
Abstract
An increasing proportion of patients with cardiovascular disease have higher acuity of disease and may have ferromagnetic implants with potential for interaction with the MRI environment. Familiarity with each device class and its potential for electromagnetic interaction is essential for radiologists and cardiologists performing MRI examinations in this population of patients. The final decision to perform MRI in patients with electronic devices or other implants is frequently made by considering the potential benefit of MRI relative to the attendant risks associated with various devices. While techniques for safe imaging with MRI in the setting of certain devices have been developed, the potential for catastrophic complications exists and dictates a high degree of vigilance to minimize patient risk.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- ECG leads
- Sternal wires
- Epicardial wires
- Stents
- Guidewires
- Swan-Ganz and thermodilution catheters
- Balloon pumps
- Gadolinium-based contrast agents
Introduction
Cardiovascular magnetic resonance imaging (MRI) has become the modality of choice in the evaluation and treatment of cardiovascular disorders. Cardiac MRI allows for a comprehensive noninvasive assessment of cardiovascular anatomy and physiology with unsurpassed soft tissue resolution and multi-planar imaging capability without the need for ionizing radiation. Cardiac MRI is a safe imaging modality without any side effects in the vast majority of patients. However, with the growing number of patients with higher acuity of disease referred to cardiac MRI, some specific safety and monitoring issues should be addressed.
First, the number of patients treated with permanently or temporarily implanted cardiovascular devices with potential electromagnetic interaction with the MRI environment is increasing. Familiarity with each device class and its potential for electromagnetic interaction is essential for radiologists and cardiologists performing cardiac MRI. There is a remarkable and ever-increasing variety of implanted medical devices, which have the potential for interaction with the powerful main and gradient magnetic fields imposed by the MRI scanner as well as the radio-frequency (RF) energy that is deposited in the patient. Therefore, the risk associated with implanted ferromagnetic devices may be lower in magnets with lower field strengths relative to high field units.
Second, the (repeated) use of gadolinium-based contrast agents (GBCAs) in cardiac MRI, especially in patients with reduced kidney function, is associated with rare though significant side effects. The most important and potentially fatal side effect is nephrogenic systemic fibrosis (NSF) which involves fibrosis of the skin and internal organs and is believed to be caused by prolonged tissue exposure to GBCAs in patients with reduced kidney function. Another only recently reported possible adverse effect involves gadolinium deposition in the brain of patients who received multiple doses of GBCAs. To date, no adverse health effects of gadolinium deposition in the brain have been identified, though further research is warranted and preventive recommendations should be adopted. Prevention of these adverse reactions involves identification of patients at risk, risk-benefit evaluation of the need for contrast administration in cardiac MRI, understanding the risks associated with different types of GBCAs, and considerations of alternative methods of diagnostic imaging (e.g., computed tomography or ultrasound) relative to MRI.
Finally, safety issues of cardiac MRI in pregnant or lactating women include possible adverse biological effects of the magnetic fields, RF energy deposition, acoustic noise, or fetal exposure to GBCAs. While there is to date no indication that CMR during pregnancy has produced deleterious effects in humans, data is limited and additional investigations are warranted to fully characterize the risks associated with CMR in pregnant patients. Cardiac MRI can generally be used on pregnant patients if other forms of diagnostic imaging are inadequate or would require exposure to ionizing radiation.
Because of the wide range of circumstances encountered in the MRI suite, the descriptions that follow should not be construed as recommendations that are appropriate for all patients. The reader is encouraged to consult dedicated handbooks or websites dedicated to MRI safety (e.g., www.mrisafety.com) that provide more specific information regarding specific safety concerns. The final decision to perform MRI is frequently made by considering the potential benefit of MRI relative to the associated risks.
Patient Condition and Monitoring
As the indications for cardiovascular MRI expand, patients with higher acuity of disease and increased dependence on monitoring and intervention are referred for MRI. Patients with cardiovascular disease are often referred for imaging in the setting of arrhythmias, hypotension, myocardial ischemia, or congestive heart failure. Contrast administration, prolonged supine imaging, and interaction with implantable or temporary devices may lead to changes in patient condition during the MRI examination. Appropriate monitoring with continuous electrocardiogram (ECG) telemetry, pulse oximetry, non-invasive blood pressure, and a well-rehearsed plan for monitoring of patient symptoms and treatment are essential for all cardiac MRI studies. To perform MRI safely, some devices may have to be disabled, and patient monitoring is limited. To increase safety, steps need to be taken to replace the disabled function.
