
Abstract
Takayasu arteritis is a rare large-vessel vasculitis that primarily affects young women. Initial evidence for a genetic component to this disease includes a preponderance of risk in East Asian, South Asian, and South American populations, as well as familial aggregation of the disease. The most reproduced genetic association with Takayasu arteritis is with the HLA-B*52 allele, with additional associations in the HLA region described across many ethnicities. Recent studies, including three large-scale genotyping array studies, have found associations with variants in regulatory regions of other immune-related genes, including IL12B, FCGR2A/FCGR3A, IL6, and LILRB3. Several of these variants have been shown to impact the regulation of their associated genes, playing a role in the incompletely understood etiology of Takayasu arteritis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
8.1 Introduction
Takayasu arteritis (TA) is an immune-mediated disease that predominately affects the aorta and its major branches. In 1990 the American College of Rheumatology proposed the presence of three of the following disease criteria for classification of TA: decreased brachial artery pulse, claudication of an extremity, blood pressure discrepancy between arms, subclavian arterial bruit, narrowing of the aorta or its major branches or proximal arteries, and an age of onset less than or equal to 40 years of age [1]. TA has been sometimes referred to as pulseless disease, due to this decrease of brachial artery pulse. Other manifestations of TA may include aortic regurgitation, hypertension, and arterial lesions, specifically in the pulmonary, left mid-common carotid, distal brachiocephalic, and thoracic arteries, as well as the abdominal aorta [1]. The specific etiology of TA is not known, though it has been shown to have a genetic component, based on geographic and familial clustering, immunogenetic analysis, candidate gene studies, and three published large genotyping array experiments.
The pathophysiology of TA is incompletely understood, but the role of infiltrating immune cells, specifically T cells, has been described. The aortic tissues of TA patients are infiltrated with CD4+ T cells, CD8+ T cells, macrophages, natural killer cells, and γδ T cells [2] (Fig. 8.1). Specifically, the infiltrating CD4+ T cells demonstrate a TH1/TH17 phenotype. Compared to healthy controls, TA patients show increased concentrations of IL-2, IL-2R, IL-12, IL-23, IFNγ, TNFα, IL-1RA, and IL-17A. Moreover, CD4+ T cells that produce either IL-17A or IFNγ are increased in active TA patients compared to both TA in remission and healthy controls [3]. Interactions between CD4+ T cells and dendritic cells have also been implicated, as co-localized CD4+ T cells and dendritic cells have been found in inflamed arteries [4]. In addition, the leukocyte adhesion molecules VCAM-1 and ICAM-1 are increased in TA patients [5]. The peripheral blood mononuclear cells (PBMCs) of patients with active TA contain a higher proportion of γδ T cells than healthy controls and a higher proportion of γδ T cells that produce IFNγ [6]. Increased apoptosis of arterial cells has been described in TA, potentially mediated by perforin, which is released by infiltrating CD8+ T cells, γδ T cells, and natural killer cells [2]. Moreover, other costimulatory molecules that enhance natural killer cell-mediated apoptosis, such as 4-1bb, 4-1 bbl, and Fas, are increased in TA patients [7]. To summarize, while inflammation in TA is predominately CD4+ T-cell mediated, CD8+ T cells, γδ T cells, natural killer cells, dendritic cells, and macrophages have been shown to also play a role in the disease etiology.

