
Abstract
In this chapter, we present an overview of various computational methods, particularly, those that are used to compute the free energy of binding to understand target site mutations that will enable us to foresee mutations that could significantly affect drug binding. We begin by looking at the driving forces that lead to drug resistance and throw some light on the various mechanisms by which drugs can be rendered ineffective. Next, we studied molecular dynamic simulations and its use to understand the thermodynamics of protein–ligand interactions. Building on these fundamentals, we discuss various methods that are available to compute the free energy binding, their mathematical formulations, the practical aspects of each these methods and finally their use in understanding drug resistance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Molecular dynamics
- Drug resistance
- MM-PB(GB)-SA
- Free energy perturbation
- Linear interaction energy
- Computational mutational scanning
- Thermodynamic integration
1 Drug Resistance Problem
Every organism attempts to survive in hostile conditions by making minor modifications in its life cycle. Though these modifications are observed phenotypically, genetic reshuffling and alterations are the underlying cause of these changes. Although we are unable to accurately explain this phenomenon and its initiation, we have been able to use this observed knowledge and empirically derive explanations for such modifications. However, it may not always be necessary to know all the details regarding genetic modifications, so long as we can correctly, at least empirically, understand such observations, and put it to effective use to predict and understand the drug resistance problem. Often the enzymes in the biochemical pathways undergo mutations to improve the survival rate of the organism by either improving the protein function or catalytic efficiency and stability to escape the inhibitory action of the drug. In the latter case, the motive for modifying the drug target is to ensure that drug binding is weakened. Moreover, the mutations are such that substrate binding is unaffected or minimally affected. Most of the computational methods employed to study the mechanism of drug resistance , attempt to understand the differences in the binding patterns of the substrate and the drug molecule, i.e. understanding the “ substrate-envelope hypothesis ”. Here, we present an overview of those computational methods that employ free energy of binding as a tool to gauge the differences in the binding of the substrate and the drug molecule before and after mutation .
In the Sect. 1, we discuss the driving force for resistant mutations and throw some light on the different mechanisms by which drug resistance can occur. In Sect. 2, we present a brief overview of molecular dynamics, thermodynamics of protein–ligand binding, and various methods for computing the free energy of binding. The last section, Sect. 3, has a detailed discussion on various free energy-based methods used to understand and predict the target site mutations leading to loss in drug binding .
1.1 Overview of the Mechanisms of Drug Resistance
The drug-induced selection pressure [1,2,3,4] is the major driving force for infectious organisms to try to evade the effects of drugs. One of the primary moves that any organism will adopt is to disrupt the action of drug molecules by one or more possible mechanisms. To show its effect, the drug must enter the cells and find its target protein. As a primary defence mechanism against drugs, the organism may down regulate the expression of influx channels that enable the entry of the drug, resulting in a decreased concentration build-up within the cell. Another strategy that hinders the build-up of the drug inside the cell is the upregulation of the expression of efflux channels/pumps that facilitate the egress of the drug molecules. These strategies are often very difficult to understand owing to the complicated pathways involved in the upregulation or downregulation of various proteins associated in the regulation of traffic to and from the cell. This attribute is difficult to study using computational techniques that use free energy -based methods . Target site mutations [5,6,7,8] that lead to disruption in the drug binding without significant loss of the protein function [9, 10] is another mechanism of drug resistance . Such mutations can be studied using computer simulations that enable us to estimate the free energy difference between the drug binding to the mutant and the wild-type protein. An essential factor to consider while understanding target site mutation is the fitness cost associated with the mutational change. This can be estimated by the change in the free energy of binding of the natural ligands /substrates; for example, a drop in their binding energy indicates that substrate binding is impeded, which this leads to increased fitness cost . This means the enzyme now must expend more energy to carry out the same reaction. Hence, we can assume that such mutations are seldom seen, and if at all they occur, a compensatory mutation(s) will be seen to counter the detrimental effects of those mutations [11, 12]. Another strategy adopted by organisms is to increase the production of drug-metabolizing enzymes that modify the drugs to their inactive form eventually leading to their elimination. A classic example of this is the inactivation of penicillin by the enzyme β-lactamase .
1.2 Overview of Computational Methods to Study Drug Resistance
Broadly , computer-assisted methods used to study drug resistance can be classified into two categories based on the information they require and the output they return. The first category of methods requires only 1D sequence data as input and the output is generally a classification type , i.e. the test sequence is classified as a resistant or a non-resistant sequence. Thus, the methods grouped under this class are collectively called as “sequence-based” methods [13]. The workflow of these methods is akin to machine learning or QSAR type classification methods. In a nutshell, sequence-based methods require sequences with the corresponding biological activity data (Ki or IC50 or any other suitable numerical value) for the drug under study. Such data can be curated from databases like HIVDB (for HIV resistance, curated and maintained by Stanford University; [14, 15]) CancerDR (for cancer resistance, curated by CSIR Institute of Microbial Technology and OSDD, India; [16]), tuberculosis resistance mutation database (curated and maintained by various departments and schools with Harvard University; [17], and many other such databases . The data is then split into training and test sets to develop and validate the predictive models . The advantage of such methods is that it is not necessary to know the tertiary structure of the protein or the drug-receptor interactions. Therefore, sequence-based methods are computationally inexpensive and large amount of data can be trained to obtain decent quality predictive models in a short time. However, they suffer from two major drawbacks; (1) a lot of a priori information on drug-resistant mutations is needed to train/develop predictive models and (2) no mechanistic insights or atomistic details can be obtained.
The drawbacks seen in the sequence-based methods are efficiently overcome by structure-based methods [13, 18, 19]. Further, structure-based methods are the methods of choice when atomistic details are desired. However, these additional details come at an added computational cost and require high-resolution protein structures to be able to make accurate and reliable predictions . However, unlike the sequence-based methods, they do not require large a priori information on mutations; on the contrary, they can be applied to systems where no data on mutation is available. To assess the binding stability which is the basis for predictions, these methods employ either empirical scoring functions that implicitly try to reflect the free energy of binding or use techniques that compute the free energy of binding per se. Molecular docking -based methods use empirical scoring functions to find the best docking conformations, and these methods are computationally less expensive. Therefore, they can be applied to assess many protein–ligand complexes. The ligand can be docked to various mutant proteins to predict their binding strength before and after mutations, and this will allow one to understand the effect of the mutation on the binding strength. The accuracy of docking-based methods relies on the accuracy of the scoring function, and they are best suited for rank ordering of compounds rather than computing the absolute free energy of binding. The major issue with docking-based methods is that most docking programs treat proteins as rigid entities, and therefore, mutations in highly flexible protein–ligand systems are poorly understood [19]. However, in recent times there have been several attempts to incorporate protein flexibility in molecular docking [20]. This has largely improved the enrichment scores. Due to the limited scope of this chapter, such docking methods will not be discussed here and have been treated elsewhere [21,22,23,24,25]. Molecular dynamics-based methods can incorporate flexibility in the protein–ligand complexes, and in most cases, are the methods of choice as a conformational sampling tool to explore the phase space accessible to the system under study. The conformations sampled are used to compute the free energy change. However, the drawback of MD-based methods is the computational cost, which is several magnitudes higher compared to docking-based methods.