Potential for Interaction with Implanted Devices
Ferromagnetic materials in a magnetic field are subject to force and torque. The potential for movement of an implanted device in the MRI environment depends on the magnetic field strength, ferromagnetic properties of the material, the implant distance from the magnet bore, and the stability of the implant [1].
The RF and pulsed-gradient magnetic fields in the MRI environment may induce electrical currents in leads and other ferromagnetic wires within the field. Implant length (vs. the RF wavelength) and conformations such as loops favor improved transition of energy to the implanted device. RF pulses may also lead to implant heating and tissue damage at the device-tissue interface.
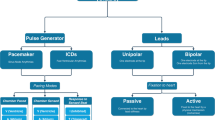
Sophisticated electronic implants, such as those in neurostimulators , pacemakers , and implantable cardioverter defibrillators (ICDs), have the potential for receiving electromagnetic interference in the MRI environment, resulting in programming changes or loss of function.
Patient Screening
Given the potential risks listed, it is essential to conduct a systematic review of the patient’s condition, implanted devices, and safety for MRI. At our institution, all patients are asked to review, answer, and sign a safety questionnaire (Fig. 9.1 ). A time-out is the last step immediately before starting the MRI to perform a final check to make sure the correct patient undergoes the correct MRI procedure. The latest data regarding the MRI safety of devices commonly used in cardiovascular patients are reviewed here.


Sample patient safety questionnaire
Devices with Potential for Interaction with Magnetic Resonance Imaging
ECG Leads
Metallic telemetry leads used routinely for patient monitoring can induce artifacts and may heat in the MRI environment, resulting in skin burns. Specially designed non-ferromagnetic ECG leads and filtered monitoring systems have been designed for the MRI environment up to 1.5 T (e.g., S/5 MRI Monitor, Datex-Ohmeda, Finland; http://www.gehealthcare.fi/kliiniset_jarjestelmat/hoitoalueet/perioperatiivinenhoito/potilasvalvonta/fi_FI/iMMMRI/_files/76784165497602959/default/MRI_8004924.pdf). Such systems also offer continuous SpO2 monitoring, which in our experience is an invaluable tool for monitoring of the cardiac rhythm, especially when the ECG signal, despite filtering, becomes unreadable in the setting of specific MRI pulse sequences.
Sternal Wires
Sternal wires used for closure after thoracotomy procedures are typically made of stainless steel, which is minimally ferromagnetic. Animal studies have suggested the safety of MRI in this setting [2]. Over the course of 15 years in a large acute care hospital setting, one patient had chest discomfort that was classified as “possibly” related to sternal wires; the MRI in this case was discontinued, with resolution of symptoms and no further complications. However, sternal wires, similar to any other metallic implant, typically induce susceptibility artifacts in the immediate area and may limit imaging of the anterior right ventricle [3].
Epicardial Wires
Temporary epicardial pacing wires are routinely cut short at the skin and left in place after cardiac surgery. There are reports of safe performance of MRI in patients with such retained temporary wires [4, 5]. Permanent epicardial pacing leads and patches placed at cardiac surgery, however, have more ferromagnetic materials and are prone to heating in the MRI environment. Unlike endovascular leads, these devices are not cooled by the blood pool, and in experimental models up to 20 °C of heating has been observed [6]. For this reason, at our institution, we do not scan patients with permanent epicardial leads and patches. Temporary epicardial wires are removed prior to MRI whenever possible.
Prosthetic Valves, Annuloplasty Rings, and Transcatheter Valves
Ex vivo studies of a variety of current prosthetic valves and annuloplasty rings , including the mechanical St. Jude, Bjork-Shiley, and Carbo-Medics and bioprosthetic (with metal struts) such as the Hancock, have shown minimal torque and heating (<0.8 °C) at 1.5 T and with specific absorption rates limited to 1.1 W/kg [7,8,9]. Artifact size correlated with the amount of metal in the device and was exaggerated on gradient echo pulse sequences.