Summary of immunologic roles of susceptibility genes associated with TA. Immune cells that have been found infiltrated in aortic tissue of TA patients are depicted. Only genes that have been identified in genotyping array studies are included in this summary. All genes that are demonstrably upregulated and all cell populations increased in PBMCs in TA are shown in green, all that are downregulated or decreased are shown in red, and all cells that have no altered prevalence are shown in blue. IL-12B, a component of both IL-23 and IL-12, is involved in TH1/TH17 cell differentiation. IL-6 is required for TH17 differentiation. HLA-DRB1 is a late activation marker of CD4+ T cells, and HLA-DRB1 expressing CD4+ T cells are increased in TA. HLA-B can be expressed by all shown cells and targeted for apoptosis by NK cells. FCGR2A and LILRB3 are both found on the surface of macrophages. LILRB3 binds to MHC class I proteins, and inhibits apoptotic activity of NK cells and γδ T cells. While decreased expression of LILRB3 has not been demonstrated in TA patients, the variant that is associated with TA is correlated to decreased LILRB3 expression in healthy controls. Abbreviations: TH1 T helper 1 cell, TH17 T helper 17 cell, aTreg activated regulatory T cell, DC dendritic cell, NK natural killer cell, T T cell, IL interleukin, CD cluster of differentiation
8.2 Geographic and Familial Localization
Evidence of a genetic component to TA stems from the varying disease prevalence in different racial and ethnic groups. TA is most commonly described in Asian and South American populations [8]. Females are more likely to develop the disease than males across all ethnicities, and it is most commonly diagnosed between the age of 25 and 30 years [9, 10]. However, the exact proportion of female TA patients varies between countries; the highest proportion is in Japan, reporting an 8:1 female to male ratio [9]. Moreover, the prevalence of specific disease manifestations varies across geographic regions. For example, ascending aorta, aortic arch, and ocular involvement is more common among Japanese patients than in the rest of the world. Meanwhile, abdominal arterial involvement, as well as secondary hypertension, is more common in Indian, Chinese, and Thai populations [8]. The geographic localization of disease incidence, as well as the distribution of specific manifestations, suggest that genetic factors contribute to disease pathogenesis and progression.
In addition to patterns in geographic localization, familial aggregation of TA has been well documented. Several studies have shown TA aggregated within families [11,12,13,14,15,16]. Moreover, TA has been reported in monozygotic twins [17, 18]. Each of these family and twin studies suggests a potential genetic association with TA. Determining the genetic differences in these families has focused on human leukocyte antigen (HLA) typing in affected patients and their families. Of the HLA alleles found in affected, but not unaffected, siblings, the HLA-B*52 and HLA-A*24 alleles have been repeatedly described [12]. However, not all family studies show consistent and unique similarities between affected siblings compared to unaffected siblings [14, 18]. This suggests a role for both non-HLA polymorphisms and environmental factors in disease progression.
8.3 Associations Within the HLA Region
Research focused on identifying specific genetic components of TA initially focused on HLA types. An association between HLA-B*52 and TA was first documented in 1978, in a Japanese population [19]. This association has since been robustly described in other ethnicities, including in northern Indian [20], Thai [21], Mexican [22], Turkish [23], European-American [24], Greek [25], and Korean [26] cohorts, in addition to other Japanese cohorts [27,28,29] (Table 8.1). Variable prevalence of these HLA allelotypes throughout the world has been repeatedly put forward as an explanation for the geographic localization of TA patients, as the HLA-B*52 allele is more common in Japan compared to the rest of the world. In more recent studies, the allelotype HLA-B*52-01 has been specifically implicated. Large population studies have also identified other associated alleles. In Japanese cohorts, HLA-B*39, HLA-DRB1*15-02, HLA-DQA1*01-03, HLA-DQB1*06-01, HLA-B*67-01, and HLA-DPB1*09-01 have also been associated with TA [30,31,32,33,34]. In a Han Chinese cohort, HLA-DRB1*07 was associated with TA, as well as a DQA1*03-01-DQB1*03-01-DRB1*07 haplotype [35]. Additional associated HLA alleles have been described in multiple ethnicities (Table 8.1). In a Korean cohort, HLA-A*30-01 and HLA-DRB1*15-02 were associated with disease risk, while HLA-A*26-02 was found to be protective [26]. In Mexican patients, HLA-B*15 was associated with TA alongside HLA-B*52 [22]. In multiple populations, HLA-DPB1*09-01 has been found to be more common in TA patients [33, 34, 36]. In Han Chinese patients, this allele and the HLA-DPB1*17-01 allele have been correlated with an earlier age of onset [36]. However, the HLA-DPB1*09-01 allele is commonly found alongside HLA-B*52, so this association may be due to a linkage disequilibrium effect and may not have an independent effect on disease etiology [36]. Because concurrent associations of allelotypes of separate genes may be due to linkage disequilibrium, the exact HLA variation that directly influences the mechanism of disease can be difficult to elucidate.