Another critical issue that must be addressed about the structure-based methods is, how fast predictions can be made, in addition to how reliable are the predictions. These methods find application in drug discovery programs , wherein additional filters can be placed to weed out molecules likely to encounter a high level of resistance or assist in suitably modifying leads to inhibit the mutant proteins. Drug discovery itself is an extremely lengthy and expensive process, and an additional filter like resistance should be economical in terms of time as well as money. Moreover, such methods should also assist medicinal chemists during lead optimization stages to identify potential groups that will help evade drug resistance and avoid late-stage failures that lead to huge financial losses.
2 Molecular Dynamics Simulations and Free Energy Calculations
2.1 Overview of MD and Conformational Sampling Methods
Computer simulations are very useful in predicting changes in molecular properties brought about by alterations in an atom or a group of atoms, particularly, amino acid residues. Therefore, they find good application in predicting the effect of mutations on drug binding at the active site or elsewhere. Protein design experiments clarify the effect of a mutation on drug or substrate binding , thereby facilitating prediction of drug-resistant mutations. This way the program can be used to select all mutations wherein drug binding is hampered and substrate binding is either improved or [26].
In case of free energy calculations, molecular dynamics (MD) simulations are the most commonly used technique to generate conformational ensembles . Hence, it is rightly called as one of the main toolkits for theoretically studying biological molecules (Hansson et al. [27], Binder et al. [28]. MD calculates the time-dependent behaviour of particles or atoms, by numerical integration of Newton’s second law of motion and predicts the future positions and momenta. MD simulations have provided detailed information on the fluctuations and conformational changes of proteins and nucleic acids upon drug/substrate binding . As a result, it is now routinely used to investigate the structure, dynamics and thermodynamics of biological molecules and their complexes. MD simulations have an advantage in that, starting from an X-ray or NMR solved structure, it can provide insights into the dynamic nature of biomolecules that are inaccessible to experiments. To accurately simulate the behaviour of molecules, one must be able to account for the thermal fluctuations and the environment-mediated interactions arising in diverse and complex systems (e.g., a protein-binding site or bulk solution). This depends on how accurately the force fields represent the atoms and treats the non-bonded interactions. A complete account of force fields can be found in the review by Pissurlenkar et al. [29]. However, most of the biological events occur at timescales that are not routinely reachable by classical MD simulations, for example, protein folding occurs in the timescale of few seconds, whereas drug binding and unbinding occur in the timescale of few microseconds to milliseconds. The routine timescale that is feasible using high-end servers equipped with graphic processing units [30,31,32] and distributed grid computing [33, 34], is few tens of microseconds, that is nearly 1/100th of the timescale required to study protein folding . Conventional MD suffers from the severe limitation that it is extremely difficult to sample high-energy regions and surmount energy barriers, leading to inaccuracies in free energy calculations.
The limitations of classical MD simulations have motivated the development of new conformational sampling algorithms that facilitate the sampling of conformational space that is inaccessible to classical MD simulation. The simplest way to encourage the system to sample the high-energy regions on the phase space is to increase the target temperature [35]. This leads to increased kinetic energy of the system that enables it to surmount these barriers. However, it has been argued by many, that such elevated temperatures (~400 K and above) lead to physiologically unrealistic states that may severely distort the results; however, such methods have been found to be advantageous in improving the sampling efficiency during MD simulations. Another method that uses elevated temperature to enhance the sampling is the replica-exchange molecular dynamics (parallel tempering, [36, 37]). In this approach, several replicas are simulated in parallel at different temperatures. At appropriate intervals, the replicas switch temperatures with the nearest replica, and this exchange is governed by the Metropolis acceptance criteria. However, all these methods do not prohibit the system from revisiting the same conformational space. This problem was resolved by adding the memory concept in molecular dynamics (local elevation method [38] Metadynamics [39]) uses Gaussian potentials that discourage the system from sampling the same conformational space. These are few of the most commonly used methods to tackle sampling problems in molecular dynamics, a complete account on enhanced sampling algorithms can be found elsewhere [40,41,42,43,44].
2.2 An Overview of Thermodynamics of Protein–Ligand Binding
Molecular interactions , between the ligand and receptor, are primarily non-covalent in nature and governed by attractive and repulsive forces. In drug design experiments, the goal is always to optimize the attractive interactions and reduce the repulsive ones [45,46,47]. Moreover, these associations are temporary, and the lifespan of such complexes are governed by the off rates (Koff) or the dissociation constant (Kd), both of which indicate the binding strength of a ligand to its protein counterpart. In the realm of thermodynamics, binding is governed by enthalpic and entropic components [48] given by Eq. 1.
where ∆G is the binding free energy ; ∆H is enthalpy ; ∆S is entropy and T is the temperature in Kelvin.
The association is favourable, i.e. spontaneous when the ∆GGibbs is negative and unfavourable otherwise. All the binding and pre-binding (recognition and pre-organization) events in biomolecular associations are either enthalpy (∆H) driven or entropy (∆S) driven. The enthalpic component represents several types of non-covalent interactions like electrostatic , van der Waals , ionic, hydrogen bonds and halogen bonds, while the entropic components reflect the contribution to binding due the dynamics or flexibility of the system. Computing the enthalpic component of binding has reached far heights, in terms of methods available for calculating the aforementioned type of interactions. However, till date, calculation of the entropic component is extremely difficult, and the algorithms are computationally very demanding.
The Gibbs equation is more relevant in biochemistry for calculating the free energy and is given by Eq. 2:
where ∆GGibbs is Gibbs free energy , R is universal gas constant , T is the temperature in Kelvin, Kd is the dissociation constant . Equations 1 and 2, along with the Born–Haber cycle [46] (Fig. 1) form the basis for the development of the methods used to compute the free energy binding. The two main methods are Free energy perturbation (FEP) and Thermodynamics Integration (TI), both of which will be dealt with in the subsequent Sect. 2.3.2. However, measuring the dissociation constants from simulations is a daunting task; nevertheless, computing the partition functions from the molecular simulations is relatively easy. Hence, the ratios of the partition functions can be used to estimate the free energy of binding, which is given by Eq. 2a,
where kB is the Boltzmann constant , T is the temperature in Kelvin, Q is the partition function with subscripts PL, P and L indicating protein–ligand complex, protein, and ligand, respectively. This section presents a summary of thermodynamics, which is imperative for understanding the application and methods developed to compute binding free energy . More elaborate discussions on the thermodynamics of protein–ligand binding can be found in the reviews by Bronowska [48], and Homans [46].