Shellock also evaluated prosthetic valve and annuloplasty ring safety at 3 T, revealing minor magnetic field interactions [10]. Studies to determine the force required to cause partial or total detachment of a heart valve prosthesis in patients with degenerative valvular disease have found that forces significantly higher than those induced at 4.7 T would be required to pull a suture through the valve annulus tissue [11].
The only prosthetic valves previously thought to have potential for experiencing enough force and torque to cause clinically concerning problems were the Star-Edwards pre-6000 series prostheses. Deflection measurements at 1.5 T revealed forces similar to peak forces exerted by the beating heart itself [7], leading to initial recommendations to exclude patients with this device series from MRI procedures. However, later studies revealed lower peak forces exerted even on this prosthetic series, and cardiac MRI is considered safe in these patients [12]. Mechanical valves do not appear to be prone to induced lead currents [9].
Although there are no reports of patient injury in the MRI environment caused by the presence of a heart valve, there are theoretical concerns about MRI at 1.5 T and higher field strengths. One such theoretical concern is the tendency of a metallic object to develop an opposing magnetic field to that through which it moves (the Lenz effect). Such a secondary magnetic field may result in a resistive pressure to opening and closing of a disk prosthesis within the valve [13]. Bjork-Shiley convex/concave heart valves (Shiley, Irvine, CA) are associated with an increased risk of mechanical failure because of outlet strut fracture. These valves are associated with large susceptibility artifacts under MRI, and such artifacts may increase in size in fractured valves [14].
Edwards et al. evaluated multiple heart valve prostheses for MRI-related forces in static fields up to 4.7 T. Most were found to be safe based on current criteria. However, valves made of Elgiloy, a Ni-Co-Cr base paramagnetic engineering material [15], such as the Carpentier-Edwards Physio Ring were found to be prone to rotational forces at such high field strengths [16].
Transcatheter-placed heart valves are sutureless and therefore could be more susceptible to magnetically induced valve dehiscence during MRI. Percutaneous aortic valves in particular have the theoretical potential for coronary occlusion with dislodgement. Limited initial reports have shown cardiac MRI to be safe in patients with pulmonary and aortic transcatheter-placed valves [17], though further studies are warranted and specific guidelines should be followed, most frequently found at the company’s website. If MRI is deemed necessary, a waiting period of 6-week post-implant to allow firm fixation by tissue ingrowth is preferred, similar to recommendations for sutureless septal occluder devices.
Coronary Stents
In vitro studies have shown minimal heating of coronary stents in the MRI environment [18]. Stent dislodgement, even micro-dislodgement, is of theoretical concern because of the potential for dissection, embolism, and thrombosis. However, most stents are made of materials with little or no ferromagnetism, such as stainless steel, nitinol, or titanium. In vitro and in vivo studies of stent movement have shown minimal movement caused by MRI [19, 20]. Despite manufacturer recommendations to wait 8 weeks after stent placement prior to imaging, no adverse effects have been noted because of MRI even in the acute post-stent period [21, 22].
A study of acute MRI after deployment of drug-eluting coronary stents (Taxus, Boston Scientific, Natick, MA; Cypher, Cordis, Johnson & Johnson, New Brunswick, NJ) revealed no acute thrombosis and 9-month adverse events comparable to that expected without MRI [23]. In vitro testing of another drug-eluting stent (Endeavor, cobalt alloy, Medtronic Vascular, Santa Rosa, CA) has also been performed, revealing minor magnetic field interactions, heating (+0.5 °C), and artifacts [24]. Electric resonating “active” coronary stents designed to act as electric resonating circuits, thus functioning as inductively coupled transmit coils to allow high-resolution imaging of in-stent restenosis, have been tested in vitro and in animal models [25]. However, thus far, no studies have examined the safety profile of these stents in clinical human cardiac MRI.
New stents are continuously developed, and each patient should be individually evaluated whether cardiac MRI can be performed safely. MRI should never be performed if there is a suspicion that a stent is not positioned properly.
Non-coronary Stents
MRI of non-ferromagnetic nickel titanium aortic stents has been safely performed in patients with minimal artifact [26]. Ex vivo studies have revealed no deflection forces and minimal heating, limited to 1.1 °C. Despite the recommendation by some authorities to delay MRI for 6 weeks after implant in the case of ferromagnetic large vessel stents [27], safe imaging has been reported in the acute post-implant period [28]. Similar to coronary stents, each patient should be individually evaluated, since, to date, not all stents have undergone MRI testing and new stents are continuously developed.