8.4 Non-HLA Candidate Gene Studies
Candidate gene studies targeting non-HLA loci have been used to identify additional genetic risk loci in TA (Table 8.2). Genes were selected for study based on the potential functional relevance of the proteins they code to disease etiology. The serum concentrations of the cytokines IL-12 and IL-6 have been shown to be increased in TA patients compared to healthy controls, while expression of IL-2 has been implicated in TA pathogenesis [3, 37]. As genetic polymorphisms can affect the rate of transcription of genes, as well as alter the activity of the translated proteins, these genes have been selected for a candidate gene study in a Turkish cohort. It was found that certain genotypes of IL12B (rs3213113, CC), IL6 (rs1800795, GG), and IL2 (rs2069762 TT) were more common in TA patients compared to healthy controls [37]. Other candidate genes that were selected based on their pro-inflammatory activity or role in other autoimmune diseases have been shown to be associated with TA. The cytokine IL-1 plays a role both in pro-inflammatory response and in vascular smooth muscle proliferation, so IL1 and IL1RN polymorphisms were analyzed in a Mexican cohort [38]. Significant associations between TA and multiple genotypes within IL1 and IL1RN were described (rs3811058 C, rs315952 C, and rs315951 G alleles; rs3811058 TC, rs419598 TT, and rs315952 TC genotypes). Another Mexican cohort demonstrated a preponderance of polymorphisms in PON1 (rs854560 and rs705379) in TA patients [39]. Furthermore, a PON1 enzyme with these polymorphisms was shown to be less active than the wild-type PON1. Nevertheless, polymorphisms in promising candidate genes are not always found to be relevant to the disease. In two studies focused on Turkish cohorts, polymorphisms around the genes PDCD1 (PD-1.3, PD-1.5, PD-1.6) [40] and PTPN22 (rs2476601) [41] were equally frequent in TA patients and healthy controls. This series of studies in Turkish and Mexican cohorts has identified several genetic polymorphisms within proteins that could be directly involved with TA etiology.
8.5 Insights from Genotyping Array Experiments
Large-scale genotyping array experiments have been used to identify novel genetic susceptibility loci in TA. One genome-wide association study (GWAS) has been performed comparing TA patients to healthy controls, using both a Turkish and a European-American cohort (Figs. 8.1 and 8.2). In a meta-analysis of both cohorts, this study found significant associations between TA and three loci: IL6 (rs2069837, meta-analysis OR, 2.1; p, 6.7 × 10−9), RPS9/LILRB3 (rs11666453, OR, 1.6; p, 2.34 × 10−8), and a locus on chromosome 21q22, near PSMG1 (rs2836878, OR, 1.8; p, 3.62 × 10−10) (Table 8.3) [42]. The cytokine IL-6 plays a role in T-cell differentiation. It is required for TH17 differentiation, and also inhibits Treg differentiation, which may play a role in the increased TH17 and decreased Treg populations seen in active TA [3, 43]. Because the associated SNP rs2069837 is found within an enhancer region of IL6, it may prove to play a role in the dysregulation of IL-6 shown in TA. Variation in LILRB3 may also directly affect pathogenesis. While LILRB3 is involved in repression of the anti-MHC class 1 immune response, the variant rs11666453 is associated with a decrease in LILRB3 expression [42]. As HLA-B, an MHC class I gene, has been repeatedly implicated in TA, this suggests that variation negatively affecting the expression of LILRB3 may play a role in this same disease pathway. Variations affecting the function of LILRB3 and IL6 may directly affect the pathways involved in disease etiology.

