
Abstract
After the first ten years of developing the large scale convective properties of larger and larger storage containers for single component cryogenic liquids of nitrogen, oxygen, hydrogen and helium, we started to be involved with multi-component cryogenic liquids and the associated industrial problems being widely met. The starting point was liquid air as a binary mixture of nitrogen and oxygen, with differing vapour and liquid compositions during their distillation by boiling. It was discovered that under low loss storage with surface evaporation, and no boiling, the differing compositions were much larger. For example the vapour from liquid air was not 79% but almost pure nitrogen, and dangerously unable to support life; while the liquid suffered oxygen enrichment with rapid increase in fire risk. Storage of liquid air is therefore unsafe as a cheap source of refrigeration at 79 K. Then we met rollover, when the liquid mixture in a storage container suddenly suffers from a boil-off rate rising to a large unexpected peak, lasting minutes or hours. Studies led to the finding that this behaviour arises from stratification, via blocking of boil-off via surface evaporation and leading to thermal overfill. This chapter discusses this mechanism and describes a number of ways for preventing stratification and rollover. Experiments with propane and butane mixing led to the discovery that mixing two liquids, which is an irreversible thermodynamic process, is also path dependent with significant differences in the large accompanied boil-offs, or vapour flashes. When hot liquid is mixed into cold liquid, the smaller boil-offs are about one half that of adding cold liquid to hot. Finally, the solubility of many components in cryogenic liquids are small but finite, in the range of 1–1000 ppm. These solubilities can decrease rapidly with decreasing temperature, leading to deposits blocking pipes and heat exchanges. One example is the problem of water in jet fuel below −40 °C, which has now been solved by recommendations of the UK Air Accident Investigation Branch in conjunction with the University of Southampton.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


5.1 Differences Between Single-Component and Multi-component Liquids
Previous chapters have all been concerned with single component liquids. We have seen how non-equilibrium states of superheating in the liquid are necessary for the surface evaporation mechanism and how they have a significant bearing on evaporation instabilities. Furthermore, the superheating can lead to density stratification with a layer of hotter, less dense liquid sitting on colder, more dense liquid in a stable fashion.
In real life, most cryogenic liquids are multi-component mixtures and can be expected to have more complicated evaporation behaviours than a single component liquid. Examples of cryogenic liquid mixtures include:
-
Liquefied air, which contains oxygen, nitrogen, argon, and traces of water, carbon dioxide and hydrocarbons,
-
Liquid natural gases, LNG , which contain nitrogen, methane, ethane, and smaller quantities of carbon dioxide and higher hydrocarbons,
-
Liquefied petroleum gases, LPG, which contain propane, butane and other hydrocarbons,
-
Industrially produced mixtures, such as synthetic natural gases, refinery off-gases, and ammonia cycle off-gases,
-
Pure liquids, such as nitrogen, argon and oxygen, which all contain small but finite traces of dissolved impurities such as carbon dioxide and hydrocarbons. On exposure to atmospheric air, liquid nitrogen will condense air and pick up oxygen and carbon dioxide in solution, and water as a particulate.
It is therefore important, when storing and handling cryogenic liquids, to understand the different evaporation behaviour of mixtures compared with that of pure liquids, and how this can affect the build-up and release of thermal overfill. Thermodynamically, the equilibrium state of a single component fluid is defined by two out of the three variable parameters, pressure P, density ρ, and temperature T, the third parameter being defined by an equation of state.
With a multi-component fluid, an equilibrium state is defined by three or more parameters respectively out of four or more, including P, ρ, T, and x(1), x(2), etc. (where x(1), x(2), etc. are the molar fractions of component (1), (2), etc. in the fluid). A further complication is that vapour and liquid phases in equilibrium have different compositions.
This is illustrated in Fig. 5.1a by the typical equilibrium (T-x) diagram for a 2-component or binary mixture at constant pressure P, where equilibrium is achieved by a freely boiling liquid in contact with its boil-off vapour. “Freely boiling” is the boiling produced by a submerged or wall heater with a sufficiently large heat flux to generate nucleate boiling and multiple streams of bubbles rising through the liquid to the surface.

a Typical vapour and liquid (T, x) curves during equilibrium (free boiling) and non-equilibrium surface evaporation. b Deviation of vapour composition y(11) from free boiling value y(1) with increasing evaporative mass flux of liquid mixture
At constant pressure, the equilibrium concentration y(1) in the vapour of the lower boiling point component (1), which is in contact with a freely boiling binary mixture, is higher than its composition x(1) in the liquid.
This difference between vapour and liquid compositions is widely used in fractional distillation for separating a mixture into a lower boiling-point component and a higher boiling-point component.
Figure 5.2 shows the particular (T-x) diagram for oxygen-nitrogen mixtures at one atmosphere pressure, in which the wide difference in composition of vapour and liquid is demonstrated. It can be seen that the vapour in equilibrium with liquid air, (79% nitrogen 21% oxygen), contains less than 7% oxygen, and therefore presents a serious hazard from asphyxia as a breathing gas. Likewise, the condensation of air starts at 81 K and produces a liquid containing up to 48% oxygen, which presents a combustion hazard. Thus, the increasingly general use of liquid air as a refrigerant involves additional hazards over those associated with using LIN as a refrigerant (see Sect. 8.7.1 on safety procedures with liquid air).

Temperature-composition diagram for oxygen-nitrogen mixtures
5.2 The Difference Between Free-Boiling and Surface Evaporation (T-x) Data
It may not be widely appreciated that, under storage conditions with surface evaporation only, the well-known equilibrium (T-x) diagrams may not apply. The vapour composition y(11), y(22), etc. will not be the same as y(1), y(2), etc. as defined by measuring the liquid composition x(1), x(2), etc. together with T and P, and using the published equilibrium (T-x) data, (again, see Fig. 5.1a).
The difference arises because, unlike nucleate boiling with vapour bubbles rising rapidly through the bulk liquid and coming into equilibrium according to the (T-x) data, the surface evaporation of each molecular species is differentially controlled by its own diffusion mechanisms, across the surface sub-layer between the bulk liquid and the liquid-vapour interface.
Evaporation-limiting molecular diffusion rates will be determined in a complex manner by the double diffusion of each molecular species under both local thermal and concentration gradients; within region 1 of the surface sub-layer largely by concentration gradients, and within region 2 by the enormous temperature gradients of 5–10,000 K/m.
Because different molecular species in a mixture have different diffusion rates (inversely proportional to their respective molecular weights), as well as different evaporation coefficients, the relative concentration of species in region 1, the molecular evaporation region, will deviate from the bulk liquid with an expected relative increase in the lower molecular weight component (generally the lower boiling point component). In contrast, with nucleate or free boiling conditions, the local concentration of molecular species in the bulk liquid through which the bubbles are rising is the controlling mechanism for the vapour composition, rather than the surface sub-layer.
The difference in concentration of each molecular species in the surface sublayer will depend on the product of its own thermal diffusion coefficient and the temperature gradient across region 2, together with the product of its own self diffusion coefficient and the concentration gradient across the same thermal conduction layer. The heat flow through this layer can also be expected to be a factor in determining the temperature gradient and thickness of this layer.
Consequently, when the heat flows and associated evaporative mass flows increase, the concentration difference between interface and bulk liquid will increase. As a result, the vapour composition y(11) will deviate progressively from the equilibrium value y(1) as the evaporation rate increases, and will almost certainly contain more of the lower boiling point component (as indicated in Fig. 5.1b).
This change in vapour composition with surface evaporation rate has been studied in experiments at the Institute of Cryogenics, Southampton, after the effect had first been observed.
The consequences of non-equilibrium (T-x) values being different to published equilibrium values have significance in many areas. For example:
-
When designing distillation columns with separation trays, or structured packing, where surface evaporation contributes to the fractional distillation.
-
When using the composition of the liquid from a liquid mixture as the control variable for the vapour composition, such as trying, unwisely, to maintain operationally the composition of Breathing Air using a liquid oxygen-nitrogen mixture. The vapour will tend to be much richer in nitrogen than the equilibrium (T-x) value, thereby creating an unexpected asphyxiation hazard.
-
When comparing published experimental vapour-liquid data from different authors. For example, liquid air data can show variations in compositions by up to 30% depending on the method of measurement.
-
When using LIN in contact with atmospheric air. The air will condense, the nitrogen will evaporate preferentially and the oxygen concentration in the LIN will rise quite quickly.