Thermodynamic or Born–Haber cycle for the receptor-ligand binding
2.3 Methods to Compute Free Energy Binding
Free energy is a quantity that can be measured for systems such as liquids or flexible macromolecules with several minimum energy configurations separated by high-energy barriers. However, its computation is far from trivial and the associated quantities such as entropy and chemical potential are also difficult to calculate. More so, the free energy cannot be accurately determined from classical molecular dynamics or Monte Carlo simulations due to their inability to sample adequately from the high-energy regions of the phase space, which also make important contributions to the free energy. However, the free energy differences (ΔΔG) are rather simple to compute. The free energy binding for the non-covalent association of two molecules (protein and ligand in this case) may be written as follows:
The binding event is an additive interaction of many events [49,50,51,52], for example solvation energy (Gsol), conformational energy (Gconf), energy due to interaction with residues in the vicinity (Gint), and energy associated with different types of motions (translational, rotational and vibrational, Gmotion). The classical binding free energy equation now can be rewritten as follows:
Directly computing the free energy from an MD or MC simulation is not trivial; hence, the following methods have been formulated. Broadly, the methods used for computing free energy are classified as partitioning-based methods or end-state free energy methods and non-partitioning-based methods. The partitioning-based methods partition the binding energy into various components as shown in Eq. 4; however, this method has been highly criticized [53] stating that it is physically unreal to partition the free energy into components.
2.3.1 End-State Free Energy Methods or Partitioning-Based Methods
The human body majorly comprises of water; hence, it is imperative to carefully include the solvation effects while computing the free energy of binding. More importantly, water plays a crucial role in ligand recognition and in the binding phenomenon. In computational chemistry, the methods for incorporation of solvent are divided into three groups: (i) continuum electrostatic methods/implicit solvent , (ii) explicit solvent models with microscopic detail and (iii) hybrid approaches. Historically, the continuum electrostatic methods were among the first to consider the solvent effect, and they still represent very popular approaches to evaluate solvation free energies , especially in quantum chemistry . Polarizable continuum model (PCM, [54]), COnductor-like Screening MOdel (COSMO, [55]) and SMD solvation model [56] are few popular models for treating solvent effects implicitly in quantum chemistry . Continuum solvation methods are computationally economical; however, the frictional drag of the solvent is highly underestimated and as a consequence may drive the system to non-physical states . Moreover, solvent–solvent and solute–solvent interactions are inadequately treated, posing a danger of underestimating the effects of such interactions. The explicit treatment of solvent enables one to consider the solvent–solvent and solute–solvent interactions . This prohibits the systems from visiting non-physical states due to the inclusion of the dampening effect shown by the solvent atoms. The principal drawback of explicit solvent models is the number of atoms to be considered in the system leading to increased computational cost. However, with the help of GPU-based acceleration, this drawback, now, is hardly any cause for worry.
The end-state free energy methods use the conformations extracted from an MD or MC simulation, wherein the system is simulated by explicitly defining the solvent. However, while solving the GB or PB equation, the solvent is implicitly treated by defining the external dielectric constant for water (for most drug design cases) and a suitable internal dielectric constant [57,58,59,60,61].
2.3.1.1 Molecular Mechanics-Poisson Boltzmann/Generalized Born Surface Area (MM-PB/GB-SA)
The MM-GBSA [62,63,64,65] approach employs molecular mechanics -based energy calculations and the generalized Born model to account for the solvation effects in the calculation of the free energy . Similarly, the MM-PBSA [66,67,68] approach solves the linear or nonlinear Poisson–Boltzmann equation [69,70,71], to account for the solvation electrostatics , whereas the MM part is calculated as in MM-GBSA from the derivative of the force field equations. Both these approaches are parameterized such that they partition the energy components into various terms, and the net free energy change is the sum of these individual terms (Coulomb, vdW, solvation, etc.). MM-PBSA has gained considerable attention for estimating the binding free energies of molecular complexes due to its exhaustive nature of computing the solvation electrostatics by iteratively solving the PB equation, whereas the GB method does not involve any rigorous and iterative procedure and hence is faster. However, this does not necessarily guarantee that the MM-PBSA method always outperforms MM-GBSA method. In MM-PB(GB)SA methods, MD- or MC-derived conformational ensembles are used to compute the “average” free energy of a state and this is approximated as follows:
where the angular bracket <> indicates average over the MD/MC conformations, EMM is the molecular mechanics energy that typically includes bond, angle, torsion, van der Waals , and electrostatic terms (see Eqs. 7c and 7d) and is evaluated with no or extremely large (virtually infinite) non-bonded cut-off limit. The second term is solved as mentioned in the preceding stanza and it forms the crux of this method. The last term T <SMM>, is the solute entropy , which is estimated by quasi-harmonic analysis [72, 73] of the trajectory or by normal mode analysis [74,75,76].
The following equation (Eq. 6) shows how the binding free energy is computed from the energies of the ligand , protein, and its complex over all the MD or MC snapshots. However, the snapshots can be obtained in two possible ways—one is called the single trajectory approach and other is the multiple trajectory approach. In the single trajectory approach, only the protein–ligand complex is simulated, and the snapshots for the protein, ligand and the complex are extracted by defining appropriate atom numbers from the parameter and coordinate file. However, in the multiple trajectory approach, three separate simulations are performed, one each for the protein, ligand and protein–ligand complex.
Furthermore, Eq. 1 is modified to accommodate solvation electrostatics and hydrophobic terms as shown in Eq. 5. Here, Eqs. 7a–7d give the computation of the individual terms,
Here, ∆EMM is computed in the gas phase using classical force fields , ∆Gsol is computed using PBSA or GBSA method, ∆Gsol-elect is computed using PB or the GB method, and the ∆Gnonpolar is computed by the solvent accessible surface area (SA). While employing the single trajectory approach, Eq. 7d generally cancels out and hence makes negligible contribution to the binding energy .