Coils
In vitro tests of non-ferromagnetic platinum microcoils have revealed no coil migration and minimal susceptibility artifacts [29]. Three-dimensional time-of-flight MRI has been performed for follow-up of patients with Guglielmi detachable coils within a week post-deployment for treatment of intracranial aneurysms [30]. Substitution of digital subtraction angiography by MRI in this patient population was safe, produced minimal artifact, and helped identify thromboembolic events associated with balloon-assisted deployment [31, 32]. Chronic studies have revealed that time-of-flight MRI is not only safe but also may indeed be more sensitive at identifying residual flow in coiled aneurysms than traditional plain radiographs and digital subtraction angiography [33, 34]. In a case series of diffusion and perfusion MRI in patients with ruptured and unruptured intracranial aneurysms treated by intravascular coiling, no MRI-related complications were reported, and stents and platinum coils added negligible effects on the quality of images [35]. Multiple coils have been evaluated at 3-Tesla, and though magnetic field interactions are stronger than at 1.5 Tesla, similar to other implants, fixation by tissue ingrowth is sufficient to prevent dislocation [36]. Therefore, postponing MRI until at least 6 weeks after implantation is prudent. If there is a clinical indication for MRI in the acute setting, the diagnostic benefits of MRI probably outweigh the risks of MRI.
Filters
Initial testing of Greenfield filters for deflection at 1.5 T found large variations in the amount of deflection experienced by each device [37]. However, in vivo studies showed no evidence of migration [38]. Although the stainless steel filters such as Tulip and Bird’s Nest filters cause extensive signal voids, the susceptibility artifacts associated with most filters appear to be minimal [39]. Imaging of the tantalum or titanium alloy filters is associated with such minimal artifact that even intraluminal tilting of the device, post-filter turbulence, and thrombi trapped within the filters can be visualized [40, 41]. A study comparing the sensitivity of MRI vs. ultrasound to assess inferior vena cava patency in the setting of Simon nitinol filters concluded that MRI is the superior modality [42]. It is recommended to wait at least 6 weeks after filter placement before MRI to allow firm tissue fixation. If clinically necessary, earlier MRI can be considered on an individual base as long as there is no suspicion of a non-properly positioned filter. Placement of vena cava filters guided by MRI has been attempted; however, it is limited by only passive visualization of the implanted device [43]. Techniques to improve real-time visualization of the vessel and interventional instruments are under development [44].
Septal Defect Closure Devices
An in vitro study to evaluate the safety of 12 different occluders used to treat patients with patent ductus arteriosus, atrial septal defects, and ventricular septal defects in a 1.5-T system was performed by Shellock and Morisoli. Occluders made of 304 stainless steels were ferromagnetic and displayed deflection forces of 248–299 dynes, whereas those made of MP35n were non-ferromagnetic. Artifacts were variable depending on the type and amount of metal used to construct the implant. The authors recommended a waiting period of 6 weeks post-implant to allow tissue growth and a stronger implant-tissue interface prior to MRI [45].
Real-time MRI guidance with the use of intravascular antenna guidewires has been used to image atrial septal defects and deploy Amplatzer Septal Occluders in swine [46]. CardioSEAL Septal Repair and STARFlex Septal Repair Implants have been tested for MRI compatibility at 3 T. Only minor translational attraction or torque was noted, leading the authors to conclude that MRI even at 3 T could be performed immediately post-implantation. Temperature rises were limited to 0.5 °C, and artifacts were minimal [47]. However, if there is any doubt about the integrity of the fixation parts of a ferromagnetic septal closure device, no MRI should be performed to prevent dislocation.
Vascular Access Ports and Catheters
Shellock and Shellock tested a variety of vascular access catheters made of titanium, polysulfone, barium sulfate, or silicone, such as the commonly used Vital-Port and Hickman catheters, for MRI safety. Vascular access ports have been assessed at 3 T, and no significant magnetic interaction was noted. Interestingly, even nonmetallic vascular access ports are seen on MRI because they contain silicone, and the signal should not be interpreted as an abnormality [48, 49].