Overlapping ontologies of genes with variants associated with TA, as identified by genotyping array studies. Listed are gene ontology (GO) terms shared between both genes in each cluster, identified using the UniProt-GOA database [57]. Every gene identified is annotated with the GO term “Immune System Processes” (GO:0002376) or “Immune Response” (GO:0006955). IL12B and IL6 are both annotated with multiple pathogen defense GO terms; the overlapping terms are defense response to gram-negative bacteria, defense response to virus, and defense response to protozoan. Each gene with variants associated with TA in a large genotyping study has an immune-related function, with specific ontologies shared between genes listed
Two targeted arrays have been performed to identify additional genetic associations with TA. A Turkish cohort and a European-American cohort were also fine mapped using an Immuno BeadChip Microarray (ImmunoChip), which targets immune-related genes and genes that have been previously associated with immune-mediated diseases (Figs. 8.1 and 8.2). Genetic associations with a GWAS level of significance were established in FCGR2A/FCGR3A (rs10919543, meta-analysis OR, 1.81; p, 5.89 × 10−12) and IL12B (rs56167332, meta-analysis OR, 1.54; p, 2.18 × 10−8) [24]. The FCGR2A/FCGR3A polymorphism is associated with increased FCGR2A expression in lymphoblastoid cells. Typically, FCGR2A is expressed on macrophages and neutrophils, and is involved in phagocytosis, so this variant may affect the activity of the infiltrating macrophages found in TA [44]. In the HLA region, a novel association with the HLADRB1/HLADQA1 locus (rs113452171, OR, 2.34; p, 3.74 × 10−9; rs189754752, OR, 2.47; p, 4.22 × 10−9) was independent of a HLA-B*52 effect. Along with HLA-B*52-01, TA patients demonstrated an increased frequency of HLA-Cw*12-02 and rs12524487, an intergenic SNP near HLA-B not associated with an HLA allelotype. However, because HLA-Cw*12-02 and rs12524487 are in linkage disequilibrium with HLA-B*52-01, these could all be part of the same genetic effect.
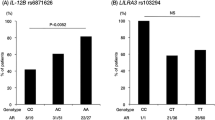
Genetic effects have been also described via genotyping arrays in two Japanese cohorts. Using an Exome BeadChip, an array targeting exonic variants, a polymorphism within the IL12B (rs6871626, meta-analysis OR, 1.76; p, 1.7 × 10−13) locus was described [27]. This genetic variant is in strong linkage disequilibrium with rs56167332, the index SNP in IL12B associated with TA in the ImmunoChip study described above [24] (Figs. 8.1 and 8.2). In Japanese patients, the IL12B locus was also found to be associated with aortic regurgitation (p, 0.0046). In addition, in these Japanese cohorts, a suggestive association with the transcription factor gene MLX was described, but this did not reach a genome level of significance (rs665268, p, 5.2 × 10−7). This same study validated the well-described association with HLA-B*52 (OR, 2.44; p, 2.8 × 10−21).
Both of these genotyping studies identified polymorphisms in IL12B that contribute to genetic risk for TA. There are multiple mechanisms by which differences of the expression of IL-12B may contribute to disease progression. The IL-12B subunit, p40, is common to both IL-12 and IL-23, and increased levels of both cytokines have been reported in TA patients [3]. IL-12 is involved in TH1 differentiation, and IL-23 is important for TH17 cell maintenance; furthermore, both cell types are increased in active TA [45, 46]. As part of the TH1 response, IL-12 activates IFNγ production and related signaling cascades; moreover, IL-12-stimulated PBMCs have been shown to inhibit angiogenesis by inhibiting endothelial cell growth in an IFNγ-dependent manner [47]. It has been suggested that this inhibition of angiogenesis leads to slower healing of aortic lesions that form during active TA [44].
The genetic variants identified from these array-based genotyping studies were subsequently examined with a candidate gene association study in Han Chinese patients and controls. The FCGR2A/FCG3A variant (rs10919543), previously identified in Turkish and European-American populations, was replicated (Table 8.3) [48]. However, this same study found no effect in the variants previously described in the MLX, HLA-B/MICA, and IL12B (rs665268, rs12524487, and rs56167332, respectively). This suggests the presence of ethnicity-specific genetic loci in TA. The genetic association between TA and the FCGR2A/FCGR3A locus was further validated in Han Chinese patients in a larger subsequent study [49].