2.3.1.2 Linear Interaction Energy (LIE)
Linear interaction energy [77,78,79] is similar to the MM-PB/GB-SA method with regard to the partitioning of the electrostatic and van der Waals terms (polar and non-polar contribution, respectively,); however, the use of the weighting parameter for electrostatic and van der Waals interactions, is unique to this method. LIE measures the binding energy by estimating the difference in the interaction energies of the ligand in the solvent (unbound state) and in the protein environment (bound state). Hence, to obtain these interactions, two separate MD simulations are performed. In one simulation, only the ligand is placed in the solvent (mostly water) and in the other, the protein–ligand complex is placed in the solvent. The formulation of this method is based on deriving the linear response approximation from converged ensemble interactions, most often extracted from well-equilibrated trajectories from the MD simulation of the ligand with its surroundings (solvent or protein).
The mathematical formula for computing free energies using LIE method is given in Eq. 8
where the angular bracket <> indicates ensemble over the MD trajectory, \( E_{\text{coul}}^{{{\text{L}} - {\text{S}}}} \) and \( E_{\text{vdW}}^{{{\text{L}} - {\text{S}}}} \) are electrostatic and van der Waals interactions between the ligand and its medium in the vicinity (PL—protein–ligand complex; L—ligand in solvent), and α is the weighting parameter for electrostatic interactions, which is most often set to 0.5 [78]. This value is assumed due to the linear response of the surroundings to the electrostatic field and was validated using more extensive computations on the ions (Na+ and Ca2+) in water [80]. β is the weighting parameter for van der Waals interactions and is set to 0.16−0.18 [81], which is a subject of much debate owing to the difficulty in estimating the vdW’s contribution to the free energy of binding. However, these values are obtained by empirical fitting the experimental binding free energies. Moreover, the linear response of the vdW term is assumed by observing the linear trend in the interaction of the hydrocarbons with the solvent (water) that depends on the number of carbons in a hydrocarbon.
2.3.2 Non-partitioning-Based Methods
In non-partitioning methods, there is no partitioning of the free energy into various components. Statistical mechanics plays a crucial role in deriving the relationship between the free energy of a system and the ensemble average of the Hamiltonian that describes the system. These methods are far more accurate than the previously mentioned end-state free energy methods, but at the same time, are computationally very demanding. Hence, while dealing with a large dataset of molecules against a particular protein target, it is worthwhile to screen the molecules using a fast method like high-throughput virtual screening [82, 83], followed by a flexible docking -based screening, then use an end-state free energy method, and finally employ the non-partitioning methods to study few tens of molecules. Here, we will present a brief discussion on FEP and TI methods along with their mathematical treatment, and then move on to explain the idea behind alchemical free energy predictions.
2.3.2.1 Free Energy Perturbation (FEP) and Thermodynamic Integration (TI)
Most of the methods for free energy calculations are generally formulated in terms of estimating the relative free energy differences, ΔG, between two equilibrium states, or binding of two similar ligands to a common target. The free energy difference between the two states I and II can be formally obtained by Zwanzig’s formula [84, 85].
Here, \( \beta = \left( {k_{B} T} \right)^{ - 1} \)
This represents a sampling of the differences in potentials (ΔV) of the two states using Monte Carlo or molecular dynamics simulation over the potential of state I. To ensure the convergence of these calculations, it is recommended that the potentials of the two systems should thermodynamically overlap. For satisfying this condition, correct conformations must be selected, which is a daunting task, and hence, to achieve this, a multistep process is usually implemented. A path between the states I and II is defined by introducing a set of intermediate potential energy functions that are constructed as linear combinations of the initial (I) and final (II) state potentials and these intermediate states are non-physical states (Eq. 10).
where the transition from one state to another is discretized into many points (m = 1,…,n), each represented by a separate potential energy function that corresponds to a given value of λ, such that λm varies from 0 to 1. Here, zero indicates the pure initial state of the system and one indicates pure final state of the system. The total free energy , thus, can be obtained by summing over the intermediate states along the λ variable.
This approach is known as free energy perturbation (FEP) where Δλm = λm−1 − λm; hence, it can be written as
Since the potential difference can also be described as the derivative of the potential with respect to λm, Eq. 12 can also be written as,
Now, expansion of the Eq. 13 by the Taylor expansion series gives Eq. 14,
wherein 0 → λ can instead be written as an integral over λ
Equation 15 is usually referred to as the thermodynamic integration (TI) method for calculating the free energy change [86, 87]. In the early days of free energy simulations, the TI approach was synonymous with the slow-growth method [88]. In the slow-growth method, the value of λ is changed at each time step during the MD simulation. While this method was claimed to be more efficient than the discrete FEP formulation, nowadays, a “non-continuous” change in λ is a better choice (50–100 discrete points are usually recommended). This facilitates equilibration at each point, the addition of extra points at any time, and use of any pattern of spacing between the λ-points, to optimize the efficiency.
2.3.2.2 Alchemical Free Energy Perturbation
Here , the free energy is computed by transforming a molecule from one state (bound-solvated) to another state (unbound-solvated) through several physically unrealistic states, that are called as alchemical states , hence the name “Alchemical Free energy ” [89, 90]. This method is regarded as one of the apt methods to study the effect of mutations on the drug binding affinity (Fig. 2). The total free energy change in a thermodynamic cycle in any alchemical transformation is equal to zero.

Thermodynamics cycle for computing alchemical free energy binding. Image reproduced from Wang et al. [91] [open-access article distributed under the terms of the Creative Commons Attribution License (CC BY)]
3 Application of Computational Methods to Understand Drug-Resistant Mutations
3.1 Computational Mutation Scanning
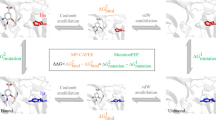
Computational mutation scanning [92] is a useful method to explore the sensitivity to changes in the composition of the amino acid in a protein-binding site (Fig. 3). In computational mutation scanning , the wild-type amino acid residue is mutated to another amino acid in the binding pocket or elsewhere. However, the most widely practised method is to mutate any amino acid residue to an alanine, since it is the simplest amino acid with a side chain (not glycine because it is devoid of a side chain). Hence, this method is equivalent to the experimental “alanine-scanning mutagenesis”, which is a powerful tool to investigate and confirm the important interactions in the protein–protein interface and protein–ligand interactions. In computational alanine scanning, all atoms from the Cβ carbon atom of the amino acid under study are replaced by three hydrogen atoms to convert it to an alanine. After the mutation , the change in the binding energy is estimated either using docking with an appropriate scoring function or by MM-PBSA or MM-GBSA to compute ΔΔG (Eq. 17c). By scanning with alanine at various positions in the binding cavity , important residues can be identified, as mutating an important amino acid will drastically decrease the binding energy.