Guidewires, Angiography, and Electrophysiology Catheters
Guidewires are typically made from stainless steel or nitinol (nickel/titanium alloy) and are prone to heating and lead currents in the MRI environment, even leading to iatrogenic skin burns in one case report [50]. However, the use of guidewires with RF decoupling to reduce heating may lead to successful MRI-guided wire and catheter placement [51]. Angiography and electrophysiology catheters with any form of internal or external conductive wire may be prone to heating or induced lead currents and are contraindicated in the MRI environment. Patients with cardiovascular catheters with internal or external conductive wires therefore should not undergo MRI, unless specific testing of the hardware has demonstrated otherwise. However, multifunctional electrophysiology catheters that also act as long loop receivers, allowing for catheter visualization, intracardiac electrogram recording, and ablation during MRI, have been developed and are currently under testing [52,53,54].
Swan-Ganz and Thermodilution Catheters
Swan-Ganz and thermodilution catheters contain long wires made of paramagnetic or magnetic materials, and the tips are not fixed. Therefore, they may be prone to movement, heating, and induction of current and are not safe in the MRI environment [27].
Implantable Monitors
The SJM Confirm (St. Jude Medical, Sylmar, CA) and Reveal LINQ (Medtronic, Minneapolis, MN) are insertable cardiac monitoring systems that can be activated by preset heart rate limits or a patient trigger. These devices are considered MRI conditional. Specific MRI scanning for these devices can be found at their respective manufacturer’s website.
Temporary Pacemakers
To our knowledge, there are no studies assessing the safety of temporary pacemakers in the MRI environment. Unlike permanent devices, temporary pacemakers typically have unfixed leads and are more prone to movement. Furthermore, the leads are longer and may be more prone to induction of lead currents. Finally, the electronic platform of external temporary pacemakers is less sophisticated and has less filtering compared to modern implantable pacemakers. Therefore, such devices are likely more susceptible to electromagnetic interference.
Permanent Pacemakers and Implantable Cardioverter Defibrillators
Because of underlying structural heart disease and accompanying conduction system disease or risk of ventricular arrhythmia, a significant proportion of patients referred for cardiac MRI have permanent pacemakers and implantable defibrillators [55,56,57]. It has been estimated that a patient with a pacemaker or implanted defibrillator has a 50–75% likelihood of having a clinically indicated MRI over the lifetime of their device [58]. The potential for movement of the device [59], programming changes, asynchronous pacing, activation of tachyarrhythmia therapies, inhibition of demand pacing [60], and induced lead currents leading to heating and cardiac stimulation [61] has led to concerns from device manufacturers [62,63,64] and MRI authorities [65, 66] regarding the performance of MRI procedures in cardiac implantable device recipients. For these reasons, a pacemaker historically has been considered a contraindication for MRI.
However, several case series have reported the safety of MRI in the setting of pacemakers [67,68,69,70,71]. A small case series has also reported neurological MRI in the setting of selected implantable cardioverter defibrillator systems (ICDs) [72]. Overall, safety has been reported, but acute changes in battery voltage, lead thresholds, and programming can be seen. More recent pacemakers have decreased ferromagnetic components and more streamlined electronic wiring and are less prone to interactions with the MRI magnetic field. The first pacemaker labeled as “MR conditional” became available in 2011 (Revo MRI SureScan by Medtronic). According to the 2013 ESC guidelines on cardiac pacing and cardiac resynchronization therapy, patients with these pacemakers can undergo MRI following strict guidelines and manufacturer’s instructions [73]. The US Food and Drug Administration (FDA) has currently approved two major groups of MRI conditional devices for safe human MRI scan (SureScan group of devices by Medtronic and ProMRI group by Biotronik).
At our institution, we have developed a protocol regarding the management of “MRI conditional” pacemakers that are FDA approved. This protocol is listed below:
-
A.
Scheduling of the exam:
-
1.
MRI should be performed six or more weeks post device implantation.
-
2.
MRI exam should only be performed on anatomical areas approved by the device manufacturer guidelines.
-
3.
Obtain cardiology consult:
-
(a)
Identify the exact make and model of the device and leads implanted.
-
(b)
Confirm device six or more weeks post implantation.