8.6 Similarities with Other Diseases
Some of the genetic associations described in TA are shared by other immune-mediated diseases. TA and ulcerative colitis (UC) have both been associated with a risk locus in FCGR2A/FCGR3A and the locus on chromosome 21q22 [24]. In a study of 470 TA patients, concurrent UC was found to be significantly higher than the prevalence of UC in Japan (6.4% vs. 0.11%; p, 1.0 × 10−10) [50], confirming earlier observations [51]. It was also found that UC patients develop TA earlier than patients with only a TA diagnosis, though there is no evidence that one disease usually precedes the other. Genetically, HLA-B*52 frequency, but not rs6871626 (IL12B) allele frequency, was increased in patients with both diseases, compared to patients with only TA (OR,12.14; p, 1.0 × 10−5) [50]. This enforces the observation that UC and TA share genetic risk loci [24], leading to the increased comorbidity of both diseases. Furthermore, TA has been shown to be genetically distinct from other diseases. The HLA-B alleles associated with TA and tuberculosis have been shown to be genetically distinct, despite the occurrence of arterial lesions in both diseases [52].
Genetic similarities between TA and giant cell arteritis (GCA) have been evaluated on a large scale using the ImmunoChip platform [53]. GCA is another large-vessel vasculitis, which similar to TA can also affect the aorta, but unlike TA, generally affects older patients. After comparing variant genotypes through a meta-analysis using ImmunoChip genetic data in both diseases, it was found that both TA and GCA share the IL12B risk locus (rs755374, p, 7.54 × 10−7). Moreover, GCA was found to be predominantly HLA class II mediated, significantly associated with the HLA-DRB1/HLA-DQA1 locus, while the HLA class I effect focused around HLA-B/MICA was again validated in TA (most significant variants, rs9405038 and rs12524487, respectively) [53].
8.7 Conclusions and Future Implications
Until very recently, the genetic association with HLA-B*52 was the only genetic risk locus confirmed for TA in multiple ethnicities. Progress in the last few years has led to the discovery and replication of several additional genetic risk loci with a GWAS level of significance for this complex disease. These findings shed light into the disease pathogenesis and implicate key immune-related mechanisms that can be potentially targeted for therapy. However, despite this progress, the functional consequences for the genetic risk variants identified in TA are still largely unknown, and the limited sample size for studies performed to date precluded from the identification of the complete genetic makeup underlying this disease.
Very little is known about how the genetic risk variants for TA contribute to disease severity or other clinical characteristics. There is some evidence that both the HLA-B*52 allele and the IL12B risk locus are associated with disease severity. HLA-B*52 has been associated with use of higher prednisone doses, higher C-reactive protein levels, early disease onset, more extensive aortic involvement, left ventricular dysfunction, and aortic regurgitation in TA [23, 54, 55]. In a Japanese cohort, individuals with the risk allele in IL12B (rs6871626-A) had a higher probability for disease relapse [56]. These observations suggest that genetic information could be developed to help in better assessing disease prognosis and outcome in TA in the future. Further, as the functional mechanisms impacted by each genetic susceptibility variant are more thoroughly understood, novel therapeutic options and individualized treatment plans for TA patients could become available.
References
Arend WP, Michel BA, Bloch DA, Hunder GG, Calabrese LH, Edworthy SM, Fauci AS, Leavitt RY, Lie JT, Lightfoot RW Jr, et al. The American College of Rheumatology 1990 criteria for the classification of Takayasu arteritis. Arthritis Rheum. 1990;33:1129–34.
Seko Y, Minota S, Kawasaki A, Shinkai Y, Maeda K, Yagita H, Okumura K, Sato O, Takagi A, Tada Y, et al. Perforin-secreting killer cell infiltration and expression of a 65-kD heat-shock protein in aortic tissue of patients with Takayasu’s arteritis. J Clin Invest. 1994;93:750–8.
Saadoun D, Garrido M, Comarmond C, Desbois AC, Domont F, Savey L, Terrier B, Geri G, Rosenzwajg M, Klatzmann D, et al. Th1 and Th17 cytokines drive inflammation in Takayasu arteritis. Arthritis Rheumatol. 2015;67:1353–60.
Inder SJ, Bobryshev YV, Cherian SM, Wang AY, Lord RS, Masuda K, Yutani C. Immunophenotypic analysis of the aortic wall in Takayasu’s arteritis: involvement of lymphocytes, dendritic cells and granulocytes in immuno-inflammatory reactions. Cardiovasc Surg. 2000;8:141–8.
Noguchi S, Numano F, Gravanis MB, Wilcox JN. Increased levels of soluble forms of adhesion molecules in Takayasu arteritis. Int J Cardiol. 1998;66(Suppl 1):S23–33; discussion S35-26.