Thermodynamic cycle for computing free energy change between mutated and wild-type protein
In the context of predicting drug-resistant mutations , one must perform alanine scanning in the binding site on two complexes, i.e. with the substrate bound complex and the inhibitor-bound complex. The change in the binding energy after mutation is computed for both the systems, viz., for inhibitor and the substrate. A decrease in the binding affinity for the inhibitor with negligible or no change in the binding affinity for the substrate indicates a hotspot amenable to resistant mutation , these spots are termed as “mutational hotspots ”. The method follows the substrate-envelope hypothesis [93,94,95], which states that there is a large fitness cost that needs to be paid if one mutates an amino acid residue that is involved in substrate binding . Mutating such amino acids could lead to impaired enzyme function resulting in the death of an organism. This can be put to appropriate use by developing inhibitors that completely overlap in the substrate binding region, leading to a lower predisposition towards developing drug resistance [96,97,98,99].
However, a major drawback in alanine scanning is that when mutating a large amino acid residue to alanine one can only study the effect of decreasing the side chain or loss of charged groups in the binding site. It is difficult to understand the resistant mutation , wherein there is a change in charged amino acid residue, for example, arginine replacing aspartate or a large amino acid replaces a small amino acid residue. Nevertheless, computational alanine scanning has been successfully used to predict mutational hotspots .
Hao et al. [100] reported a modification of computational alanine scanning (CAS), named computational mutation scanning (CMS) to study drug resistance in six HIV-1 protease inhibitors. This protocol is an improvised version of the classical CAS that enables a geometry optimization step and incorporates entropy calculations by means of normal model analysis. Using a single trajectory approach and modifying the standard MM-PBSA protocol, to allow for mutations with other amino acid residues, they computed the change in the binding affinities (ΔΔG) of 77 drug-mutant combinations (includes single and double mutants). They obtained promising results with ~83% consistency with the experimental observations, demonstrating that the prowess of the method lies in identifying the binding hotspots. However, Hao et al., do not report the change in the binding affinity for various substrates, from which they could have investigated the substrate-envelope hypothesis for the HIV-1 protease. This could have led to interesting findings facilitating our understanding about those mutations that would lead to a decrease in the enzyme function, either leading to the death of the organism or compelling a compensatory mutation to counter the lethal effects of any mutation. This information can be used to unravel the role and need for double, triple or even multiple mutations.
Tse and Verkhivker [101] used CAS along with residue interaction network to elucidate the effects of inhibitor binding on the network of residues in ABL kinase. They showed the utility of this combination in deducing the critical networks of amino acid residues and the changes that follow upon inhibitor binding, using a selective kinase inhibitor (nilotinib ) and two promiscuous (bosutinib and dasatinib) kinase inhibitors. The changes in the interaction networks in the enzyme holds key hints to unravel the mystery of how drug-resistant mutations are seen for ABL kinase inhibitors. Moreover, the mutations that occur far from the binding site can also be explained, since a mutation far off from the site can affect drug binding through a cascade of events that eventually percolate into the binding site through the changes in the residue interaction network . CAS followed by MM-PBSA added the energetic component to locate the hotspots that could lead to drug resistance in the kinase inhibitors
3.2 MM-PB(GB)-SA
MM-GBSA or MM-PBSA are two widely used free energy methods employed to understand the effects of mutations on the drug binding affinity, moreover, these methods are successful in predicting likely mutations leading to drug resistance . These methods are able to predict due to their amenability to decompose the free energy into its components at the residue level that leads to better understanding of the effect of mutations on drug binding . Lethal effects of the V82F/I84V double mutation in HIV-1 protease on amprenavir were demonstrated using MM-PBSA approach on snapshots obtained from the well-equilibrated protein–ligand complex [102]. It was reported that amprenavir lost its binding affinity due to distortions in the binding site, hence weakening many favourable interactions (ΔΔG = 3.73 kcal/mol). Such a distortion of the binding site was previously observed and attributed to the rapid flap movements seen in this double mutant which is absent in the wild-type HIV-1 protease [103]. Furthermore, newer inhibitors, that are very close structural analogues of amprenavir , like TMC126 (ΔΔG = 2.01 kcal/mol) and TMC114 (darunavir, ΔΔG = 3.45 kcal/mol) were also seen to be affected by these mutations , though to a lesser extent than amprenavir . Despite structural distortions in the binding site, it had no effect on the substrate binding , and hence, the catalytic process was unhindered.
Hou et al. [104] combined MM-GBSA with the positional variability approach, to modify Kollman’s FV value [105] to give a new scoring function also called FV (Free energy /Variability) score. Using the FV score, they evaluated the binding of six substrates that are hydrolysed by HIV-1 protease and confirmed Kollman’s [105] observation that drug-resistant mutations are more likely to occur at less conserved regions. The FV score reported by Hou et al. comprises two components, one that reflects the binding energetics at the per-residue level, obtained by MM-GBSA , and the second component is the sequence variability that represents the conservation of amino acids at each position. Using this score, one can identify amino acid residues that are crucial for substrate and inhibitor binding, and thus classify the residues that are exclusively involved in substrate binding and those that are exclusive for inhibitor binding. Such a classification when coupled with the positional variability of amino acid residues can extract those positions with low conservation and exclusivity for inhibitor binding; such positions are highly amenable to mutations leading to drug resistance . Employing this method Hou et al. confirmed their previous observation [102] that the V82F/I84V double mutations are lethal for many FDA approved HIV-1 protease inhibitors, whereas TMC126 is still active against this mutant.
3.3 Vitality Analysis
One of the primary drawbacks of the aforementioned methods to predict drug-resistant mutations is their inability to accurately estimate the binding affinity for the substrate molecule(s). The fitness cost of the mutation can be estimated by gauging the change in the binding affinity of the substrate to its enzyme target; any perturbation in the substrate binding is likely to affect the function of the enzyme. Therefore, computing the catalytic efficiency of the enzyme before and after mutation will enable us to understand the fitness cost . Pioneering work in this line was done by Gulnik et al. [1]. In this work, they have determined the catalytic efficiency (Eq. 18a) of HIV-1 protease following few active and non-active site mutations . This principle was incorporated in terms of free energy change by Warshel et al., and they employed this method (Eq. 18b) to computationally predict the likely mutations that could potentially abolish drug binding leading to drug resistance . This method is aptly named as “Vitality approach” wherein higher vitality values indicate that the resistance is more likely as there is little chance of increase in the catalytic efficiency of the enzyme. The basic workflow adopted by Warshel et al. [106, 107] is to estimate the change in the drug binding before and after mutation , depicted in the first part of Eq. 18b and then estimate the catalytic efficiency by determining the binding of the substrate by modelling the transition state (TS) conformation of the enzyme, depicted in the second part of Eq. 18b. However, the challenge of employing this method to predict likely mutations is that a thorough knowledge of the catalytic mechanism of the enzyme is essential. Nonetheless, this method is far more accurate and truly predictive in nature. This is exemplified by the fact that Warshel et al. successfully used this method on six clinical agents active against HIV-1 protease.
where Ki = inhibition constant ; kcat = constant that defines the turnover rate of an enzyme-substrate complex to the product; Km = Michaelis constant .