5.3 Stratification in Cryogenic Liquid Mixtures
From Chap. 4, it can be seen how liquid convective circulation can lead to density stratification in single component liquids. In the absence of any continuous mechanical mixing of the single component liquid, the intrinsic mechanisms of superheated wall boundary layer flow, incomplete evaporation at the surface, and central downward jet of less superheated liquid, can lead to stratification into two layers. Each layer will be at a more or less uniform but different temperature, with a hotter, less dense layer on top of a colder, more dense layer.
As a guide to the associated temperature and density differences, see Table 5.1 which contains estimates of the changes of density with temperature and pressure along the saturation P − T line for a whole range of cryogens. However, note that the density changes are for guidance only, since they relate to the equilibrium saturation condition and not to the superheated liquid from which evaporation takes place or to isenthalpic changes of liquid state.
Since the temperature difference across the liquid-liquid interface between the two layers is small, of the order of 0.1–1.0 K, the mixing effect of thermally driven molecular diffusion in the absence of any convective motion is relatively small. The associated density difference across the liquid-liquid interface acts so as to suppress local convective mixing and the stratification is therefore extremely stable.
However, the heating of the top layer by the wall boundary layer flow continues, leading to a continuing rise in temperature, or an increase in thickness accompanied by a downward migration of the interface, or a combination of the two.
When the density difference between the two layers becomes large enough, of the order of 1.0% (corresponding to an approximate ΔT of 0.05 K for LHe, 0.6 K for LH2, 1.8 K for LIN, 2.5 K for LCH4, etc. from Table 5.1), the superheated wall boundary layer flow in the denser, lower layer suddenly has insufficient buoyancy and inertia to penetrate the liquid-liquid interface. The boundary layer flow is trapped in the lower layer and no evaporation can take place to release its superheat. Instead, the wall boundary layer turns over at the interface and its motion and superheat is locked into the lower liquid causing it to heat up instead. When this happens, the evaporation rate will fall as the first indication of a stratification effect and associated increase in thermal overfill via the unstable superheated state. As described in Sect. 2.2.3, this type of thermal overfill in a single component liquid may be released by a violent QHN boiling of the lower layer, with consequences such as the ejection of vapour mixed with liquid through the vents, and possible mechanical damage to the storage vessel.
Figure 5.3 illustrates how density stratification leads to the lower layer boundary layer flow failing to penetrate the liquid-liquid interface.

Stratification with low density top layer above high density bottom layer. Density difference >1.0%
With a multi-component cryogenic liquid, the triggering and build-up of stratification can occur in a variety of ways, depending on the identity of the liquid components, and the previous history of the liquid elements.
Once again, the stratification is convectively stable; but now mixing across the liquid-liquid interface is controlled by double diffusion, with both temperature and concentration gradients contributing to density-gradient driven, liquid convective mixing.
This stratification in a cryogenic liquid mixture inevitably leads to unstable evaporation, which has acquired the name “rollover”. The unstable evaporation takes place when the stratified layers spontaneously mix, which can lead to a rapid increase in boil-off rate and hence tank pressure.
From detailed studies of unstable evaporation from stratified liquid mixtures carried out in Tokyo, MIT, Southampton, and elsewhere [1,2,3,4,5,6,7], it has been observed that every experimentally simulated rollover event was spectacular and different in terms of all measurable parameters, including the time taken to spontaneously rollover, the peak evaporation rate generated, and the total vapour produced.
5.4 Double Diffusive Convection in Multi-component Cryogenic Liquids
In multi-component liquids, density stratification can take place in the following ways:
-
(1)
by temperature differences between layers,
-
(2)
by composition differences between layers,
-
(3)
by combinations of (1) and (2).
A step-change in density across a horizontal interface between two layers will tend to disappear with time through (a) thermal diffusion across the interface tending towards the same temperature in the two layers, and (b) self-diffusion of molecular species across the interface tending towards the same composition in the two layers.
Both diffusion processes will spontaneously lead to density changes which will in turn affect convection processes and convective mixing of the two layers across the interface. Since convective mixing is much faster than diffusive mixing, it is the cross effect of diffusion on convection which is dominant.
Double diffusive convection has been widely studied from the observation of local convective motion in the sea by oceanographers, where heat and salt content are the two contributors. The standard analysis is given in, for example, references by Turner in 1965 [8] and 1975 [9], and by Huppert in 1971 [10].
Since the buoyancy or convective force on an element dm of liquid is dm g Δρ, convection across the interface between 2 layers is governed by the sign and magnitude of Δρ, equal to ρ2 − ρ1, the density difference across the interface between the lower layer 1 and the upper layer 2.
The density difference is given analytically from the double diffusion equation by
In the limit,
For convective stability, i.e. stable stratification with no convective mixing, dρ/dz should remain negative in sign and therefore analytically less than zero. If during double diffusion across the interface, the negative value of dρ/dz becomes smaller, approaches zero, and then becomes positive, the interface becomes convectively unstable and triggers convective mixing between the two layers.
In general, term I is negative and stabilising, provided dT/dz is positive, since (dρ/dT)x,P is always negative; but is positive and destabilising if dT/dz is negative.
Term II can be positive (destabilising) or negative (stabilising).
Term III is always negative and stabilising.
For cryogenic liquid mixtures, the magnitudes of terms I and II are similar, while term III is much smaller (since the compressibilities of cryogenic liquids, except helium, are small).
For most liquid mixtures, including most LNG mixtures, term II is positive and destabilising convectively against stratification. For example, preferential evaporation of less dense methane from LNG leaves a less volatile, more dense (e.g. ethane rich) surface layer which mixes spontaneously with the bulk, and there is no stratification.
On the other hand, when the more volatile (lower boiling point) component is more dense than the less volatile components, preferential evaporation of the more volatile component makes term II positive and stabilising. This will lead to spontaneous stratification, or autostratification, the consequences of which may be serious.
The most important examples of autostratification are LNG mixtures containing nitrogen, and liquid argon-oxygen mixtures (see Sect. 5.6.2).
5.5 Storage Behavior of Two Layers of Liquid Mixtures
5.5.1 The Dynamic Storage Behaviour of 2 Liquid Layers with Different Density Under Constant Isobaric Pressure
If two multi-component liquid layers, with different densities and/or different temperatures and a common interface, are allowed to stand in a thermally isolated tank, then there is no convection; equilibration of composition and temperature takes place relatively slowly through molecular and thermal diffusion across the interface.
When heat enters the liquid space through the walls, the heat is convectively absorbed by the upward moving wall boundary layer flow which feeds into the evaporating surface. However, when there is a density reduction across the liquid-liquid interface (in the upward direction of the convection at the wall) the wall boundary flow in the lower liquid layer may have insufficient buoyancy to penetrate this density drop. Instead, the boundary layer flow turns over through 90° at the interface to become a radial inflow just below the liquid/liquid interface and then joins a central downward jet to be mixed into the lower layer. Heat entering the lower liquid via the boundary layer cannot then be released by surface evaporation, and remains trapped in the lower layer.
Looking analytically at this lack of penetration by the boundary layer flow, the quantity (dρ/dT)/ρ for LIN/LOX mixtures and LNG mixtures, is of the order of 0.5% per degree K, at their respective boiling points (see Table 5.1).
Experimental measurements on these mixtures indicate that the local temperature rise in the boundary layer is about 1 K. Thus, the buoyancy or upward force Δρg on unit element driving the boundary layer flow is of the order 0.005ρg and this leads to a vertical velocity in the boundary layer of the order 0.1 m/s, as is observed.
If the density drop across the interface is greater than 0.01ρ, (or 1% of the mean liquid density) the buoyancy force driving the boundary layer upwards disappears. However, the momentum of the boundary layer will carry it beyond the interface a vertical distance of about 0.01 m, or 1 cm, before the boundary flow turns over.
This is what is observed experimentally; viz. the boundary layer flow turns through 90° away from the wall to join a radial inward flow while its vertical velocity component oscillates about the interface with an exponential decay and an initial amplitude of the order of 0.01 m.
We now have a convective mechanism whereby heat entering the lower liquid layer through the walls and floor of the storage tank cannot be released by the normal process of surface evaporation. Instead, this heat is mixed convectively throughout the lower layer, via the central downward jet.
The mean temperature of the lower layer rises, and the mean density falls with time. Eventually, the lower layer density becomes less than that of the upper layer and a convective instability is reached.
