86/21 (De Mot et al. 1997), pAN12 (6.3 Kb) from R. erythropolis AN12 and pKNR01 (4.4 Kb) from R. opacus B4 are some of the small circular cryptic plasmids, described in the literature, that have been used to construct E. coli-Rhodococcus shuttle plasmids (Kostichka et al. 2003). The first two plasmids require the activity of two replication proteins, RepA and RepB, to replicate. As these two replication proteins resemble replication proteins of the theta-type replicating Mycobacterium plasmid pAL5000 (Ventura et al. 2007), pKA22 and pFAJ2600 are categorized as replicons of pAL5000 family. The plasmid pAN12 is of the pIJ101/pJV1 family, which usually replicates thanks to a single replication. Detection of ssDNA intermediates in several of the pIJ101/pJV1 family of plasmids suggested that they replicate by rolling circle mechanism (Kostichka et al. 2003).
5 Towards Genome-Scale Modification of Rhodococcus Through System and Synthetic Biology
Genetic manipulation (or genetic engineering) methods have been applied to Rhodococcusstrains in order to obtain new strains that express additional genetic properties to acquire new catabolic capacities (Xiong et al. 2012, 2016; Venkataraman et al. 2015; Rodrigues et al. 2001; Hirasawa et al. 2001), to perform promoter activity studies (van der Geize et al. 2008; Cappelletti et al. 2011) and to introduce genetic mutations or deletions to determine the phenotypic alterations and the function of a specific gene or gene cluster (van der Geize et al. 2008; Amara et al. 2016).
Genetic manipulation methods are based on the development of efficient Rhodococcus transformation procedure, as biochemical and genetic characteristics of this genus have for long time limited the genetic manipulation of this strain (Sallam et al. 2006; Cappelletti 2010). Although protoplast-mediated transformation methods have also been used for some Rhodococcus strains (Singer and Finnerty 1988; Duran 1998), the use of electroporation procedure showed the highest efficiency, and it is presently the method that is most widely used for Rhodococcus transformation (Shao et al. 1995; Sekizaki et al. 1998; Kalscheuer et al. 1999). Several genetic manipulation strategies during the last three decades led to the development of (1) E. coli-Rhodococcus shuttle vectors from cryptic plasmids of Rhodococcus strains (Kostichka et al. 2003; Na et al. 2005; Matsui et al. 2006), (2) expression vectors for heterologous gene expression and protein production (Kalscheuer et al. 1999; Nakashima and Tamura 2004a, b), (3) random mutagenesis methods using transposons or spontaneous illegitimate recombination (Desomer et al. 1991; Sallam et al. 2006; Crespi et al. 1994) and (4) targeted gene disruption methods based on unmarked mutagenesis deletion systems with SacB as counter-selection (van der Geize et al. 2001).
Recently, advances in genomics and genome editing offered new opportunities to engineer Rhodococcus in a directed and combinatorial manner through a genomic-scale rational design of the cell through system and synthetic biology approaches.
With the aim of developing synthetic biology platforms for Rhodococcus engineering, a first BioBrick™-compatible plasmid system was designed for R. opacus PD630 by Ellinger and Schmidt-Dannert (2017). Generally, the BioBrick™ toolbox allows convenient and reproducible assembly of multigene pathways into series of vectors and the possibility to quickly mobilize any cloned gene into vectors with different features for gene expression and protein purification (Vick et al. 2011). Currently, the majority of the available BioBricks were designed for the Gram-negative model organism Escherichia coli. However, recently, novel BioBrick™ tools have been designed for the engineering of other bacteria, e.g. Bacillus (Radeck et al. 2013). The first BioBrick™-compatible vector designed for a Rhodococcus strain pSRKBB derived from the backbone of the E. coli-Rhodococcus shuttle vector pSRK21, which was modified by removing all BioBrick™-incompatible restriction, while it retained the capacity to replicate in R. opacus PD630 (Ellinger and Schmidt-Dannert 2017) (Fig. 6). This BioBrick-compatible plasmid was demonstrated to enable robust heterologous protein expression in R. opacus PD630 from the lac promoter, giving a first demonstration of the use of Rhodococcus as platform for synthetic biology.

Diagram of BioBrick™-compatible vector pSRKBB for Rhodococcus. The enlarged region includes the lac promoter and the ribosome binding site (RBS) upstream of the multi-cloning site where coding sequences can be cloned for constitutive expression. Further details are provided by Ellinger and Schmidt-Dannert (2017)
Moreover, in order to expand the molecular toolkit for Rhodococcus genetic engineering, a recent work analysed the genetic tools to obtain gene expression control in R. opacus PD630 by characterizing a constitutive promoter library, optimizing antibiotic resistance markers and defining the copy numbers of different gene expression plasmids (DeLorenzo et al. 2018). The same work also reported methods for genome editing by using recombinase-based technique for site-specific genomic modifications and by describing the methodology to identify neutral integration sites for stable heterologous expression. Lastly, a tunable and targeted gene repression system was developed by utilizing for the first time in Rhodococcus the CRISPR interference method (DeLorenzo et al. 2018). Taken together, the heterologous gene expression tools along with the CRISPRi method open new prospective for the optimization of the bioconversion abilities of R. opacus and for the possible development of molecular tools for other Rhodococcus species with relevant environmental and/or industrial characteristics.