4 Concluding Remarks
This chapter describes important computational methods that have been proven extremely helpful in gaining insights into mutations leading to drug resistance . We have attempted to introduce methods used to compute the free energy of binding along with their mathematical formulations, practical implementation and pros and cons of such methods. Finally, we have discussed a few applications of such methods to study drug resistance.
References
Gulnik SV, Suvorov LI, Liu B, Yu B, Anderson B, Mitsuya H, Erickson JW (1995) Kinetic characterization and cross-resistance patterns of HIV-1 protease mutants selected under drug pressure. Biochem 34(29):9282–9287
Schliekelman P, Garner C, Slatkin M (2001) Natural selection and resistance to HIV. Nature 411(6837):545–546
Toprak E, Veres A, Michel J-B, Chait R, Hartl DL, Kishony R (2012) Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nature Genet 44(1):101–105
Yang Z, Nielsen R, Goldman N, Pedersen A-MK (2000) Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155(1):431–449
Blanchard JS (1996) Molecular mechanisms of drug resistance in Mycobacterium tuberculosis. Annu Rev Biochem 65(1):215–239
Borst P, Ouellette M (1995) New mechanisms of drug resistance in parasitic protozoa. Annu Rev Microbiol 49(1):427–460
Longley D, Johnston P (2005) Molecular mechanisms of drug resistance. J Pathol 205(2):275–292
Walsh C (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406:775–781
Andersson DI, Levin BR (1999) The biological cost of antibiotic resistance. Curr Opin Microbiol 2(5):489–493
Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ (2006) The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946
Böttger EC, Springer B, Pletschette M, Sander P (1998) Fitness of antibiotic-resistant microorganisms and compensatory mutations. Nature Med 4(12):1343–1344
Sander P, Springer B, Prammananan T, Sturmfels A, Kappler M, Pletschette M, Böttger EC (2002) Fitness cost of chromosomal drug resistance-conferring mutations. Antimicrob Agents Chemother 46(5):1204–1211
Cao ZW, Han LY, Zheng CJ, Ji ZL, Chen X, Lin HH, Chen YZ (2005) Computer prediction of drug resistance mutations in proteins. Drug Discov Today 10(7):521–529
Rhee S-Y, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW (2003) Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res 31(1):298–303
Shafer RW (2006) Rationale and uses of a public HIV drug-resistance database. J Infect Dis 194(Supplement 1):S51–S58
Kumar R, Chaudhary K, Gupta S, Singh H, Kumar S, Gautam A, Kapoor P, Raghava GP (2013) CancerDR: cancer drug resistance database. Sci Rep 3:1445
Sandgren A, Strong M, Muthukrishnan P, Weiner BK, Church GM, Murray MB (2009) Tuberculosis drug resistance mutation database. PLoS Med 6(2):e1000002
Carbonell P, Trosset J-Y (2014) Overcoming drug resistance through in silico prediction. Drug Discov Today Technol 11:101–107
Hao G-F, Yang G-F, Zhan C-G (2012) Structure-based methods for predicting target mutation-induced drug resistance and rational drug design to overcome the problem. Drug Discov Today 17(19):1121–1126
Martis EAF, Joseph B, Gupta SP, Coutinho EC, Hdoufane I, Bjij I, Cherqaoui D (2017) Flexibility of important HIV-1 targets and in silico design of anti-HIV drugs. Curr Chem Biol 12(1):23–39
Chandrika B-R, Subramanian J, Sharma SD (2009) Managing protein flexibility in docking and its applications. Drug Discov Today 14(7):394–400
Coupez B, Lewis R (2006) Docking and scoring-theoretically easy, practically impossible? Curr Med Chem 13(25):2995–3003
Davis IW, Baker D (2009) RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol 385(2):381–392
Lin J-H (2011) Accommodating protein flexibility for structure-based drug design. Curr Top Med Chem 11(2):171–178
Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL (2005) Docking: successes and challenges. Curr Pharm Des 11(3):323–333
van Gunsteren WF (1988) The role of computer simulation techniques in protein engineering. Protein Eng 2(1):5–13
Hansson T, Oostenbrink C, van Gunsteren WF (2002) Molecular dynamics simulations. Curr Opin Struct Biol 12(2):190–196
Binder K, Horbach J, Kob W, Paul W, Varnik F (2004) Molecular dynamics simulations. J Phys Condens Matter 16:S429
Pissurlenkar RR, Shaikh MS, Iyer RP, Coutinho EC (2009) Molecular mechanics force fields and their applications in drug design. AntiInfect Agents Med Chem 8(2):128–150
Anderson JA, Lorenz CD, Travesset A (2008) General purpose molecular dynamics simulations fully implemented on graphics processing units. J Comput Phys 227(10):5342–5359
Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC (2012) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J Chem Theory Comput 8(5):1542–1555
Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. explicit solvent particle mesh Ewald. J Chem Theory Comput 9(9):3878–3888
Beberg AL, Ensign DL, Jayachandran G, Khaliq S, Pande VS (2009) Folding@ home: lessons from eight years of volunteer distributed computing. In: IEEE international symposium on parallel & distributed processing, 2009. IPDPS 2009. IEEE
Larson SM, Snow CD, Shirts M, Pande VS (2009) Folding@ Home and Genome@ Home: Using distributed computing to tackle previously intractable problems in computational biology. DOI: arXiv preprint arXiv:0901.0866
Bruccoleri RE, Karplus M (1990) Conformational sampling using high-temperature molecular dynamics. Biopolymers 29(14):1847–1862
Earl DJ, Deem MW (2005) Parallel tempering: theory, applications, and new perspectives. Phys Chem Chem Phys 7(23):3910–3916
Sugita Y, Okamoto Y (1999) Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett 314(1):141–151
Huber T, Torda AE, van Gunsteren WF (1994) Local elevation: a method for improving the searching properties of molecular dynamics simulation. J Comput Aided Mol Des 8(6):695–708
Laio A, Parrinello M (2002) Escaping free-energy minima. Proc Natl Acad Sci USA 99(20):12562–12566
Belubbi AV, Martis EAF (2017) Advanced techniques in bimolecular simulations. In: Bharati SK (ed) Handbook of research on medicinal chemistry, Apple Academic Press (in Press)
Berne BJ, Straub JE (1997) Novel methods of sampling phase space in the simulation of biological systems. Curr Opin Struct Biol 7(2):181–189