Mixing models, using double diffusive convection, are able to predict the onset of the convective instability [11] with reasonable precision, but they do not explain why the peak evaporation can be as large as 100–200 times the normal boil-off. An additional model is needed (see Sect. 5.6.2 below).
5.5.2 The Dynamic Storage of 2 Layers with Different Density Under Constant Isochoric Volume with Rising Pressure and Zero Boil-Off
In storage at constant pressure the stratification is unstable, as a result of surface evaporation from the top layer and heat trapped in the lower layer of an LNG mixture, leading to a decreasing density with time.
On the other hand, with storage at a constant volume under a rising pressure, with zero boil-off, the stratification into 2 layers is stable with no rollover. The top layer absorbs all the heat influx and warms up as the pressure rises, while the density rises isochorically. The bottom layer absorbs little heat and remains close to the initial temperature, while the density rises, also isochorically, as the pressure rises. The density difference between the 2 layers remains small and continues to remain small in a stable form as the pressure continues to rise. There appears to be no development with time of density differences able to promote ‘rollover’, unlike in isobaric storage.
5.6 Rollover
5.6.1 Basic Description of Rollover
The convective instability reached by the 2 liquid layers with different densities in the last section was originally believed to cause the stratified layers to reverse position by rolling over.
What happens in practice is that there is an unexpectedly rapid rise in evaporation rate, associated with releasing thermal overfill in the tank. If the vent valves on the tank are insufficiently sized, they will not be able to release the evaporated gas, the tank ullage pressure will rise and may lead to rupture of the tank.
Rollover is therefore a dangerous event and steps must be taken to avoid stratification building up with significant density discontinuities between layers. These steps involve the continuous mechanical mixing of the contents during all custody management operations, including filling, emptying and long-term storage [7]. In addition, the precursor of rollover, namely the development of density stratification, needs to be monitored and detected with appropriate instrumentation.
But first of all, what do we now know about the phenomenon of rollover, and how does a convective instability cause such a dramatic event?
The first recorded rollover event took place at La Spezia, and was described in detail by Sarsten in 1972 [12]. Briefly, the tank held a heel of 9500 m3 of LNG with a density of 541.12 kg/m3 at 114.36 K. A cargo of 33,700 m3 of weathered LNG (it had been in the tanker for over a month), with a density of 544.9 kg/m3 at 118.99 K, was bottom-filled into the tank with minimum mixing. The density difference was therefore 0.694%.
Eighteen hours after the filling was completed, the tank experienced a sudden rise in pressure, which marked the beginning of the rollover. This rollover event was accompanied by the evolution of a huge amount of LNG vapour lasting one and a quarter hours, with the tank vent valves open to the atmosphere. The tank took another two hours, after the vent valves had closed, to reach equilibrium with the normal boil-off rate. Some 200 tonnes of LNG were evaporated in the three and a quarter hours, with the peak boil-off estimated to be over 250 times the normal rate. Fortunately the vented gas did not catch fire, or explode.
5.6.2 Penetrative, Oscillating Convection Across the Interface, and Surface Evaporation Increase, During a Rollover
In fluid dynamical terms, as the densities of two cryogenic liquid layers approach the same value with time, there comes a point at which double diffusive convection manifests itself in the following remarkable way, which has been observed about 100 times experimentally, and also recorded on video tape [13].
Since double diffusive convection is not unique to LNG or cryogenic liquids, experimental rollover simulations have been studied with LA/LIN, LOX/LIN, and liquid refrigerant mixtures, as well as LNG mixtures.
In all cases, the liquid-liquid interface is observed to become unstable whereby upwards penetrative oscillations of the interface build up in vertical amplitude. Elements of the lower layer are mixed into the upper layer, and the interface migrates slowly downwards.
Figure 5.4 shows a sequence of flow visualisation photographs, of an experimental rollover between stratified layers of R 11/113 mixtures, which illustrate the downward migration of the interface over a period of 172 min followed by it’s disappearance after total mixing by penetrative convective oscillations after 182 min [5].

Sequence of flow visualisation photographs showing spontaneous mixing (rollover) of two stratified layers (with lateral and base heating) of R11/R113 mixtures differing in initial density by 1%. Exposure time was 4 s to show trajectories of seed particles
In the case of LOX/LIN rollovers, when the amplitude of the convective penetrative oscillations increases so that the oscillations reach the surface, the evaporation rate rises very rapidly by 10–50 fold.
The convective oscillations are believed to break up the morphology of the surface sub-layer, particularly the thermal conduction region, and cause the unimpeded and much higher molecular rate of evaporation to be realised (see Sect. 2.4.2 on molecular evaporation rates).
This high evaporation rate is then maintained so long as the convective oscillations continue to disturb the surface—up to the time when the mixing of the two layers is complete. Then the rate drops to an intermediate high value corresponding to the excess superheat temperature across the “new” surface layers of the now self-mixed (by convection) liquid of homogeneous density, but not yet homogeneous in temperature and composition. Subsequent diffusive (compositional and thermal) mixing towards homogeneous temperature and composition is then accompanied by the intermediate high evaporation rate before the final mixture, homogeneous in temperature, composition and density, is arrived at naturally.
Optical observations of the transition through the critical point in a critical point cell (when the density of vapour and liquid phases equalise) show similar oscillatory, penetrative convection across the phase boundary.
This model, whereby penetrative convective oscillations reach the surface and break up the surface sub-layer, enables the very high peak boil-off rates to be predicted. While the estimated evaporation coefficient for LIN is 10−3, the value for LNG is not known; it could be larger by a factor of 10, thereby allowing the predicted peak boil-off rate to rise 190 fold, close to that observed in the La Spezia incident.
5.6.3 Release of Thermal Overfill During Rollover
In terms of thermal overfill, we have seen how the heat flow entering the lower liquid layer by thermal conduction from ambient through the insulation is trapped in the lower layer, causing its temperature and energy content (superheat or thermal overfill) to rise with time. The build up of thermal overfill within the lower layer is clearly triggered if, or when, the density difference between the two layers exceeds some critical value, which appears to be the order of 1.0% (0.694% for the La Spezia event).
The occurrence of two stratified layers is then the forerunner of the sequence of mechanisms leading to rollover.
When rollover takes place, it is the thermal overfill energy in the tank, not just the lower layer, which is released via the latent heat of the venting vapour. The maximum rate of generation of vapour is determined by the molecular surface evaporation limit—it is not infinitely great like an explosion.
From the 100+ instrumented, experimental rollover events made at Southampton, the following conclusions can be made:
-
There was never any nucleate boiling—only surface evaporation.
-
The envelope of the evaporation rate-time graph is different for every rollover.
-
The total additional evaporated mass equates to the total thermal overfill energy of the two layers released during the rollover.
-
Once a stratification of less dense layer on top of a more dense layer is set up, and heat energy is supplied to the lower layer, then this energy will only be released spontaneously by an inevitable rollover event.
-
The peak evaporation rate may be the order of 50–250 times the normal storage rate, but it is not infinite as in an explosion.
5.6.4 Experimental Studies: The Two Modes or Types of Rollover
Experimental observations on laboratory generated rollovers have been made by several groups, including those at Southampton using video recording of the flow visualisations during the rollover mixing of previously stratified layers. These studies showed that there is a whole spectrum of variations of evaporation rate with time between the extremes of two Modes of rollover. These Modes were identified by dramatically different evaporation behaviours, and by somewhat different rates of migration of the liquid-liquid interface separating the layers.
Figures 5.5 and 5.6 show summaries of two experimental runs following the setting up of two stratified layers, which demonstrated Mode 1 and Mode 2 rollovers respectively. The difference is very significant [4].

Mode 1 rollover with LOX/LIN mixtures. Initial density difference 19 kg/m3 or 2.5%. High heat flux of 67 W/m2 into lower layer only

Mode 2 rollover with LOX/LIN mixtures. Initial density difference 8.7 kg/m3 or 1%. Low heat fluxes of 3.6 and 4.2 W/m2 into upper and lower layers respectively
In the first case, Mode 1, the evaporation rate increased relatively slowly with time to a peak value over a period of 60 min or more, and then subsided equally slowly back to the normal storage value. The excess evaporation tracked the rise and fall of liquid superheat in the upper layer, as the mixing proceeded spontaneously to completion and as the thermal overfill was released. The interface also moved down very slowly. All was very peaceful and controllable, without any emergency venting to the atmosphere.