Chauhan SK, Tripathy NK, Sinha N, Nityanand S. T-cell receptor repertoire of circulating gamma delta T-cells in Takayasu’s arteritis. Clin Immunol. 2006;118:243–9.
Seko Y, Sugishita K, Sato O, Takagi A, Tada Y, Matsuo H, Yagita H, Okumura K, Nagai R. Expression of costimulatory molecules (4-1BBL and Fas) and major histocompatibility class I chain-related A (MICA) in aortic tissue with Takayasu’s arteritis. J Vasc Res. 2004;41:84–90.
Numano F. Differences in clinical presentation and outcome in different countries for Takayasu’s arteritis. Curr Opin Rheumatol. 1997;9:12–5.
Numano F, Okawara M, Inomata H, Kobayashi Y. Takayasu’s arteritis. Lancet. 2000;356:1023–5.
Phillip R, Luqmani R. Mortality in systemic vasculitis: a systematic review. Clin Exp Rheumatol. 2008;26:S94–104.
Jeeva I, Sajid J, Ali O, Bonthron DT, Frossard PM. Atypical Takayasu arteritis: a family with five affected siblings. Med Sci Monit. 2007;13:CS101–5.
Morishita KA, Rosendahl K, Brogan PA. Familial Takayasu arteritis—a pediatric case and a review of the literature. Pediatr Rheumatol Online J. 2011;9:6.
Numano F. Hereditary factors of Takayasu arteritis. Heart Vessels Suppl. 1992;7:68–72.
Makino N, Senda Y, Yamaguchi Y. Takayasu’s disease in two brothers. Analysis of HLA types. Br Heart J. 1981;46:446–8.
Kodama K, Kida O, Morotomi Y, Tanaka K. Male siblings with Takayasu’s arteritis suggest genetic etiology. Heart Vessel. 1986;2:51–4.
Heo JH, Kim M. Familial Takayasu’s arteritis in female siblings. Rheumatol Int. 2011;31:815–8.
Numano F, Isohisa I, Kishi U, Arita M, Maezawa H. Takayasu’s disease in twin sisters. Possible genetic factors. Circulation. 1978;58:173–7.
Enomoto S, Iwasaki Y, Bannai S, Nara Y, Matsuoka A, Aizawa Y, Shibata A. Takayasu’s disease in twin sisters. Jpn Heart J. 1984;25:147–52.
Isohisa I, Numano F, Maezawa H, Sasazuki T. HLA-Bw52 in Takayasu disease. Tissue Antigens. 1978;12:246–8.
Mehra NK, Jaini R, Balamurugan A, Kanga U, Prabhakaran D, Jain S, Talwar KK, Sharma BK. Immunogenetic analysis of Takayasu arteritis in Indian patients. Int J Cardiol. 1998;66(Suppl 1):S127–32; discussion S133.
Charoenwongse P, Kangwanshiratada O, Boonnam R, Hoomsindhu U. The association between the HLA antigens and Takayasu’s arteritis in Thai patients. Int J Cardiol. 1998;66(Suppl 1):S117–20.
Vargas-Alarcon G, Flores-Dominguez C, Hernandez-Pacheco G, Zuniga J, Gamboa R, Soto ME, Granados J, Reyes PA. Immunogenetics and clinical aspects of Takayasu’s arteritis patients in a Mexican Mestizo population. Clin Exp Rheumatol. 2001;19:439–43.
Sahin Z, Bicakcigil M, Aksu K, Kamali S, Akar S, Onen F, Karadag O, Ozbalkan Z, Ates A, Ozer HT, et al. Takayasu’s arteritis is associated with HLA-B*52, but not with HLA-B*51, in Turkey. Arthritis Res Ther. 2012;14:R27.
Saruhan-Direskeneli G, Hughes T, Aksu K, Keser G, Coit P, Aydin SZ, Alibaz-Oner F, Kamali S, Inanc M, Carette S, et al. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am J Hum Genet. 2013;93:298–305.