At the same time, genomic information have been used to develop genome-scale metabolic models, i.e. mathematical representations of the stoichiometry of the biochemical networks occurring in Rhodococcus cell. In this regard, an in silico genome-scale metabolic model (iMT1174) was developed to make metabolic predictions on the behaviour of R. jostii RHA1 in relation to the accumulation of three types of carbon storage (i.e. glycogen, polyhydroxyalkanoate and triacylglycerols), using different carbon sources (glucose or acetate) and under growth conditions typically occurring in activated sludge bioreactor systems for wastewater recovery (Tajparast et al. 2015, 2018). The simulations were compared with the experimentally measured metabolic fluxes through 13C-labelling assays, and the predictive capacity of the model was established. These works represent the first steps towards systems biology approaches enabling to simulate and predict Rhodococcus metabolic fluxes leading to the production of industrially interesting metabolites. More efforts are needed to extend system and synthetic biology tools for genome-scale engineering of Rhodococcus species different from the model ones, i.e. R. opacus PD630 and R. jostii RHA1, in order to get deep into the extraordinary physiological and metabolic diversity featuring different Rhodococcus strains and to provide new opportunities for their utilization in targeted bioconversion and biodegradation processes.
6 Conclusions
Due to the biotechnological significance of this genus, a dramatic increase of sequenced Rhodococcus genomes has been published and become available in the database. The comparative and functional analyses of these genome data provided the opportunity to get insight into the genetic basis of the extraordinary metabolic diversity and versatility of Rhodococcus genus members. The high versatility and adaptability of members of Rhodococcus was found to be the reflection of the size and complexity of their genomes. The co-existence of linear and circular replicons (chromosome and plasmids) and the presence of large catabolic plasmids have strong implications in rhodococcal genome plasticity, fluidity and metabolic diversity. Further, the genetic and functional redundancy is a general characteristic of Rhodococcus genus, which supports metabolic robustness and versatility towards the degradation of different classes of substrates and organic compounds. The specific type of genomic redundancy was found to reflect the peculiar niche adaptation need of each Rhodococcus strain, and, at least in part, it was related to the size of the genome.
Lastly, advances in Rhodococcus genomics and genome editing provided new opportunities for the application of directed and combinatorial approaches to Rhodococcus engineering. Recent breakthroughs in genetic engineering of Rhodococus have included the use of synthetic biology platforms and new approaches for genome editing (CRISPR/Cas9 tool) of R. opacus PD630. Systems biology approaches have also been applied to R. jostii RHA1 to develop genome-scale metabolic models to predict cell metabolic fluxes for the production of relevant metabolites and storage compounds. Despite more efforts are needed to expand system and synthetic biology tools for genome-scale engineering of Rhodococcus species different from the model ones, i.e. R. opacus PD630 and R. jostii RHA1, the novel molecular toolkits along with the extended knowledge on genomic and functional genomics have largely contributed in making Rhodococcus strains relevant for future novel industrial applications.
References
Ali A (2013) Microbial comparative genomics: an overview of tools and insights into the genus Corynebacterium. J Bacteriol Parasitol 04:1–16. https://doi.org/10.4172/2155-9597.1000167
Alvarez HM, Mayer F, Fabritius D et al (1996) Formation of intracytoplasmic lipid inclusions by Rhodococcus opacus strain PD630. Arch Microbiol 165:377–386
Amara S, Seghezzi N, Otani H et al (2016) Characterization of key triacylglycerol biosynthesis processes in rhodococci. Sci Rep 6:24985. https://doi.org/10.1038/srep24985
Aminov RI (2011) Horizontal gene exchange in environmental microbiota. Front Microbiol 2:158. https://doi.org/10.3389/fmicb.2011.00158
Anastasi E, MacArthur I, Scortti M (2016) Pangenome and phylogenomic analysis of the pathogenic actinobacterium Rhodococcus equi. Genome Biol Evol 8(10):3140–3148
Bentley SD, Chater KF, Cerdeño-Tárraga AM et al (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417(6885):141–147
Cappelletti M (2010) Monooxygenases involved in the n-alkanes metabolism by Rhodococcus sp. BCP1. PhD Thesis Dissertation, University of Bologna