In the second case, Mode 2, after 350 min, the evaporation rate rose very sharply within a few seconds to a high peak value, 20 times normal, before subsiding. At the same time, the interface moved downward much more rapidly than in Mode 1.
Mode 2 behaviour had features similar to that of a vapour explosion described in Chap. 3.
The studies at Southampton used binary mixtures of nitrogen and oxygen in two stratified layers and included measurements of the vertical temperature profiles, vertical composition profiles and evaporation rates, all as a function of time up to and after the rollover. However, the studies were unable to pinpoint any measurable factors which might help to predict whether the inevitable rollover would be Mode 1 or Mode 2.
5.6.5 Experimental Studies: The Two Convective Mixing Mechanisms of Rollover
There are two mixing mechanisms, reported in the literature, as the density difference diminishes with time to zero. These are:
-
(a)
wall boundary flow penetration, and
-
(b)
entrainment mixing via vertically oscillating convective plumes simultaneously across the whole of the liquid-liquid interface.
Both mechanisms cause upward entrainment mixing of the bottom layer into the top layer, and downward motion of the interface.
From the laboratory rollover simulations and numerical modelling at Southampton [2, 5], both wall-heating and base-heating of the bottom layer generally led to entrainment mixing by oscillating convective plumes only. The oscillating motion of the convective plumes, upwards into the top layer and downwards into the bottom layer, increased in amplitude with time as the spontaneous rollover proceeded; while the average position of the interface moved slowly down.
When the amplitude of the oscillating convective plumes built up slowly from small oscillations and never reached the top layer, then Mode 1 rollover behaviour was observed both in the experimental simulations and in the modelling (Fig. 5.7).

Mode 1 rollover
With slight wall-heating only, it was possible to induce wall boundary flow penetration and mixing, but this was a special case; mixing by convective plumes was generally the dominant mechanism in the experimental rollovers.
For Mode 2 events, the amplitude of the oscillating plumes increased very quickly and almost immediately penetrated through the surface sub-layer structure into the molecular evaporating region at the liquid-vapour interface. This penetration appeared to disturb the delicate morphology of the surface layers, so that superheated liquid from the bottom layer replaced the surface evaporating layer enabling the evaporation rate to rise 20–50 fold, i.e. to the molecular limit, just like a vapour explosion (Fig. 5.8).

Mode 2 rollover
Because the entrainment mixing was so intense, the interface moved quickly downwards at the same time as the evaporation rate rose to a peak. Once the mixing was complete, the convective plumes disappeared, the disturbance of the surface layers stopped, and the evaporation rate fell relatively quickly to the intermediate value determined by the liquid superheat, as the surface sub-layer repaired itself into its normal morphology.
In some experimental rollovers. Mode 1 behaviour converted into Mode 2 when the penetrative oscillating convective plumes increased in amplitude with time and penetrated the evaporating surface sub-layer.
One feature of the numerical modelling was that the intensity of the entrainment mixing was influenced by the ratio of base to wall heat fluxes. Generally, a higher heat flux ratio led to more intense final mixing. It should be noted that in the La Spezia incident, the ratio of base to wall heat flux was high at 3.0. It may therefore be concluded that the peak boil-off during a rollover is significantly reduced if the base heat flux is no larger than the wall heat flux.
It should be additionally noted from the experiments that:
-
all stratifications of two layers led to naturally occurring, spontaneous rollovers,
-
the peak evaporation mass fluxes observed during Mode 2 rollovers, and some Mode 1 rollovers, could, on scaling up to storage tank dimensions, exceed the venting capacity of the safety valves on some storage tanks, and lead to structural damage.
During all the experimental studies at Southampton, at no time did the layers mix or exchange position by “rolling over”. In every case, the mixing occurred via oscillating penetrative convective plumes across the whole of the liquid-liquid interface separating the two layers.
The term “rollover” is therefore not a correct description of the spontaneous mixing, but since the term is in common usage to describe this spectacular and worrying event by a single dramatic word, we shall continue to use the term also.
5.7 Factors Leading to Stratification and Hence Rollover
There are several ways in which a cryogenic liquid, whether a single component liquid, or a multi-component mixture, can become stratified in a storage tank.
5.7.1 Custody Management Creating Two Layers
Rollover was first met in the storage of LNG, so let us consider an example assuming LNG is a two-component or binary mixture of methane and ethane. One consequence of the difference in composition between vapour and liquid is that the vapour is richer in the lower boiling point component—methane, normal boiling point 112 K, liquid density 424 kg/m3—leaving the liquid richer in the higher boiling point component—ethane, normal boiling point 184 K, liquid density 544 kg/m3.
It follows that, as surface evaporation continues under normal, well-insulated, storage conditions, the liquid will become progressively richer in ethane, and the density and boiling temperature, at constant tank pressure, of the liquid mixture will increase steadily with time.
Let us look more closely at an element of superheated liquid mixture at the liquid/vapour interface during surface evaporation. The spent liquid, after evaporating a methane rich vapour, has increased in density due to both evaporative latent heat of cooling and an increase in more dense ethane composition. The spent element is convectively unstable and sinks away from the surface.
Together with all the similarly spent elements, it mixes with the bulk liquid producing a uniform density and composition. There is no stratification.
Now, if a fresh supply of the same LNG binary mixture is added to the “old” or “weathered” liquid, or “heel”, without being mixed with it, the fresh liquid will have a lower density and lower temperature and will tend to sit on top of the more dense heel in a very stable fashion. Thus a stratified top layer has been accidentally, or unknowingly, established.
If the density difference between layers is greater than about 1.0%, corresponding to about 5.0% difference in methane concentration, and if any difference in temperature is ignored, then the stratification will be convectively stable against mixing and will trigger the following sequence of events. This sequence has been observed in our experimental studies.
Firstly, the step in density will prevent the wall boundary flow in the lower layer from reaching the surface; the heat inflow into the lower layer will be trapped, causing the layer to heat up (Fig. 5.3). Its thermal overfill energy will increase with time and its density will decrease with time.
Secondly, and at the same time, the upper layer will be able to evaporate freely with methane-rich vapour being generated, so that the upper layer density-increases with time as the ethane concentration rises in the liquid phase.
Thirdly, the stabilising influence of the density difference between the layers will diminish with time until it approaches zero.
Fourthly, when the convective stability disappears, the layers start to mix spontaneously, or to rollover, by the penetrative mixing process. This penetration breaks up the surface sub-layer enabling the much faster molecular evaporation to take place.
Fifthly, after density equilibration and the peak evaporation has passed, the temperature difference between elements of the two layers is still large, of the order of 1–2 K. The associated thermal mixing and release of thermal overfill energy continues to generate further heating which can only be released by additional evaporation.
Sixthly, a further additional energy release arises from the heat of mixing associated with compositional equilibration, and this will add to the evaporation.
Finally, the evaporation rate decreases to its original normal value, balancing the total heat in-leak to the tank.
This sequence of events is exactly parallel to the observed sequence of events in the La Spezia incident.
Many other rollover events have been reported, or can be expected, with LNG, LPG, LIN/LOX mixtures and other industrial cryogenic liquid mixtures.
5.7.2 Auto-stratification in Mixtures
5.7.2.1 Mechanisms Due to Density Differences
Since stratification is the necessary precursor of a rollover event, it is important to consider the various ways in which stratification can occur, can be anticipated, and can be prevented.
We have already noted that density differences between layers can occur from temperature differences, composition differences, and combinations of the two. Also, that wall boundary layer flow cannot penetrate a density difference greater than about 1% between the layers, in which case the wall boundary layer flow is trapped in the bottom layer.
Table 5.1 shows that a 1% density difference corresponds to a temperature difference of about 1.8 K in liquid nitrogen and 2.5 K in liquid methane at their respective normal boiling points.
Likewise at constant temperature, a 1% density difference corresponds to a 2.5% change in nitrogen concentration in liquid air as a binary mixture of nitrogen and oxygen, and a 5% change in methane concentration in LNG as a binary mixture of methane and ethane.
Thus, stratification can be expected to be a common occurrence, since the necessary density changes are so small. Using the free-boiling (T-x) diagrams, the preferential evaporation of nitrogen from liquid air containing 80% nitrogen can lead to a spent liquid containing down to only 56% nitrogen with a density increase of perhaps 10%, while the preferential evaporation of methane from LNG in storage containing 90% methane leads to a spent liquid containing perhaps less than 70% methane with a density increase greater than 4%.