Karageorgaki ZT, Bertsias GK, Mavragani CP, Kritikos HD, Spyropoulou-Vlachou M, Drosos AA, Boumpas DT, Moutsopoulos HM. Takayasu arteritis: epidemiological, clinical, and immunogenetic features in Greece. Clin Exp Rheumatol. 2009;27:S33–9.
Lee SW, Kwon OJ, Park MC, Oh HB, Park YB, Lee SK. HLA alleles in Korean patients with Takayasu arteritis. Clin Exp Rheumatol. 2007;25:S18–22.
Terao C, Yoshifuji H, Kimura A, Matsumura T, Ohmura K, Takahashi M, Shimizu M, Kawaguchi T, Chen Z, Naruse TK, et al. Two susceptibility loci to Takayasu arteritis reveal a synergistic role of the IL12B and HLA-B regions in a Japanese population. Am J Hum Genet. 2013;93:289–97.
Yajima M, Numano F, Park YB, Sagar S. Comparative studies of patients with Takayasu arteritis in Japan, Korea and India—comparison of clinical manifestations, angiography and HLA-B antigen. Jpn Circ J. 1994;58:9–14.
Yoshida M, Kimura A, Katsuragi K, Numano F, Sasazuki T. DNA typing of HLA-B gene in Takayasu’s arteritis. Tissue Antigens. 1993;42:87–90.
Kobayashi Y, Numano F. 3. Takayasu arteritis. Intern Med. 2002;41:44–6.
Terao C, Yoshifuji H, Ohmura K, Murakami K, Kawabata D, Yurugi K, Tazaki J, Kinoshita H, Kimura A, Akizuki M, et al. Association of Takayasu arteritis with HLA-B 67:01 and two amino acids in HLA-B protein. Rheumatology (Oxford). 2013;52:1769–74.
Takamura C, Ohhigashi H, Ebana Y, Isobe M. New human leukocyte antigen risk allele in Japanese patients with Takayasu arteritis. Circ J. 2012;76:1697–702.
Kimura A, Kitamura H, Date Y, Numano F. Comprehensive analysis of HLA genes in Takayasu arteritis in Japan. Int J Cardiol. 1996;54(Suppl):S61–9.
Dong RP, Kimura A, Numano F, Yajima M, Hashimoto Y, Kishi Y, Nishimura Y, Sasazuki T. HLA-DP antigen and Takayasu arteritis. Tissue Antigens. 1992;39:106–10.
Lv N, Wang Z, Dang A, Zhu X, Liu Y, Zheng D, Liu G. HLA-DQA1, DQB1 and DRB1 alleles associated with Takayasu arteritis in the Chinese Han population. Hum Immunol. 2015;76:241–4.
Lv N, Dang A, Wang Z, Zheng D, Liu G. Association of susceptibility to Takayasu arteritis in Chinese Han patients with HLA-DPB1. Hum Immunol. 2011;72:893–6.
Saruhan-Direskeneli G, Bicakcigil M, Yilmaz V, Kamali S, Aksu K, Fresko I, Akkoc N, Kiraz S, Ozer HT, Tunc E, et al. Interleukin (IL)-12, IL-2, and IL-6 gene polymorphisms in Takayasu’s arteritis from Turkey. Hum Immunol. 2006;67:735–40.
Soto Lopez ME, Gamboa Avila R, Hernandez E, Huesca-Gomez C, Castrejon-Tellez V, Perez-Mendez O, Reyes PA, Fragoso-Lona JM, Vargas-Alarcon G, Cruz-Robles D. The interleukin-1 gene cluster polymorphisms are associated with Takayasu’s arteritis in Mexican patients. J Interf Cytokine Res. 2013;33:369–75.
Huesca-Gomez C, Soto ME, Castrejon-Tellez V, Perez-Mendez O, Gamboa R. PON1 gene polymorphisms and plasma PON1 activities in Takayasu’s arteritis disease. Immunol Lett. 2013;152:77–82.
Direskeneli H, Tuna-Erdogan E, Gunduz F, Bandurska-Luque A, Alparslan B, Kebe M, Uyar FA, Bicakcigil M, Aksu K, Kamali S, et al. PDCD1 polymorphisms are not associated with Takayasu’s arteritis in Turkey. Clin Exp Rheumatol. 2012;30:S11–4.