spiepr A3B2 twb=.25w?>Cappelletti M, Fedi S, Frascari D et al (2011) Analyses of both the alkB gene transcriptional start site and alkB promoter-inducing properties of Rhodococcus sp. strain BCP1 grown on n-alkanes. Appl Environ Microbiol 77:1619–1627
Cappelletti M, Frascari ZD et al (2012) Microbial degradation of chloroform. Appl Microbiol Biotechnol 96:1395–1409
spiepr A3B2 tlsb=-.02w?>Cappelletti M, Di Gennaro P, D’Ursi P et al (2013) Genome sequence of Rhodococcus sp. strain BCP1, a biodegrader of alkanes and chlorinated compounds. Genome Announc 1(5):e00657–e00613. https://doi.org/10.1128/genomeA.00657-13
Cappelletti M, Presentato A, Milazzo G et al (2015) Growth of Rhodococcus sp. strain BCP1 on gaseous n-alkanes: new metabolic insights and transcriptional analysis of two soluble di-iron monooxygenase genes. Front Microbiol 6:393. https://doi.org/10.3389/fmicb.2015.00393
Cappelletti M, Fedi S, Zampolli J et al (2016) Phenotype microarray analysis may unravel genetic determinants of the stress response by Rhodococcus aetherivorans BCP1 and Rhodococcus opacus R7. Res Microbiol 167:1–9
Cappelletti M, Pinelli D, Fedi S (2017) Aerobic co-metabolism of 1,1,2,2-tetrachloroethane by Rhodococcus aetherivorans TPA grown on propane: kinetic study and bioreactor configuration analysis. J Chem Technol Biotechnol 93:155–165
Carroll AC, Wong A (2018) Plasmid persistence: costs, benefits, and the plasmid paradox. Can J Microbiol 64:293–304
Castro AR, Rocha I, Alves MM et al (2016) Rhodococcus opacus B4: a promising bacterium for production of biofuels and biobased chemicals. AMB Express 6:35. https://doi.org/10.1186/s13568-016-0207-y
Ceniceros A, Dijkhuizen L, Petrusma M et al (2017) Genome-based exploration of the specialized metabolic capacities of the genus Rhodococcus. BMC Genomics 18:593. https://doi.org/10.1186/s12864-017-3966-1
Chen CW, Huang CH, Lee HH et al (2002) Once the circle has been broken: dynamics and evolution of Streptomyces chromosomes. Trends Genet 18:522–529
Chen Y, Ding Y, Yang L et al (2014) Integrated omics study delineates the dynamics of lipid droplets in Rhodococcus opacus PD630. Nucleic Acids Res 42(2):1052–1064
Choi KY, Kim D, Sul WJ et al (2005) Molecular and biochemical analysis of phthalate and terephthalate degradation by Rhodococcus sp. strain DK17. FEMS Microbiol Lett 252:207–213
Choi KY, Kim D, Chae JC et al (2007) Requirement of duplicated operons for maximal metabolism of phthalate by Rhodococcus sp. strain DK17. Biochem Biophys Res Commun 357:766–771
Ciavarelli R, Cappelletti M, Fedi S et al (2012) Chloroform aerobic cometabolism by butane-growing Rhodococcus aetherovorans BCP1 in continuous-flow biofilm reactors. Bioprocess Biosyst Eng 35:667–681
Costa JSD, Herrero OM, Alvarez HM et al (2015) Label-free and redox proteomic analyses of the triacylglycerol-accumulating Rhodococcus jostii RHA1. Microbiology 161:593–610
Creason AL, Vandeputte OM, Savory EA et al (2014a) Analysis of genome sequences from plant pathogenic Rhodococcus reveals genetic novelties in virulence loci. PLoS One 9(7):e101996. https://doi.org/10.1371/journal.pone.0101996
Creason AL, Davis EW II, Putnam ML et al (2014b) Use of whole genome sequences to develop a molecular phylogenetic framework for Rhodococcus fascians and the Rhodococcus genus. Front Plant Sci 5:406. https://doi.org/10.3389/fpls.2014.00406
Crespi M, Vereecke D, Temmerman W et al (1994) The fas operon of Rhodococcus fascians encodes new genes required for efficient fasciation of host plants. J Bacteriol 176:2492–2501
Crombie TA, Rhodius VA, Miller MC et al (2015) Regulation of plasmid-encoded isoprene metabolism in Rhodococcus, a representative of an important link in the global isoprene cycle. Environ Microbiol 17:3314–3329
de Carvalho CCCR, da Fonseca MMR (2005) The remarkable Rhodococcus erythropolis. Appl Microbiol Biotechnol 67:715–726
de Carvalho CCCR, Costa SS, Fernandes P et al (2014) Membrane transport systems and the biodegradation potential and pathogenicity of genus Rhodococcus. Front Physiol 5:133. https://doi.org/10.3389/fphys.2014.00133
spiepr A3B2 tlsb=-.02w?>De Mot R, Nagy I, De Schrijver A et al (1997) Structural analysis of the 6 kb cryptic plasmid pFAJ2600 from Rhodococcus erythropolis NI86/21 and construction of Escherichia coli – Rhodococcus shuttle vectors. Microbiology 143:3137–3147
DeLorenzo DM, Rottinghaus AG, Henson WR et al (2018) Molecular toolkit for gene expression control and genome modification in Rhodococcus opacus PD630. ACS Synth Biol 7(2):727–738
Denis-Larose C, Labbe D, Bergeron H et al (1997) Conservation of plasmid-encoded dibenzothiophene desulfurization genes in several rhodococci. Appl Environ Microbiol 63:2915–2919
Desomer J, Crespi M, Van Montagu M (1991) Illegitimate integration of non-replicative vectors in the genome of Rhodococcus fascians upon electrotransformation as an insertional mutagenesis system. Mol Microbiol 5:2115–2124