While it is important to realise that auto-stratification can occur in both single component liquids as well as the more usual multi-component mixtures, let us first consider several examples of auto-stratification particular to cryogenic mixtures. Then, in Sect. 5.7.3, we will consider additional examples applicable to both single component liquids and mixtures.
5.7.2.2 High MW Volatile Component in the Mixture
During surface evaporation of a mixture, the more volatile, lower boiling point component evaporates preferentially, leaving a higher concentration of the less volatile, higher boiling point components in the “spent” surface layer. If the evaporating component has a density (or molecular weight, MW) greater than the higher boiling point components remaining behind, then the “spent” layer has a reducing density as surface evaporation proceeds and will spontaneously grow into an upper stratified layer, or auto-stratify.
Two important examples are LNG with a significant amount (>1.0%) of nitrogen, and liquid oxygen with a significant amount (again >1.0%) of argon. In the first case of LNG with, say, 5% nitrogen, preferential evaporation of nitrogen with MW = 28, liquid density 808 kg/m3, from LNG with MW ~16, liquid density ~430 kg/m3, would result in a liquid density reduction of 3.0%, which is more than enough for auto-stratification.
In the second case of LOX with, say, 5% argon, preferential evaporation of argon with MW = 40, liquid density 1394 kg/m3, from liquid oxygen with MW = 32, liquid density 1141 kg/m3, would result in a density reduction of about of 1.0%, which is probably sufficient for auto-stratification to occur.
In both examples, it should be noted that the enhanced composition of the vapour with the lower boiling point constituent, due to the surface evaporation effect discussed in Sect. 5.2, will further enhance the density reduction required for auto-stratification to take place.
5.7.2.3 Non-volatile Impurities in the Surface
If carbon dioxide, water, or any other non-volatiles are present in solution, which may be at concentrations of 1 ppm or considerably less, then on evaporation, they come out of their solution phase and may subsequently remain at the surface as a monomolecular film. This film may be continuous, or it may consist of discontinuous floating rafts of impurity; but when it forms, it will provide an additional significant impedance to the surface evaporation process and literally switch off the evaporative mass flow.
A triggering mechanism has again been provided whereby the superheated liquid from the wall boundary flow cannot evaporate and a superheated layer forms immediately below the surface.
Non-volatile films may also arise from condensation of carbon dioxide, water, etc. entering the ullage vapour space during custody management operations, or from leakage of atmospheric air.
Laboratory studies with liquid nitrogen have witnessed on many occasions the switching off of evaporative mass flows for periods of many minutes, and the subsequent rapid rise in evaporation beyond the upper limit of the flowmeter. However, the introduction of carbon dioxide, either into solution in liquid nitrogen, or into the nitrogen vapour, in order to stimulate, or simulate, the formation of a film and switch off the evaporation, proved to be inconclusive. So, there are no systematic studies or results to support the idea of monomolecular films suppressing the evaporation of cryogenic liquids.
These monomolecular films do, of course, exist at ambient temperature and can be used to reduce the evaporation of water stored in reservoirs. They are also responsible for the stability of smog particles in the atmosphere.
The Schleiren photographs of evaporating cryogenic liquid surfaces sometimes show areas where there appears to be no evaporation.
5.7.2.4 Marangoni Film Flow Effect
It may not be commonly known that the tank wall above the liquid level in a LNG tank is wetted by surface tension driven film flows up the tank wall. This effect, an example of the so-called Marangoni effect, was first studied at Southampton in 1974 [14]. Look into a container of LNG and see for yourself!! Then compare what you see with the tear-drops round the edge of a glass of sherry or port.
Due to surface tension differences between methane and ethane, a film of ethane-rich liquid is drawn up the wall, losing methane by evaporation, until pure ethane droplets build up at the 185 K (NBP of ethane) temperature level. The ethane droplets run back down the wall, and being more dense than LNG, they collect over a long period of time at the tank bottom as a thin, warm, dense, stratified layer. Above the 185 K level, a film of propane rich liquid is drawn up the warmer levels of the wall and propane droplets appear at the 231 K level (NBP of propane) if there is sufficient concentration in the LNG (Fig. 5.9).

Marangoni effect. Schematic diagram showing appearance of transparent Dewar wall above LNG together with observed temperatures
However, studies of the mass dynamics of the surface tension driven separation process show that the mass flows in the films are not great enough to be commercially viable for extracting ethane or propane from LNG [15].
Another point to watch is that the wetted walls and cold exit piping, due to the Marangoni effect, make the monitoring of vapour composition particularly difficult.
5.7.3 Auto-stratification in Both Single Component Liquids and Mixtures
Auto-stratification is not confined to cryogenic liquid mixtures, and can occur in a number of other ways in both single component and multi-component liquids.
5.7.3.1 Self-pressurising Storage Tank
If a tank is allowed to pressurise, then the evaporation is progressively suppressed as the ullage pressure rises, and a surface layer is created with a rising temperature in equilibrium with the increasing pressure.
The superheated wall boundary flow feeds into this surface without evaporating. When the density difference between bulk liquid and surface layer reaches the critical value of about 1%, the wall boundary flow can no longer feed into the surface layer, and the heat is trapped in a lower layer of bulk liquid as thermal overfill.
5.7.3.2 Tall, Thin Storage Tank, Freely Venting
In a tall, thin tank, the surface area is insufficient to enable the superheated wall boundary flow, feeding into the surface sub-layer, to lose all its superheat by evaporation before entering the central downward jet.
An upper layer becomes heated by the central jet until the density difference exceeds a critical value of the order of 1%, when the wall boundary flow becomes trapped and switches into a lower layer of bulk liquid, resulting in a drop in boil-off rate.
5.7.3.3 Passing Atmospheric Weather Fronts
A falling atmospheric pressure with time, from say an approaching storm front, will cause the evaporation rate to rise from a freely venting tank, and the bulk temperature to fall by up to about 1 K, until the storm front passes.
A rising atmospheric pressure with time, after say the passing of a storm front, will tend to suppress evaporation and lead to the mechanism of stratification outlined in 5.7.3.1 above. The surface layer temperature will rise to maintain equilibrium with the rising atmospheric pressure, while the bulk liquid temperature will remain constant. The associated density difference between the two layers, even for a severe storm with a pressure drop of 100 millibars, is unlikely to exceed 0.5% for most liquids, except for LHe with an estimated 2% density difference (see Table 5.1). The trapping and switching of wall boundary flow into the lower bulk liquid is therefore unlikely, except perhaps for LHe.
5.7.4 Custody Management Filling with Subcooled Liquid Creating Thermal Underfill
If a tank is topped up with subcooled liquid—a dangerous practice—then auto-stratification will take place immediately (see Fig. 5.10).

Autostratification by addition of subcooled liquid. For propane, when density difference between layers >1% or subcooling of lower layer >5 K. This will lead to a sub-atmospheric pressure in ullage space and b rollover with rapid increase in boil-off. Conclusion: avoid, particularly in LPG and LNG sea tankers
Subcooled liquid, i.e. liquid cooled to below its boiling point at tank pressure, will be more dense than the heel of old liquid. If mixing is incomplete, the old liquid will rise above the in-coming subcooled liquid and collect as a stratified upper layer.
The thermal overfill energy associated with this type of stratification is negative, hence “thermal underfill”.
Thermal underfill is dangerous to have in a storage tank because mixing will lead to sub-atmospheric pressure in the tank and the consequential ingress of atmospheric air, or the collapse of the tank which may well not be designed to withstand a negative pressure differential.
Filling an empty tank with subcooled liquid will also lead to auto-stratification. Some of the subcooled liquid will absorb heat from the tank walls, piping, etc. to produce significant quantities of saturated liquid at the tank operating pressure, which partially evaporates as cooldown vapour. This saturated liquid will form a warmer, less dense stratified layer on top of the subcooled liquid.
Filling a partly filled tank with subcooled liquid may result in possibly three layers, with a top saturated layer, a middle layer of heel liquid, and a bottom layer of subcooled liquid.
Subcooled liquid must not be loaded into sea-going LNG or LPG tankers. Otherwise, all will appear to be well when the tanker sets sail. However, when the sea gets rough outside the harbour, the motion of the tanker will induce mixing and generate a negative pressure in the tanks, requiring emergency purging with nitrogen or propulsion engine exhaust gas.