Sahin N, Aksu K, Kamali S, Bicakcigil M, Ozbalkan Z, Fresko I, Ozer H, Akar S, Onat AM, Cobankara V, et al. PTPN22 gene polymorphism in Takayasu’s arteritis. Rheumatology (Oxford). 2008;47:634–5.
Renauer PA, Saruhan-Direskeneli G, Coit P, Adler A, Aksu K, Keser G, Alibaz-Oner F, Aydin SZ, Kamali S, Inanc M, et al. Identification of Susceptibility Loci in IL6, RPS9/LILRB3, and an Intergenic Locus on Chromosome 21q22 in Takayasu Arteritis in a Genome-Wide Association Study. Arthritis Rheumatol. 2015;67:1361–8.
Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6:a016295.
Terao C, Yoshifuji H, Mimori T. Recent advances in Takayasu arteritis. Int J Rheum Dis. 2014;17:238–47.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–44.
Strasly M, Cavallo F, Geuna M, Mitola S, Colombo MP, Forni G, Bussolino F. IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J Immunol. 2001;166:3890–9.
Qin F, Wang H, Song L, Lu XL, Yang LR, Liang EP, Wang W, Zou YB, Bian J, Wu HY, et al. Single nucleotide polymorphism rs10919543 in FCGR2A/FCGR3A region confers susceptibility to Takayasu arteritis in Chinese population. Chin Med J. 2016;129:854–9.
Chen S, Wen X, Li J, Li Y, Li L, Tian X, Yuan H, Zhang F, Li Y. Association of FCGR2A/FCGR3A variant rs2099684 with Takayasu arteritis in the Han Chinese population. Oncotarget. 2017;8:17239–45.
Terao C, Matsumura T, Yoshifuji H, Kirino Y, Maejima Y, Nakaoka Y, Takahashi M, Amiya E, Tamura N, Nakajima T, et al. Takayasu arteritis and ulcerative colitis: high rate of co-occurrence and genetic overlap. Arthritis Rheumatol. 2015;67:2226–32.
Watanabe R, Ishii T, Nakamura K, Shirai T, Fujii H, Saito S, Harigae H. Ulcerative colitis is not a rare complication of Takayasu arteritis. Mod Rheumatol. 2014;24:372–3.
Soto ME, Vargas-Alarcon G, Cicero-Sabido R, Ramirez E, Alvarez-Leon E, Reyes PA. Comparison distribution of HLA-B alleles in mexican patients with takayasu arteritis and tuberculosis. Hum Immunol. 2007;68:449–53.
Carmona FD, Coit P, Saruhan-Direskeneli G, Hernandez-Rodriguez J, Cid MC, Solans R, Castaneda S, Vaglio A, Direskeneli H, Merkel PA, et al. Analysis of the common genetic component of large-vessel vasculitides through a meta-Immunochip strategy. Sci Rep. 2017;7:43953.
Origuchi T, Fukui S, Umeda M, Nishino A, Nakashima Y, Koga T, Kawashiri SY, Iwamoto N, Ichinose K, Tamai M, et al. The severity of Takayasu arteritis is associated with the HLA-B52 allele in Japanese patients. Tohoku J Exp Med. 2016;239:67–72.
Renauer P, Sawalha AH. The genetics of Takayasu arteritis. Presse Med. 2017;46(7–8 Pt 2):e179–87.
Matsumura T, Amiya E, Tamura N, Maejima Y, Komuro I, Isobe M. A novel susceptibility locus for Takayasu arteritis in the IL12B region can be a genetic marker of disease severity. Heart Vessel. 2016;31:1016–9.
Huntley RP, Sawford T, Mutowo-Meullenet P, Shypitsyna A, Bonilla C, Martin MJ, O’Donovan C. The GOA database: gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015;43:D1057–63.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gensterblum, E., Sawalha, A.H. (2019). Takayasu Arteritis. In: Martín, J., Carmona, F. (eds) Genetics of Rare Autoimmune Diseases. Rare Diseases of the Immune System. Springer, Cham. https://doi.org/10.1007/978-3-030-03934-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-03934-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-03933-2
Online ISBN: 978-3-030-03934-9
eBook Packages: MedicineMedicine (R0)