Di Canito A, Zampolli J, Orro A et al (2018) Genome-based analysis for the identification of genes involved in o-xylene degradation in Rhodococcus opacus R7. BMC Genomics 19:587
Di Gennaro P, Rescalli E, Galli E et al (2001) Characterization of Rhodococcus opacus R7, a strain able to degrade naphthalene and o-xylene isolated from polycyclic aromatic hydrocarbon-contaminated soil. Res Microbiol 152:641–651
Di Gennaro P, Terreni P, Masi G et al (2010) Identification and characterization of genes involved in naphthalene degradation in Rhodococcus opacus R7. Appl Microbiol Biotechnol 87(1):297–308
Di Gennaro P, Zampolli J, Presti I et al (2014) Genome sequence of Rhodococcus opacus strain R7, a biodegrader of mono- and polycyclic aromatic hydrocarbons. Genome Announc 2(4):e00827–e00814. https://doi.org/10.1128/genomeA.00827-14
Dib JR, Wagenknecht M, Farías ME et al (2015) Strategies and approaches in plasmidome studies—uncovering plasmid diversity disregarding of linear elements? Front Microbiol 6:463. https://doi.org/10.3389/fmicb.2015.00463
Dodge AG, Wackett LP, Sadowsky MJ (2011) Plasmid localization and organization of melamine degradation genes in Rhodococcus sp strain Mel. Appl Environ Microbiol 78(5):1397–1403. https://doi.org/10.1128/AEM.06468-11
Dueholm MS, Albertsen M, D’Imperio S (2014) Complete genome of Rhodococcus pyridinivorans SB3094, a methyl-ethyl-ketone-degrading bacterium used for bioaugmentation. Genome Announc 2(3):e00525-14. https://doi.org/10.1128/genomeA.00525-14
Duran R (1998) New shuttle vectors for Rhodococcus sp. R312 (formerly Brevibacterium sp. R312), a nitrile hydratase producing strain. J Basic Microbiol 38:101–106
Ellinger J, Schmidt-Dannert C (2017) Construction of a BioBrick™ compatible vector system for Rhodococcus. Plasmid 90:1–4. https://doi.org/10.1016/j.plasmid.2017.01.004
Fang H, Xu T, Cao D et al (2016) Characterization and genome functional analysis of a novel metamitron-degrading strain Rhodococcus sp. MET via both triazinone and phenyl rings cleavage. Sci Rep 6:32339. https://doi.org/10.1038/srep32339
Francis I, De Keyser A, De Backer P et al (2012) pFiD188, the linear virulence plasmid of Rhodococcus fascians D188. MPMI 25:637–647
Francis IM, Stes E, Zhang Y et al (2016) Mining the genome of Rhodococcus fascians, a plant growth-promoting bacterium gone astray. New Biotechnol 33:706–717
Frascari D, Pinelli D, Nocentini M et al (2006) Chloroform degradation by butane-grown cells of Rhodococcus aetherovorans BCP1. Appl Microbiol Biotechnol 73(2):421–428
Fukuda M, Shimizu S, Okita N et al (1998) Structural alteration of linear plasmids encoding the genes for polychlorinated biphenyl degradation in Rhodococcus strain RHA1. Antonie Leeuwenhoek 74:169–173
Gao B, Gupta RS (2012) Phylogenetic framework and molecular signatures for the main clades of the phylum Actinobacteria. Microbiol Mol Biol Rev 76(1):66–112. https://doi.org/10.1128/MMBR.05011-11
Gonçalves ER, Hara H, Miyazawa D et al (2006) Transcriptomic assessment of isozymes in the biphenyl pathway of Rhodococcus sp. strain RHA1. Appl Environ Microbiol 72:6183–6193
Goodfellow M, Alderson G, Chun J (1998) Rhodococcal systematics: problems and developments. Antonie Van Leeuwenhoek 74:3–20
Goordial J, Raymond-Bouchard I, Zolotarov Y et al (2016) Cold adaptive traits revealed by comparative genomic analysis of the eurypsychrophile Rhodococcus sp. JG3 isolated from high elevation McMurdo Dry Valley permafrost, Antarctica. FEMS Microbiol Ecol 92. https://doi.org/10.1093/femsec/fiv154
Goris J, Konstantinidis KT, Klappenbach JA (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91
Gravouil K, Ferru-Clément R, Colas S et al (2017) Transcriptomics and lipidomics of the environmental strain Rhodococcus ruber point out consumption pathways and potential metabolic bottlenecks for polyethylene degradation. Environ Sci Technol 51:5172–5181. https://doi.org/10.1021/acs.est.7b00846
Grӧning JAD, Eulberg D, Tischler D et al (2014) Gene redundancy of two-component (chloro)phenol hydroxylases in Rhodococcus opacus 1CP. FEMS Microbiol Lett 361:68–75
Grzeszik C, Lubbers M, Reh M et al (1997) Genes encoding the NAD-reducing hydrogenase of Rhodococcus opacus MR11. Microbiology 143:1271–1286
Guo C, Wu ZL (2017) Construction and functional analysis of a whole-cell biocatalyst based on CYP108N7. Enzym Microb Technol 106:28–34
Gürtler V, Mayall BC, Seviour R (2004) Can whole genome analysis refine the taxonomy of the genus Rhodococcus? FEMS Microbiol Rev 28:377–403
Hara H, Eltis LD, Davies JE et al (2007) Transcriptomic analysis reveals a bifurcated terephthalate degradation pathway in Rhodococcus sp. strain RHA1. J Bacteriol 189:1641–1647