5.8 Prevention and Avoidance of Rollover
The prevention and avoidance of stratification and inevitable rollover or vapour explosions during cryogenic storage, can be achieved by:
-
(1)
appropriate instrumentation to detect and monitor stratification,
-
(2)
adequate design of tank auxiliaries, fill nozzles, tank vents, vapour lines, etc.,
-
(3)
correct custody management and early removal of thermal overfill,
-
(4)
possible use of internal convective devices.
5.8.1 Detection of Stratification
High precision instruments are required to detect density, temperature , and composition changes of the order of 1%, while location of the liquid-liquid interface requires multi-point measuring heads, or a vertically traversing head, all measuring to 0.1% precision. This level of precision is a challenge to achieve under industrial conditions.
The measurement of temperature to 0.1% can be achieved in the laboratory, but considerable effort will be required to meet this level of precision under industrial conditions; the precision of normal industrial thermometers is in the range of 1–2%.
The measurement of density with in situ instruments to the required precision of 0.1% will be difficult to achieve—the advent of cold electronics should help [16].
Measurement of composition by liquid withdrawal through a capillary tube, followed by total evaporation and gas phase analysis, is extremely time consuming and expensive for any continuous monitoring programme to be maintained.
The interface may be detectable using ultrasonic or electromagnetic (optical, radio, etc.) waves. It is therefore possible that a cryogenic sonar/radar system, using low temperature electronics, could be devised to detect stratification.
5.8.2 Adequate Design of Tank Auxiliaries
Tank systems must enable the operations to be carried out for dealing with and removing a build-up of thermal overfill through stratification, before a rollover or vapour explosion occurs. All tanks should therefore have bottom-fill capability via angled nozzle injectors, top-fill capability via spray nozzles sited well above the highest liquid level, and bottom-entry emptying (pumping) lines feeding from a sump below the bottom of the tank.
The angle of the bottom-fill nozzle injector must be such as to induce both horizontal and vertical swirl motions to the fresh liquid to achieve satisfactory mixing. The vertical swirl must be strong enough to reach the surface of liquid in the tank right up to the point when the filling operation is complete. For top filling, the spray nozzles must be widely distributed so that the fresh liquid falls evenly across the whole surface of the liquid content.
When a rollover occurs, the emergency vent valves must be large enough for the expected maximum evaporation rate not to raise the tank pressure above the design pressure. A necessary comfort for liquid management!!
5.8.3 Avoidance and Early Removal of Stratification
Stratification can be avoided by correct and adequate mixing of the tank contents, whether LNG, LPG , or liquid air, during filling operations. Mixing of fresh, lower density liquid with old weathered liquid can be achieved by bottom filling through upward angled nozzles set to induce vertical and radially inward mixing flows [7]. Instrumental monitoring is needed to test the effectiveness of filling and mixing procedures; otherwise guessing is totally inadequate and possibly dangerous.
Once stratification has occurred, the extra thermal overfill will have to be removed with some significant additional evaporation, the vapour volume generated being possibly path-dependent. This requires careful and safe tank management by mixing, either internally, or preferentially by liquid transfer between two cold tanks. If the mixing is too rapid, the additional evaporated gas flow may exceed the capacity of the vapour lines, the pressure will rise and the safety vents will open allowing large quantities of flammable gas into the environment.
If the vents cannot cope, when the flow through them approaches the velocity of sound and becomes choked with the formation of internal shock waves, the pressure will rise until structural damage occurs.
If the tank roof is lifted by the excess pressure generated, the roof may then collapse back into the liquid causing a surge of the contents, over the surviving primary containment and secondary walls, into the local environment.
This type of tank failure happened to a very large, 100 m diameter tank of LPG in the Arabian Gulf, a few years ago, with many casualties.
The practice of adding fresh liquid without mixing, so as to store two stratified layers with different density (for example, two different LPGs) in a single container or tank, is extremely dangerous, and should be stopped.
The real problem arises in deciding what to do if stratification is suspected. If nothing is done, then a rollover will undoubtedly occur.
On a tank farm with several storage tanks, one effective, and popular, procedure is to transfer liquid intermittently or continuously from tank to tank via bottom emptying and top spray filling. Passage through the spray nozzles results in some pressure drop and partial evaporation of the liquid. The liquid spray droplets have increased density and consequently sink through the top layer liquid so as to promote mixing. The additional vapour generated arises from the heat in-leak during the tank-to-tank transfer, and also from the release of thermal overfill energy arising from stratification by custody management or by auto processes.
The tank ullage pressure is determined by the rate of production of this additional vapour and can therefore be controlled, in principle, by the rate of liquid transfer. Should the ullage pressure rise too high, the liquid pumps should be stopped to try and reduce the rate of dissipation of thermal overfill energy, and allow the ullage pressure to fall back.
The safest procedure is to carry out tank-to-tank transfers continuously, when the overall rate of vapour generation should become constant. Initially, inter-tank mixing will produce a high rate of evaporation as any thermal overfill introduced during a fill with fresh, dense liquid is dissipated.
With a single tank in service, the only effective option is to circulate liquid from the bottom and in through the top fill sprays. Again, the tank ullage pressure rise is proportional to the additional evaporation produced by heat in-leak during the liquid transfer and the rate of dissipation of thermal overfill. It can again, in principle, be controlled by the rate of liquid transfer, and the pumps stopped if the pressure rises out of control. However, the circulation must be restarted, perhaps slowly at first, in order to totally remove the thermal overfill energy that has built up. There is no other option!!
If vapour is lost through the vents as a result of recirculating liquid, then this is a relatively small penalty to pay for removing a potential rollover when a much larger quantity of vapour would be lost.
Finally, one word of warning when handling a tank or vessel which is full to the brim with a cryogenic liquid: remember the significant volume compressibility of all cryogenic liquids. Under no circumstance must an excess tank pressure be controlled by suddenly switching on vapour compressors to absorb an increasing vapour flow. The vapour line pressure and hence the tank ullage pressure may be inadvertently reduced so as to cause liquid to expand and boil up into the vapour lines. The subsequent uncontrolled increase in evaporation rate and rise in pressure may lead to serious mechanical damage. Switching on vapour compressors should be carried out very gently when the vessel is brimful and then only intermittently to start with.
5.8.4 Possible Use of Internal Convective Devices to Destabilise Stratification
With a density difference of the order of 1% between stratified layers, it is possible to envisage the use of convective devices to reduce the rate of build-up of thermal overfill in the lower layer and to encourage convective mixing. Bearing in mind that once stratification occurs, the ‘A’ heat in-flow to the lower layer through the tank wall and floor insulation is trapped within the layer. The purpose of a convective device would then be to funnel this ‘A’ heat in-flow up to the top layer, thereby reducing the build-up of thermal overfill in the lower layer and delaying the onset of rollover. The question is whether the device is effective and practicable.
In tanks with a depth/diameter ratio greater than 1, the wall boundary layer suction is strong enough to pull the heated liquid in contact with the tank floor into the wall flow. Experimental attempts to increase the momentum of natural convective wall boundary layer flows in an open vessel have not been successful in the past. It is therefore unlikely that any convective device would be successful in such tanks.
In tanks with a depth/diameter ratio less than 0.5 (as in very large LNG and LPG tanks), the wall boundary layer flow may not be strong enough, and some of the heat inflow from the tank floor may be concentrated by natural convection into a number of thermals rising through the lower layer. As mentioned in Sects. 3.2 and 4.6.4, these thermals are the centres of large convection cells with horizontal spacings of the same order as their vertical dimension, namely the depth of the lower layer. Their number and position relative to the tank floor will therefore also depend on the depth of the lower layer.
If these convection thermals can be caught in large conical apertures leading into chimneys extending through the lower/upper layer interface up to the free liquid surface, then in principle the thermal catcher and chimney device could be effective in reducing thermal overfill in the lower layer [17]. The devices would have to be positioned around the tank floor and not in the centre where the central downward jet from the upper layer surface would act in opposition.
Another device includes the possible multiple concentration of the heat inflow through the floor to provide hot-spots for generating hot thermals with high buoyancy capable of passing through the lower/upper layer interface to the free surface without the aid of convective chimneys.
Devices using the introduction of gas-bubble streams are unlikely to be efficient mixers in large tanks with liquid depths of 10–50 m.