Hamelberg D, Mongan J, McCammon JA (2004) Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys 120(24):11919–11929
Lei H, Duan Y (2007) Improved sampling methods for molecular simulation. Curr Opin Struct Biol 17(2):187–191
Zuckerman DM (2011) Equilibrium sampling in biomolecular simulation. Annu Rev Biophys 40:41–62
Böhm HJ, Klebe G (1996) What can we learn from molecular recognition in protein-ligand complexes for the design of new drugs? Angew Chem Int Ed 35(22):2588–2614
Homans S (2007) Dynamics and thermodynamics of ligand–protein interactions. In: Peters T (ed) Bioactive Conformation I. Springer, Berlin, Heidelberg, pp 51–82
Whitesides GM, Krishnamurthy VM (2005) Designing ligands to bind proteins. Q Rev Biophys 38(4):385–396
Bronowska, A. K. (2011). Thermodynamics of ligand-protein interactions: implications for molecular design. In: Moreno-Pirajan JC (ed) Thermodynamics—Interaction Studies—Solids, Liquids and Gases. INTECH Open Access Publisher, Croatia, pp 1–48
Datar PA, Khedkar SA, Malde AK, Coutinho EC (2006) Comparative residue interaction analysis (CoRIA): a 3D-QSAR approach to explore the binding contributions of active site residues with ligands. J Comput Aided Mol Des 20(6):343–360
Martis EA, Chandarana RC, Shaikh MS, Ambre PK, D’Souza JS, Iyer KR, Coutinho EC, Nandan SR, Pissurlenkar RR (2015) Quantifying ligand–receptor interactions for gorge-spanning acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease. J Biomol Struct Dyn 33(5):1107–1125
Verma J, Khedkar VM, Prabhu AS, Khedkar SA, Malde AK, Coutinho EC (2008) A comprehensive analysis of the thermodynamic events involved in ligand–receptor binding using CoRIA and its variants. J Comput Aided Mol Des 22(2):91–104
Wang T, Wade RC (2001) Comparative binding energy (COMBINE) analysis of influenza neuraminidase—inhibitor complexes. J Med Chem 44(6):961–971
van Gunsteren WF (1993) Molecular dynamics studies of proteins. Curr Opin Struct Biol 3(2):277–281
Mennucci B (2012) Polarizable continuum model. Wiley Interdisc Rev Comput Mol Sci 2(3):386–404
Klamt A, Schuurmann G (1993) COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J Chem Soc Perkin Trans 2(5):799–805
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phy Chem 113(18):6378–6396
Hou T, Wang J, Li Y, Wang W (2010) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. the accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 51(1):69–82
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J Comput Chem 32(5):866–877
Sun H, Li Y, Shen M, Tian S, Xu L, Pan P, Guan Y, Hou T (2014) Assessing the performance of MM/PBSA and MM/GBSA methods. 5. improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys 16(40):22035–22045
Sun H, Li Y, Tian S, Xu L, Hou T (2014) Assessing the performance of MM/PBSA and MM/GBSA methods. 4. accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys 16(31):16719–16729
Xu L, Sun H, Li Y, Wang J, Hou T (2013) Assessing the performance of MM/PBSA and MM/GBSA methods. 3. the impact of force fields and ligand charge models. J Phy Chem B 117(28):8408–8421
Dominy BN, Brooks CL (1999) Development of a generalized born model parametrization for proteins and nucleic acids. J Phy Chem B 103(18):3765–3773
Jayaram B, Sprous D, Beveridge D (1998) Solvation free energy of biomacromolecules: parameters for a modified generalized born model consistent with the AMBER force field. J Phy Chem B 102(47):9571–9576
Onufriev A, Bashford D, Case DA (2000) Modification of the generalized Born model suitable for macromolecules. J Phy Chem B 104(15):3712–3720
Onufriev A, Bashford D, Case DA (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct Funct Bioinf 55(2):383–394
Homeyer N, Gohlke H (2012) Free energy calculations by the molecular mechanics Poisson—Boltzmann surface area method. Mol Inform 31(2):114–122
Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 33(12):889–897
Srinivasan J, Cheatham TE, Cieplak P, Kollman PA, Case DA (1998) Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J Am Chem Soc 120(37):9401–9409
Edinger SR, Cortis C, Shenkin PS, Friesner RA (1997) Solvation free energies of peptides: comparison of approximate continuum solvation models with accurate solution of the Poisson-Boltzmann equation. J Phy Chem B 101(7):1190–1197
Gilson MK, Davis ME, Luty BA, McCammon JA (1993) Computation of electrostatic forces on solvated molecules using the Poisson-Boltzmann equation. J Phy Chem 97(14):3591–3600
Im W, Beglov D, Roux B (1998) Continuum solvation model: computation of electrostatic forces from numerical solutions to the Poisson-Boltzmann equation. Comput Phys Commun 111(1):59–75
Baron R, van Gunsteren WF, Hünenberger PH (2006) Estimating the configurational entropy from molecular dynamics simulations: anharmonicity and correlation corrections to the quasi-harmonic approximation. Trends Phys Chem 11:87–122
Harris S, Laughton C (2007) A simple physical description of DNA dynamics: quasi-harmonic analysis as a route to the configurational entropy. J Phys: Condens Matter 19(7):076103
Case DA (1994) Normal mode analysis of protein dynamics. Curr Opin Struct Biol 4(2):285–290
Karplus M, Kushick JN (1981) Method for estimating the configurational entropy of macromolecules. Macromolecules 14(2):325–332
Tidor B, Karplus M (1993) The contribution of cross-links to protein stability: a normal mode analysis of the configurational entropy of the native state. Proteins Struct Funct Bioinf 15(1):71–79
Aqvist J, Marelius J (2001) The linear interaction energy method for predicting ligand binding free energies. Comb Chem High Throughput Screen 4(8):613–626
Åqvist J, Medina C, Samuelsson J-E (1994) A new method for predicting binding affinity in computer-aided drug design. Protein Eng 7(3):385–391
Hansson T, Marelius J, Åqvist J (1998) Ligand binding affinity prediction by linear interaction energy methods. J Comput Aided Mol Des 12(1):27–35
Åqvist J (1990) Ion-water interaction potentials derived from free energy perturbation simulations. J Phys Chem 94(21):8021–8024
Wang W, Wang J, Kollman PA (1999) What determines the van der waals coefficient β in the LIE (linear interaction energy) method to estimate binding free energies using molecular dynamics simulations? Proteins Struct Funct Bioinf 34(3):395–402