Hernández MA, Mohn WW, Martínez E et al (2008) Biosynthesis of storage compounds by Rhodococcus jostii RHA1 and global identification of genes involved in their metabolism. BMC Genomics 9:600. https://doi.org/10.1186/1471-2164-9-600
Hirasawa K, Ishii Y, Kobayashi M et al (2001) Improvement of desulfurization activity in Rhodococcus erythropolis KA2-5-1 by genetic engineering. Biosci Biotechnol Biochem 65:239–246
Holder JW, Ulrich JC, DeBono AC et al (2011) Comparative and functional genomics of Rhodococcus opacus PD630 for biofuels development. PLoS Genet 7(9):e1002219. https://doi.org/10.1371/journal.pgen.1002219
Honda K, Yamashita S, Nakagawa H et al (2008) Stabilization of water-in-oil emulsion by Rhodococcus opacus B-4 and its application to biotransformation. Appl Microbiol Biotechnol 78:767–773
Honda K, Imura M, Okano K et al (2012) Identification of replication region of a 111-kb circular plasmid from Rhodococcus opacus B-4 by λ red recombination-based deletion analysis. Biosci Biotechnol Biochem 76:1758–1764
Hori K, Kobayashi A, Ikeda H et al (2009) Rhodococcus aetherivorans IAR1, a new bacterial strain synthesizing poly(3-hydroxybutyrate-co-3-hydroxyvalerate) from toluene. J Biosci Bioeng 107:145–150
Hülter N, Ilhan J, Wein T et al (2017) An evolutionary perspective on plasmid lifestyle modes. Curr Opin Microbiol 38:74–80
Irvine VA, Kulakov LA, Larkin MJ (2000) The diversity of extradiol dioxygenase (edo) genes in cresol degrading rhodococci from a creosote-contaminated site that express a wide range of degradative abilities. Antonie Van Leeuwenhoek 78:341–352
Iwasaki T, Miyauchi K, Masai E et al (2006) Multiple-subunit genes of the aromatic-ring-hydroxylating dioxygenase play an active role in biphenyl and polychlorinated biphenyl degradation in Rhodococcus sp. strain RHA1. Appl Environ Microbiol 72:5396–5402
Iwasaki T, Takeda H, Miyauchi K et al (2007) Characterization of two biphenyl dioxygenases for biphenyl/PCB degradation in a PCB degrader, Rhodococcus sp. strain RHA1. Biosci Biotechnol Biochem 71:993–1002
Kalkus J, Dorrie C, Fischer D et al (1993) The giant linear plasmid pHG207 from Rhodococcus sp. encoding hydrogen autotrophy: characterization of the plasmid and its termini. J Gen Microbiol 139:2055–2065
Kalscheuer R, Arenskötter M, Steinbüchel A (1999) Establishment of a gene transfer system for Rhodococcus opacus PD630 based on electroporation and its application for recombinant biosynthesis of poly(3-hydroxyalkanoic acids). Appl Microbiol Biotechnol 52:508–515
Khairy H, Meinert C, Wübbeler JH et al (2016) Genome and proteome analysis of Rhodococcus erythropolis MI2: elucidation of the 4,4′-dithiodibutyric acid catabolism. PLoS One 11(12):e0167539. https://doi.org/10.1371/journal.pone.0167539
Kikuchi Y, Hirai K, Gunge N et al (1985) Hairpin plasmid-a novel linear DNA of perfect hairpin structure. EMBO J 4:1881–1886
Kim D, Kim YS, Kim SK et al (2002) Monocyclic aromatic hydrocarbon degradation by Rhodococcus sp. strain DK17. Appl Environ Microbiol 6:3270–3278
Kim D, Choi KY, Yoo M (2018) Biotechnological potential of Rhodococcus biodegradative pathways. J Microbiol Biotechnol 28(7):1037–1051
Konig C, Eulberg D, Groning J et al (2004) A linear megaplasmid, p1CP, carrying the genes for chlorocatechol catabolism of Rhodococcus opacus 1CP. Microbiology 150:3075–3087
Konishi M, Nishi S, Fukuoka T et al (2014) Deep-sea Rhodococcus sp. BS-15, lacking the phytopathogenic fas genes, produces a novel glucotriose lipid biosurfactant. Mar Biotechnol 16:484–493
Konstantinidis KT, Tiedje JM (2005) Towards a genome-based taxonomy for prokaryotes. J Bacteriol 187:6258–6264. https://doi.org/10.1128/JB.187.18.6258-6264.2005
Kosono S, Maeda M, Fuji F et al (1997) Three of the seven bphC genes of Rhodococcus erythropolis TA421, isolated from a termite ecosystem, are located on an indigenous plasmid associated with biphenyl degradation. Appl Environ Microbiol 63:3282–3285
Kostichka K, Tao L, Bramucci M et al (2003) A small cryptic plasmid from Rhodococcus erythropolis: characterization and utility for gene expression. Appl Microbiol Biotechnol 62:61–68
Kulakov LA, Larkin MJ (2002) Genomic organization of Rhodococcus. In: Danchin A (ed) Genomics of GC-rich gram-positive bacteria. Caister Academic Press, Norfolk, pp 15–46
Kulakov LA, Larkin MJ, Kulakova AN (1997) Cryptic plasmid pKA22 isolated from the naphthalene degrading derivative of Rhodococcus rhodochrous NCIMB13064. Plasmid 38:61–69
Kulakov LA, Chen S, Allen CC et al (2005) Web-type evolution of Rhodococcus gene clusters associated with utilization of naphthalene. Appl Environ Microbiol 71:1754–1764
Kulakova AN, Stafford TM, Larkin MJ et al (1995) Plasmid pRTL1 controlling 1-chloroalkane degradation by Rhodococcus rhodochrous NCIMB13064. Plasmid 33:208–217