As far as we know, while all the internal convective mixing devices may work well on a small scale in the laboratory, no-one has used them in large scale storage tanks.
5.9 Path Dependent Mixing of Boiling Cryogenic Liquids, with Evaporation
So far in this chapter , we have not considered the volumes of vapour generated when two boiling liquids are mixed together. Let us now look more closely at the mixing process, particularly when the two liquids have widely separated boiling points.
At temperatures above ambient, the homogeneous mixing of boiling liquids is not an everyday experience. Adding immiscible liquids, like boiling oil into water, and vice versa, are probably the closest we meet, when the results are spectacularly explosive and hazardous because of homogeneous nucleate boiling of the water.
On the other hand, at temperatures below ambient, the forced convection mixing of miscible boiling cryogenic liquids, such as LPGs, LNGs, other hydrocarbon liquids, and liquid air components, is commonly carried out. Intuitively, one might expect the vapour generated to contain more of the lower boiling point component, but hardly that the volume of vapour might vary significantly with the mixing profile.
The mixing process is, of course, irreversible, and is accompanied by an irreversible increase in entropy. Mixing with evaporation is particularly difficult to model because, in addition to a possible heat of mixing , the process is significantly path dependent. A considerable volume of vapour is produced by the usual positive heat of mixing and by thermal contact between colder and hotter components (no homogeneous nucleate boiling has been observed) before the final equilibrium state of the mixture is achieved.
However, because the mixing is path-dependent, the volume of vapour produced is a variable path-dependent phenomenon. This path dependence is clearly demonstrated when liquid propane and liquid butane are mixed.
It follows that path-dependent mixing of other cryogenic liquids can be expected to produce significantly large variations in the volume of vapour generated.
5.9.1 Propane-Butane Mixing
The variable vapour volume, path-dependent mixing phenomenon is, in fact, widely met in the LPG industry when mixing liquid propane. NBP −42 °C, density 581 kg/m3, with liquid butane, NBP −0.5 °C, density 601 kg/m3.
For example, when liquid propane is added to liquid butane, the volume of vapour generated is observed to be considerably greater, by a factor of up to 2, than when liquid butane is added to liquid propane.
Our attention was drawn to this mixing phenomenon by the problem met on board large 30,000 m3, multi-tank, fully-refrigerated LPG tankers, when tank loads (5000–10,000 m3) of propane-rich liquids are required to be mixed with similar tank loads of butane-rich liquid during sea-passages. (This on-board mixing is not allowed in port).
Total re-liquefaction of the evolved vapour is required to conserve the hydrocarbon cargo, and since the on-board excess refrigeration capacity is limited, the mixing operation rate is determined by the rate of vapour evolved. At the same time, the pressures within the tanks have to be maintained within narrow margins above and below atmospheric pressure. Hence the mixing operation has to be carefully monitored so that the vapour is re-liquefied at the same rate as it is produced and so that tank working pressures are not exceeded.
When propane is added to butane, the mixing operation takes twice as long to complete because of the unexpectedly large amount of vapour generated. The transfer pumps have to be throttled, or periodically stopped to allow the mixing tank pressure to fall back to its normal range.
We carried out some experimental mixing of C3 propane and C4 butane liquids at Southampton, which demonstrated the phenomenon is not an artefact. A mixing model was then developed which showed how a correlation might be produced to predict the volumes of vapour produced [18].
5.9.2 Experimental Conclusions on the Forced Mixing of Propane and n-Butane
-
1.
Liquid propane is about 3% less dense than liquid butane and stratification is a major problem to be faced when mixing the two liquids. The (T-x) curves for propane/n-butane at 1 bar pressure are shown in Fig. 5.11 [19]. There is a wide composition difference between liquid and vapour with, for example, vapour containing 85 mol% propane in equilibrium with the liquid containing 50 mol%. The heat of mixing cannot be ignored and is calculated to be a maximum of 23 kJ/kg for a 50 mol% mixture, corresponding to the evaporation or “flash” of 5.75% of the liquid.
Fig. 5.11 
Temperature-composition diagram for propane/n-butane at 1 bar
-
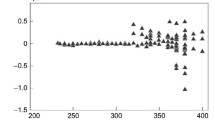
2.
Experimental mixing results are summarised in Fig. 5.12 in terms of the percentage vapour flash, and are plotted together with the vapour flash from the estimated heat of mixing.
Fig. 5.12 
Experimental vapour flash results from mixing propane into n-butane, and vice versa, as flash volume versus final mixture composition. The continuous curve represents the vapour flash from the estimated heat of mixing
-
3.
The results showed clearly that the vapour flash from adding propane C3 into normal butane n-C4 was much larger than the flash due to the heat of mixing. The vapour flash from adding n-C4 to C3 was close to the expected flash from the heat of mixing.
-
4.
The ratio of the two vapour flash volumes was between 1.5 and 2.0. The ratio increased when the mixing time was reduced, but was largely independent of whether the propane or the butane was initially transferred to the top or the bottom.
-
5.
Strangely, the vapour flash is richer in n-butane when propane is added, than when n-butane is added.
-
6.
Experimental mixing results with commercial samples of liquid propane containing 8.5% ethane and 5.5% n-butane, and liquid butane containing 1% propane and 34% iso-butane, produced less flash, but the ratio remained about the same.
-
7.
A path-dependent mixing model was developed which theoretically carried out the mixing by adding liquid fractions in a sequence of stages, and decoupling the vapour generation and liquid mixing processes at each stage. This model indicated how C3 added to n-C4 generates up to twice the vapour flash when compared with adding n-C4 to C3.


5.9.3 Some Consequences of Path-Dependent Mixing
The experiments on propane/butane mixing have shown conclusively that vapour flash volumes are path-dependent, and may be expected to be much larger than those predicted from heats of mixing. It follows that all cryogenic liquid mixing can be expected to be path-dependent, with noticeable consequences on operations at large scales, including the following examples:
-
1.
Adding and mixing fresh liquid to the existing heel, to prevent stratification. If cold is added to hotter liquid, the vapour flash volume may be greater than if hot is added to cold liquid. This applies to any pair of cryogenic liquids being mixed, whether LPG, LNG, LIN/LOX/Lair, etc.
-
2.
When removing stratification by mechanically mixing the 2 layers, bottom to top mixing may generate more vapour flash than top to bottom mixing.
-
3.
When a rollover occurs, the mixing is irreversible and therefore path-dependent. The penetrative mixing is from bottom to top, and the vapour flash volume can therefore be expected to be larger than predicted from the heats of mixing of the 2 layers.
5.10 Low Solubility Impurities in the Range 1–10 to 100 ppm
Until now, this chapter has been largely concerned with totally miscible components in a liquid mixture. However, there are many common substances which dissolve in cryogenic liquids up to relatively low limits of solubility in the 1–10 to 100 ppm range.
It is quite clear that, provided the concentration in the solution phase of these minority substances, or impurities, remains well below their solubility limit, there is no problem. Indeed, their presence will probably go unnoticed. Provided there is adequate flushing of the impurity laden cryogen through the system, then an approach to the solubility limit never happens and all is well.
However, when the cryogen is boiling as part of a separation process, such as LOX in a reboiler/condenser, and there is inadequate flushing, then a mechanism exists whereby the impurity concentration can build up and exceed the solubility limit. The impurity will then pass out of the solution phase and form a solid, (the liquid solvent is at a temperature well below the triple point of the solute), firstly as microcrystals in the liquid, and then as a solid deposit, (for acetylene in LOX, see Sect. 8.8.3).
A number of these impurity solubilities have been studied as a function of temperature, using FTIR spectroscopy and gravimetric measurements, and they all vary in the standard way as shown in Fig. 5.13 for carbon dioxide, nitrous oxide and organic compounds in liquid nitrogen, oxygen and argon [20,21,22,23,24].

a Solubility of solutes in liquid nitrogen as a function of temperature. b Solubility of solutes in liquid oxygen as a function of temperature. c Solubility of solutes in liquid argon as a function of temperature
It can be seen that the solubility decreases rapidly with decreasing temperature, falling by some two orders of magnitude between the critical temperature and normal boiling point of the solvent liquid.
In other words, a liquid under pressure and at an elevated temperature can dissolve considerably more impurity than at its normal boiling point. If a liquid solution at high pressure and elevated temperature is expanded isenthalpically through a valve to a lower pressure and temperature, the solubility limit at the lower temperature may well be exceeded and the impurity will pass out of solution.