Bajorath J (2002) Integration of virtual and high-throughput screening. Nat Rev Drug Discov 1(11):882–894
Shoichet BK (2004) Virtual screening of chemical libraries. Nature 432(7019):862–865
Zwanzig RW (1954) High-temperature equation of state by a perturbation method. i. nonpolar gases. J Chem Phy 22(8):1420–1426
Zwanzig RW (1955) High-temperature equation of state by a perturbation method. II. polar gases. J Chem Phy 23(10):1915–1922
van Gunsteren WF (1989) Methods for calculation of free energies and binding constants: successes and problems. In: van Gunsteren WF, Weiner PK (eds) Computer simulation of biomolecular systems: theoretical and experimental applications. Escom, Leiden, pp 27–59
van Gunsteren WF, Berendsen HJ (1987) Thermodynamic cycle integration by computer simulation as a tool for obtaining free energy differences in molecular chemistry. J Comput Aided Mol Des 1(2):171–176
Kollman P (1993) Free energy calculations: applications to chemical and biochemical phenomena. Chem Rev 93(7):2395–2417
Chodera JD, Mobley DL, Shirts MR, Dixon RW, Branson K, Pande VS (2011) Alchemical free energy methods for drug discovery: progress and challenges. Curr Opin Struct Biol 21(2):150–160
Shirts MR, Mobley DL, Chodera JD (2007) Alchemical free energy calculations: ready for prime time? Annu Rep Comput Chem D A Dixon 3:41–59
Wang Q, Edupuganti R, Tavares CD, Dalby KN, Ren P (2015) Using docking and alchemical free energy approach to determine the binding mechanism of eEF2K inhibitors and prioritizing the compound synthesis. Front Mol Biosci 2:9
Massova I, Kollman PA (1999) Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J Am Chem Soc 121(36):8133–8143
Chellappan S, Kairys V, Fernandes MX, Schiffer C, Gilson MK (2007) Evaluation of the substrate envelope hypothesis for inhibitors of HIV-1 protease. Proteins Struct Funct Bioinf 68(2):561–567
Nalam MN, Ali A, Altman MD, Reddy GKK, Chellappan S, Kairys V, Özen A, Cao H, Gilson MK, Tidor B (2010) Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J Virol 84(10):5368–5378
Shen Y, Altman MD, Ali A, Nalam MN, Cao H, Rana TM, Schiffer CA, Tidor B (2013) Testing the substrate-envelope hypothesis with designed pairs of compounds. ACS Chem Biol 8(11):2433–2441
Chellappan S, Kiran Kumar Reddy G, Ali A, Nalam MN, Anjum SG, Cao H, Kairys V, Fernandes MX, Altman MD, Tidor B (2007). Design of mutation‐resistant HIV protease inhibitors with the substrate envelope hypothesis. Chem Biol Drug Des 69(5): 298–313
Kairys V, Gilson MK, Lather V, Schiffer CA, Fernandes MX (2009) Toward the design of mutation-resistant enzyme inhibitors: further evaluation of the substrate envelope hypothesis. Chem Biol Drug Des 74(3):234–245
Nalam MN, Ali A, Reddy GKK, Cao H, Anjum SG, Altman MD, Yilmaz NK, Tidor B, Rana TM, Schiffer CA (2013) Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem Biol 20(9):1116–1124
Nalam MN, Schiffer CA (2008) New approaches to HIV protease inhibitor drug design II: testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr Opin HIV AIDS 3(6):642
Hao G-F, Yang G-F, Zhan C-G (2010) Computational mutation scanning and drug resistance mechanisms of HIV-1 protease inhibitors. J Phy Chem B 114(29):9663–9676
Tse A, Verkhivker GM (2015) Molecular determinants underlying binding specificities of the ABL kinase inhibitors: combining alanine scanning of binding hot spots with network analysis of residue interactions and coevolution. PLoS ONE 10(6):e0130203
Hou T, Yu R (2007) Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: mechanism for binding and drug resistance. J Med Chem 50(6):1177–1188
Perryman AL, Lin JH, McCammon JA (2004) HIV-1 protease molecular dynamics of a wild-type and of the V82F/I84V mutant: possible contributions to drug resistance and a potential new target site for drugs. Protein Sci 13(4):1108–1123
Hou T, McLaughlin WA, Wang W (2008) Evaluating the potency of HIV-1 protease drugs to combat resistance. Proteins: Struct, Funct, Bioinf 71(3):1163–1174
Wang W, Kollman PA (2001) Computational study of protein specificity: the molecular basis of HIV-1 protease drug resistance. Proc Natl Acad Sci USA 98(26):14937–14942
Ishikita H, Warshel A (2008) Predicting drug-resistant mutations of HIV protease. Angew Chem Int Ed 47(4):697–700
Singh N, Frushicheva MP, Warshel A (2012) Validating the vitality strategy for fighting drug resistance. Proteins Struct Funct Bioinf 80(4):1110–1122
Acknowledgements
E. A. F. Martis and E. C. Coutinho are grateful to Ian R. Craig, Ph.D. (BASF, Ludwigshafen) for his critical comments and feedback on this chapter. The authors are grateful to Department of Science and Technology (DST), Department of Biotechnology (DBT) and Council of Scientific and Industrial Research (CSIR) for their financial support to build the High-Performance Computing system at the Department of Pharmaceutical Chemistry, Bombay College of Pharmacy. E. A. F. Martis and E. C. Coutinho are also thankful to nVIDIA Corporation for their hardware support grant. E. A. F. Martis is indebted to BASF, Ludwigshafen, Germany for the Ph.D. fellowship and the MCBR4 (2015) consortium (Prof. Dr. P. Comba, University of Heidelberg; Prof. Dr H. Zipse LMU, Munich and Prof. Dr. G. N. Sastry, IICT, Hyderabad for MCBR visiting fellowship to Heine-Heinrich University of Düsseldorf, Germany). E. A. F. Martis would also like to thank Prof. Dr. Holger Gohlke, Heine-Heinrich University of Düsseldorf for his guidance during the sabbatical in his CPCLab. Gratitude is expressed to Sandhya Subash, Ph.D. (Bristol-Meyer-Squibb, India), for her assistance in preparing and proofreading the drafts of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Martis, E.A.F., Coutinho, E.C. (2019). Free Energy-Based Methods to Understand Drug Resistance Mutations. In: Mohan, C. (eds) Structural Bioinformatics: Applications in Preclinical Drug Discovery Process. Challenges and Advances in Computational Chemistry and Physics, vol 27. Springer, Cham. https://doi.org/10.1007/978-3-030-05282-9_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-05282-9_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-05281-2
Online ISBN: 978-3-030-05282-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)