Kwasiborski A, Mondy S, Teik-Min C et al (2015) Core genome and plasmidome of the quorum-quenching bacterium Rhodococcus erythropolis. Genetica 143:253–261
Laczi K, Kis A, Horváth B et al (2015) Metabolic responses of Rhodococcus erythropolis PR4 grown on diesel oil and various hydrocarbons. Appl Microbiol Biotechnol 99:9745–9759
Larkin MJ, DeMot R, Kulakov LA et al (1998) Applied aspects of Rhodococcus genetics. Antonie Van Leeuwenhoek 74:133–153
Larkin MJ, Kulakov LA, Allen CCR (2010) Genomes and plasmids in Rhodococcus. In: Alvarez HM (ed) Biology of Rhodococcus. Springer, Berlin, pp 73–90
LeBlanc JC, Gonçalves ER, Mohn WW (2008) Global response to desiccation stress in the soil actinomycete Rhodococcus jostii RHA1. Appl Environ Microbiol 74:2627–2636
Letek M, González P, MacArthur I et al (2010) The genome of a pathogenic Rhodococcus: cooptive virulence underpinned by key gene acquisitions. PLoS Genet 6(9):e1001145. https://doi.org/10.1371/journal.pgen.1001145
Martìnkovà L, Uhnàkovà B, Pàtek M et al (2009) Biodegradation potential of the genus Rhodococcus. Environ Int 35:162–177
Matsui T, Saeki H, Shinzato N et al (2006) Characterization of Rhodococcus – E. coli shuttle vector pNC9501 constructed from the cryptic plasmid of a propene-degrading bacterium. Curr Microbiol 52:445–448
McLeod MP, Warren RL, Hsiao WW et al (2006) The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc Natl Acad Sci U S A 103(42):15582–15587
Meinhardt F, Schaffrath R, Larsen M (1997) Microbial linear plasmids. Appl Microbiol Biotechnol 47:329–336
Moran NA (2002) Microbial minimalism: genome reduction in bacterial pathogens. Cell 108:583–586
Na K, Kuroda A, Takiguchia N (2005) Isolation and characterization of benzene-tolerant Rhodococcus opacus strains. J Biosci Bioeng 99:378–382
Nakashima N, Tamura T (2004a) A novel system for expressing recombinant proteins over a wide temperature range from 4 to 35°C. Biotechnol Bioeng 86:136–148
Nakashima N, Tamura T (2004b) Isolation and characterization of a rolling-circle-type plasmid from Rhodococcus erythropolis and application of the plasmid to multiple-recombinant-protein expression. Appl Environ Microbiol 70:5557–5568
National Center for Biotechnology Information (NCBI) (2018) U.S. National Library of Medicine, Rockville Pike. www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=1827 Accessed July 2018
Orro A, Cappelletti M, D’Ursi P et al (2015) Genome and phenotype microarray analyses of Rhodococcus sp. BCP1 and Rhodococcus opacus R7: genetic determinants and metabolic abilities with environmental relevance. PLoS One 10(10):e0139467. https://doi.org/10.1371/journal.pone.0139467
Pathak A, Chauhan A, Blom J et al (2016) Comparative genomics and metabolic analysis reveals peculiar characteristics of Rhodococcus opacus strain M213 particularly for naphthalene degradation. PLoS One 17:1–32
Patrauchan MA, Florizone C, Dosanjh M et al (2005) Catabolism of benzoate and phthalate in Rhodococcus sp. strain RHA1: redundancies and convergence. J Bacteriol 187:4050–4063
Patrauchan MA, Florizone C, Eapen S et al (2008) Roles of ring-hydroxylating dioxygenases in styrene and benzene catabolism in Rhodococcus jostii RHA1. J Bacteriol 190(1):37–47
Patrauchan MA, Miyazawa D, LeBlanc JC et al (2012) Proteomic analysis of survival of Rhodococcus jostii RHA1 during carbon starvation. Appl Environ Microbiol 78(18):6714–6725
Pérez-Pérez JM, Candela H, Micol JL (2009) Understanding synergy in genetic interactions. Trends Genet 8:368–376
Petrusma M, Hessels G, Dijkhuizen L et al (2011) Multiplicity of 3-ketosteroid-9α-hydroxylase enzymes in Rhodococcus rhodochrous DSM43269 for specific degradation of different classes of steroids. J Bacteriol 193(15):3931–3940. https://doi.org/10.1128/JB.00274-11
Presentato A, Piacenza E, Anikovskiy M et al (2016) Rhodococcus aetherivorans BCP1 as cell factory for the production of intracellular tellurium nanorods under aerobic conditions. Microb Cell Factories 15:204. https://doi.org/10.1186/s12934-016-0602-8
Presentato A, Cappelletti M, Sansone A et al (2018a) Aerobic growth of Rhodococcus aetherivorans BCP1 using selected naphthenic acids as the sole carbon and energy sources. Front Microbiol 9:672. https://doi.org/10.3389/fmicb.2018.00672
Presentato A, Piacenza E, Anikovskiy M et al (2018b) Biosynthesis of selenium-nanoparticles and -nanorods as a product of selenite bioconversion by the aerobic bacterium Rhodococcus aetherivorans BCP1. New Biotechnol 41:1–8
Presentato A, Piacenza E, Darbandi A et al (2018c) Assembly, growth and conductive properties of tellurium nanorods produced by Rhodococcus aetherivorans BCP1. Sci Rep 8:3923. https://doi.org/10.1038/s41598-018-22320-x
Priefert H, O’Brien XM, Lessard PA et al (2004) Indene bioconversion by a toluene inducible dioxygenase of Rhodococcus sp. I24. Appl Microbiol Biotechnol 65:168–176