Initially, this will be as submicroscopic crystals throughout the solvent phase, but thermo-diffusiophoresis in local temperature gradients downstream of the valve will aid mechanical deposition on to the walls of the pipework or vessel, where the impurity will collect. The submicron crystals are unable to grow because the temperature is too low, and they remain as a finely powdered solid with a very large surface area/volume ratio.
Some of these cold, finely powdered solid impurities are benign, e.g. water and CO2, but some finely powdered solid impurity/cryogen combinations may be pyrophoric or spontaneously combustible in the cryogenic liquid. The latter represent a safety hazard and are discussed in Chap. 8 on Safety.
5.11 Water/Ice in Jet Fuel
As a mixture of hydrocarbons, jet fuel may absorb water from the atmosphere and other sources up to a limit of 100 ppm. The variation in solubility of water in jet fuel is similar to other low solubility impurities, and decreases rapidly with falling temperature according to the standard cryogenic relation, log s = A/T + B where s is the solubility at temperature T and A and B are constants which have to be measured by experimental data.
The 100 ppm limit appears to indicate water will come out of solution below the temperature range −35 to −45 °C, and freeze into small particles of ice. Now, the wing tanks of the latest passenger aircraft, like the Boeing 777, are exposed to the cold air at the cruising height of 40,000 ft at the bottom of the stratosphere. The static temperatures at this level are in the range −65 to −75 °C, and after allowing for temperature rise from skin friction, the tank walls/wing surface may be as cold as −50 to −65 °C depending on the unpredictable temperatures in the lower stratosphere.
If the solubility limit is exceeded at these temperatures, then the dissolved water will freeze out as ice on the inside of the tank walls (see Fig. 5.14).

Solubility of water in jet fuel. Predicted rapid fall in solubility “s” of water in jet fuel, with decreasing temperature
Figure 5.14 Predicted rapid fall in solubility of water “s” in jet fuel, with decreasing temperature.
When the aircraft descends into warmer air towards the end of the flight, the ice starts to melt and becomes detached from the tank wall. The ice particles may then start to block the fuel lines and heat exchangers thereby restricting the flow to the jet engines. This is believed to have happened to a Boeing 777, which crash landed at London Heathrow when both engines failed on its landing approach.
The Air Accident Investigation Branch accepted the cryogenic explanation and therefore recommended that all high flying passenger jets should start descending at least 30 min before landing: they should also descend to warmer air in the top of the troposphere at 30,000–35,000 ft if the static air temperature is observed to fall to −70 °C. The result of these recommendations, which have been accepted by all aircrews, is that no further engine failures due to ice in jet fuel have been reported, to the time of writing (see Ref. [25]).
5.12 Summary on Mixtures
-
1.
(T-x) data for surface evaporation of mixtures may not be the same as the (T-x) free-boiling data used for distillation. The deviation in vapour composition from the equilibrium data is proportional to the surface evaporation mass flux.
-
2.
While stratification in a single component liquid may lead to QHN boiling, the consequences of density stratification are more common in multi-component liquid mixtures. The density varies with both temperature and composition, and stratified layers experience double diffusive convection instabilities and spontaneous mixing or rollover.
-
3.
The inevitable consequence of stratification in a mixture is a rollover incident when the 2 layers mix spontaneously by vertical penetrative oscillating convection across the whole of the liquid/liquid interface.
-
4.
The peak boil-off during rollover is determined by:
-
(a)
the increased superheat of the surface layer in Mode 1 rollover, or
-
(b)
the penetrative convection reaching the surface sub-layer in Mode 2 rollover, and breaking down its morphology so that the full unimpeded mass flux of molecular evaporation is reached at 20–250 times the normal boil-off, depending on the magnitude of the evaporation coefficient.
-
(a)
-
5.
There are several ways in which autostratification can occur.
-
6.
Monitoring of tanks for the occurrence of stratification is necessary, otherwise continuous pumping of the liquid should be applied to promote mixing as a preventative measure.
-
7.
Impurities with limited solubilities, which all decrease rapidly with decreasing pressure and temperature, present problems through coming out of solution during long term storage or transfer of impure liquids.
-
8.
This summary applies to all cryogenic liquid mixtures, whether LPG, LNG, jet fuels or LIN/LOX/Lair mixtures.
References
Agbabi, T., Beduz, C., Scurlock, R.G., Shi J.Q.: Evaporation stability of cryogenic liquids under storage. In: Proceedings of LTEC 90, 1.5 (1990)
Shi, J.Q., Beduz, C., Scurlock, R.G.: Numerical modelling and flow visualisation of mixing of stratified layers and rollover in LNG. Cryogenics 33, 1116 (1993)
Scurlock, R.G.: Stability of cryogenic liquids under storage. In: Proceedings of Kryogenika 90, Kosice (1990)
Agbabi, T.: Rollover and interfacial studies of LNG mixtures. Ph.D. thesis, Southampton University (1987)
Shi, J.Q.: Numerical modelling and experimental study of rollover. Ph.D. thesis, Southampton University (1990)
Sugawara, Y., Kubota, A., Muraki, S.: Rollover test in LNG storage tank and simulation model. Adv. Cryog. Eng. 29, 805 (1983)
Smith, K.A., Lewis, J.P., Randall, G.A., Meldon, J.H.: Mixing and rollover in LNG storage tanks. Adv. Cryog. Eng. 20, 124 (1974)
Turner, J.S.: Buoyancy effects in fluids. Cambridge University Press, Cambridge (1979)
Turner, J.S.: The complex turbulent transport of salt and heat across a sharp density interface. Int. J. Heat Transfer 8, 759 (1965)
Huppert, H.E.: On the stability of a series of double-diffusive layers. Deep Sea Res. Oceanogr. Abstr. 1005, (1971)
Germales, A.E.: A model of LNG roll-over. Adv. Cryog. Eng. 21, 330 (1975)
Sarsten, J.A.: LNG stratification and roll-over. Pipelines Gas J. 199, 37 (1972)
Beduz, C., Scurlock, R.G.: Spontaneous convective mixing or “rollover” between two stratified layers of LIN/LOX mixture. Video tape demonstrated at Heat Transfer Conference, San Francisco (1986)
Booth, D.A., Bulsara, A., Joyce, F.G., Morton, I.P., Scurlock, R.G.: Wall film flow effects with LNG. Cryogenics 14, 562 (1974)
San Roman, O.: The dynamics of methane/ethane separation by differential surface tension driven flows; or the Marangoni effect. Ph.D. thesis, Southampton University (1978)
Scurlock, R.G.: On-line instrumentation of cryogenic systems and plant to an accuracy of 0.01% using cold electronics. In: Proceedings of ISA, Houston, 32, 139 (1993)
Voyteshonok, V.: Safe storage vessels for low-boiling liquids. Cold Facts 21, 33 (2005)
Tchikou, A.: Mixing of propane and butane. Ph.D. thesis, Southampton University (1985)
Williams, A.F., Lorn, W.L.: Liquefied Petroleum Gases. Ellis Horwood (1982)
Rest, A.J., Scurlock, R.G., Wu, M.F.: Criteria for assessing the solubility of solutes in cryogenic liquids. Cryogenics 25, 591 (1985)
Rest, A.J., Scurlock, R.G., Wu, M.F.: The solubilities of N2O, CO2, aliphatic ethers, alcohol, and water in cryogenic liquids. Chem. Eng. J. 43, 25 (1990)
Rebiai, R.: Solubility of non-volatile impurities in cryogenic liquids. Ph.D. thesis, Southampton University (1985)
Wu, M.F.: Solubilities in cryogenic liquids. Ph.D. thesis, Southampton University (1986)
Yun, S.: Phase equilibria for CO2 in LNG components. Ph.D. thesis, Southampton University (1988)
Scurlock, R.G.: Cryogenic problems of flying long-haul at 40,000 ft. (12,200 m) in stratosphere at −57 °C. In: Proceedings of IIR Cryogenic Conference, Prague (2010)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Bostock, T.D., Scurlock, R.G. (2019). Multi-component Liquids. In: Low-Loss Storage and Handling of Cryogenic Liquids. International Cryogenics Monograph Series. Springer, Cham. https://doi.org/10.1007/978-3-030-10641-6_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-10641-6_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-10640-9
Online ISBN: 978-3-030-10641-6
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)