Puglisi E, Cahill MJ, Lessard PA et al (2010) Transcriptional response of Rhodococcus aetherivorans I24 to polychlorinated biphenyl-contaminated sediments. Microb Ecol 60(3):505–515. https://doi.org/10.1007/s00248-010-9650-5
Qu J, Miao L-L, Liu Y et al (2015) Complete genome sequence of Rhodococcus sp. Strain IcdP1 shows diverse catabolic potential. Genome Announc 3(4):e00711-15. https://doi.org/10.1128/genomeA.00711-15
Radeck J, Kraft K, Bartels J (2013) The Bacillus BioBrick Box: generation and evaluation of essential genetic building blocks for standardized work with Bacillus subtilis. J Biol Eng 7:29
Redenbach M, Altenbuchner J (2002) Why do some bacteria have linear chromosomes and plasmids. BIOspektrum 8:158–163
Richter M, Rosselló-Móra R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131
Rodrigues JLM, Maltseva OV, Tsoi TV et al (2001) Development of a Rhodococcus recombinant strain for degradation of products from anaerobic dechlorination of PCBs. Environ Sci Technol 35:663–668
Sallam KI, Mitani Y, Tamura T (2006) Construction of random transposition mutagenesis system in Rhodococcus erythropolis using IS1415. J Biotechnol 121:13–22
Sameshima Y, Honda K, Kato J et al (2008) Expression of Rhodococcus opacus alkB genes in anhydrous organic solvents. J Biosci Bioeng 106:199–203
Sangal V, Jones AL, Goodfellow M et al (2014) Comparative genomic analyses reveal a lack of a substantial signature of host adaptation in Rhodococcus equi (“Prescottella equi”). Pathog Dis 71:352–356
Sekine M, Tanikawa S, Omata S et al (2006) Sequence analysis of three plasmids harboured in Rhodococcus erythropolis strain PR4. Environ Microbiol 8:334–346
Sekizaki T, Tanoue T, Osaki M (1998) Improved electroporation of Rhodococcus equi. J Vet Med Sci 60:277–279
Shao Z, Dick WA, Behki RM (1995) An improved Escherichia coli – Rhodococcus shuttle vector and plasmid transformation in Rhodococcus spp. using electroporation. Lett Appl Microbiol 21:261–266
Sheng HM, Gao HS, Xue LG et al (2011) Analysis of the composition and characteristics of culturable endophytic bacteria within subnival plants of the Tianshan Mountains, northwestern China. Curr Microbiol 62:923–932
Shevtsov A, Tarlykov P, Zholdybayeva E et al (2013) Draft genome sequence of Rhodococcus erythropolis DN1, a crude oil biodegrader. Genome Announc 1:e00846–e00813. https://doi.org/10.1128/genomeA.00846-13
Shimizu S, Kobayashi H, Masai E et al (2001) Characterization of the 450-kb linear plasmid in a polychlorinated biphenyl degrader, Rhodococcus sp. strain RHA1. Appl Environ Microbiol 67:2021–2028
Singer ME, Finnerty WR (1988) Construction of an Escherichia coli–Rhodococcus shuttle vector and plasmid transformation in Rhodococcus spp. J Bacteriol 170:638–645
Swain K, Casabon I, Eltis LD et al (2012) Two transporters essential for the reassimilation of novel cholate metabolites by Rhodococcus jostii RHA1. J Bacteriol 194(24):6720–6727
Szőköl J, Rucká L, Šimčíková M et al (2014) Induction and carbon catabolite repression of phenol degradation genes in Rhodococcus erythropolis and Rhodococcus jostii. Appl Microbiol Biotechnol 98:8267–8279
Taguchi K, Motoyama M, Kudo T (2004) Multiplicity of 2, 3-dihydroxybiphenyl dioxygenase genes in the Gram-positive polychlorinated biphenyl degrading bacterium Rhodococcus rhodochrous K37. Biosci Biotechnol Biochem 68:787–795
Taguchi K, Motoyama M, Iida T et al (2007) Polychlorinated biphenyl/biphenyl degrading gene clusters in Rhodococcus sp. K37, HA99, and TA431 are different from well-known bph gene clusters of rhodococci. Biosci Biotechnol Biochem 71:1136–1144
Tajparast M, Frigon D (2015) Genome-scale metabolic model of Rhodococcus jostii RHA1 (iMT1174) to study the accumulation of storage compounds during nitrogen-limited condition. BMC Syst Biol 9:43. https://doi.org/10.1186/s12918-015-0190-y
Tajparast M, Frigon D, Virolle MJ (2018) Predicting the accumulation of storage compounds by Rhodococcus jostii RHA1 in the feast-famine growth cycles using genome-scale flux balance analysis. PLoS One 13:e0191835. https://doi.org/10.1371/journal.pone.0191835
Takeda H, Shimodaira J, Yukawa K et al (2010) Dual two-component regulatory systems are involved in aromatic compound degradation in a polychlorinated-biphenyl degrader, Rhodococcus jostii RHA1. J Bacteriol 192:4741–4751
Takei D, Washio K, Morikawa M (2008) Identification of alkane hydroxylase genes in Rhodococcus sp. strain TMP2 that degrades a branched alkane. Biotechnol Lett 30:1447–1452
Taketani RG, Zucchi TD, Soares de Melo I et al (2013) Whole-genome shotgun sequencing of Rhodococcus erythropolis strain P27, a highly radiation-resistant actinomycete from Antarctica. Genome Announc 1(5):e00763–e00713. https://doi.org/10.1128/genomeA.00763-13
Táncsics A, Benedek T, Szoboszlay S et al (2015) The detection and phylogenetic analysis of the alkane 1-monooxygenase gene of members of the genus Rhodococcus. Syst Appl Microbiol 38:1–7
Tao F, Zhao P, Li Q et al (2011) Genome sequence of Rhodococcus erythropolis XP, a biodesulfurizing bacterium with industrial potential. J Bacteriol 193:6422–6423
Tomás-Gallardo L, Canosa I, Santero E et al (2006) Proteomic and transcriptional characterization of aromatic degradation pathways in Rhodococcus sp. strain TFB. Proteomics 6:S119–S132. https://doi.org/10.1002/pmic.200500422
Treadway SL, Yanagimachi KS, Lankenau E (1999) Isolation and characterization of indene bioconversion genes from Rhodococcus strain I24. Appl Microbiol Biotechnol 51:786–793
Valero-Rello A, Hapeshi A, Anastasi E et al (2015) An invertron-like linear plasmid mediates intracellular survival and virulence in bovine isolates of Rhodococcus equi. Infect Immun 83:2725–2737. https://doi.org/10.1128/IAI.00376-15
Van Beilen JB, Smits THM, Whyte LG et al (2002) Alkane hydroxylase homologues in gram-positive strains. Environ Microbiol 4:676–682
Van der Geize, Dijkhuize L (2004) Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr Opin Microbiol 7:255–261
Van der Geize R, Hessels GI, van Gerwen R (2001) Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid Δ1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counters electable marker. FEMS Microbiol Lett 205:197–202
Van der Geize R, Yam K, Heuser T et al (2007) A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A 104:1947–1952
Van der Geize R, de Jong W, Hessels GI et al (2008) A novel method to generate unmarked gene deletions in the intracellular pathogen Rhodococcus equi using 5-fluorocytosine conditional lethality. Nucleic Acids Res 36:e151
Venkataraman H, Evelien MP, Rosłoniec KZ et al (2015) Biosynthesis of a steroid metabolite by an engineered Rhodococcus erythropolis strain expressing a mutant cytochrome P450 BM3 enzyme. Appl Microbiol Biotechnol 99:4713–4721
Ventura M, Canchaya C, Tauch A et al (2007) Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Biol Rev 71:495–548
Vick J, Johnson E, Choudhary S et al (2011) Optimized compatible set of BioBrick™ vectors for metabolic pathway engineering. Appl Microbiol Biotechnol 92:1275–1286
Villalba MS, Hernandéz MA, Silva RA et al (2013) Genome sequences of triacylglycerol metabolism in Rhodococcus as a platform for comparative genomics. J Mol Biochem 2:94–105
Volff JN, Altenbuchner J (2000) A new beginning with new ends: linearization of circular chromosomes during bacterial evolution. FEMS Microbiol Lett 186:143–150. https://doi.org/10.1111/j.1574-6968.2000.tb09095.x
Voss I, Steinbuchel A (2001) High cell density cultivation of Rhodococcus opacus for lipid production at a pilot-plant scale. Appl Microbiol Biotechnol 55:547–555
Warren R, Hsiao WW, Kudo H et al (2004) Functional characterization of a catabolic plasmid from polychlorinated-biphenyl degrading Rhodococcus sp. strain RHA1. J Bacteriol 186:7783–7795
Watcharakul S, Röther W, Birke J (2016) Biochemical and spectroscopic characterization of purified Latex Clearing Protein (Lcp) from newly isolated rubber degrading Rhodococcus rhodochrous strain RPK1 reveals novel properties of Lcp. BMC Microbiol 16:92. https://doi.org/10.1186/s12866-016-0703-x
Whyte LG, Smits THM, Labbe’ D et al (2002) Gene cloning and characterization of multiple alkane hydroxylase systems in Rhodococcus strains Q15 and NRRL B-16531. Appl Environ Microbiol 68:5933–5942
Xiong X, Wang X, Chen S (2012) Engineering of a xylose metabolic pathway in Rhodococcus strains. Appl Environ Microbiol 78(16):5483–5491. https://doi.org/10.1128/AEM.08022-11
Xiong X, Lian J, Yu X et al (2016) Engineering levoglucosan metabolic pathway in Rhodococcus jostii RHA1 for lipid production. J Ind Microbiol Biotechnol 43:1551–1560
Xu JL, He J, Wang ZC et al (2007) Rhodococcus qingshengii sp. nov., a carbendazim-degrading bacterium. Int J Syst Evol Microbiol 57:2754–2757
Zampolli J, Collina E, Lasagni M et al (2014) Biodegradation of variable-chain-length n-alkanes in Rhodococcus opacus R7 and the involvement of an alkane hydroxylase system in the metabolism. AMB Express 4:73
Zhang J (2012) Genetic redundancies and their evolutionary maintenance. Evol Syst Biol 751:279–300
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Cappelletti, M., Zampolli, J., Di Gennaro, P., Zannoni, D. (2019). Genomics of Rhodococcus . In: Alvarez, H. (eds) Biology of Rhodococcus. Microbiology Monographs, vol 16. Springer, Cham. https://doi.org/10.1007/978-3-030-11461-9_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-11461-9_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11460-2
Online ISBN: 978-3-030-11461-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)