-
(c)
Contact vendor as necessary to verify the patients’ pacing system.
-
(d)
If required, perform and evaluate screening x-ray to confirm appropriate device and device position.
-
(a)
-
4.
Contact radiology MRI chief technologist/MRI physician leads.
-
(a)
Confirm the device MRI safety with the manufacturer manual.
-
(b)
Confirm anatomic coverage.
-
(c)
Discuss scheduling considerations and staff availability.
-
(d)
Assist with scheduling options on 1.5 T scanner.
-
(a)
-
5.
Schedule MRI examination. Order must include the following information:
-
(a)
Body part to be examined.
-
(b)
Precise device type/model number.
-
(c)
Statement that cardiology consult has been obtained.
-
(a)
-
1.
-
B.
Performance of the MRI scan:
-
1.
Interface with manufacturer representative to program the device to MRI safe program mode before the scan.
-
2.
A certified health care provider will be present during the entire scan (e.g., online training for SureScan devices: http://www.medtronic.com/mrisurescan-us/radiologytutorial.html).
-
3.
MRI is performed during normal business hours, so that the code team is expected to be available (i.e., availability in MRI expected to be less than 2 min.)
-
4.
MRI technologist:
-
(a)
Confirm device type.
-
(b)
Perform standard MRI safety screening.
-
(c)
Insure that manufacturer-specific conditions are met (correct slew rate and SAR).
-
(d)
Perform the exam on 1.5 T scanners.
-
(e)
Continuous monitoring of the patient during the exam is mandatory; verbal contact, ECG monitoring, and pulse oximetry.
-
(f)
Discontinue scan/remove patient from scan room in the case of patient distress, loss of verbal contact, ECG monitoring, or pulse oximetry.
-
(g)
Radiology or other physician staff needs to be physically present in the department.
-
(a)
-
C.
On completion of scan, consult with cardiology if reprogramming of the device following completion of the scan is necessary.
Steps detailed above are related to FDA-approved devices only and not all the conditional devices. MRI scanning of devices that are not FDA approved or under conditions that are not FDA approved is not covered by this policy and is not to be electively performed.
Non-FDA-Approved Devices
For specialized centers with the staff expertise in electrophysiology, a number of centers have developed approaches to performing MRI scans in patients with non-FDA-approved pacemakers and ICDs. An outline at our institution with programming steps to reduce the risk of inappropriate pacemaker inhibition or activation or inappropriate activation of tachyarrhythmia functions is shown in Fig. 9.2 [74]. Note that extensive device experience is required to safely evaluate non-FDA-approved devices, and on-site programming capability is required.

Safety protocol for MRI of patients with non-FDA-approved permanent pacemakers and implantable defibrillators [74]. Reprinted from Nazarian et al. [[74]], http://circ.ahajournals.org/content/114/12/1277 with permission from Wolters Kluwer Health
Devices should be programmed to minimize inappropriate activation or inhibition of brady-/tachyarrhythmia therapies, and MRI sequences should be used with a specific absorption rate (SAR) of maximum 2.0 W/kg. It is important to note, however, that because of poor correlation of heating at different specific absorption rates across different scanners even within those of the same manufacturer, the specific absorption rates from the authors’ results should not be directly applied to other MRI systems [75].
In general, ICDs tend to be more problematic than conventional pacemakers as they contain more ferromagnetic components and electrodes placed inside the myocardium. Strict adherence to manufacturer guidelines therefore remains essential when scanning these patients, as potential problems are still reported [76, 77].
Neurostimulation Systems
In vitro studies of a chronic deep brain stimulation system (Soletra model 7426, Medtronic) at 1.5 T have revealed temperature rises as high as 25 °C depending on the type of RF coil used, positioning of the electrodes, and the specific absorption rate of sequences [66, 78]. Such excessive heating was thought to be avoidable by using send/receive head RF coils and limiting the SAR of sequences to 2.4 W/kg using a 1.5-T/64-mHz Vision MR imaging system (Siemens Medical Systems, Iselin, NJ) [79]. Another in vitro study using a 1.5-T Sonata MRI system (Siemens Medical Systems) to scan bilateral neurostimulation systems (Soletra 7246, 7495, and 3389, Medtronic) revealed temperature rises limited to 2.1 °C [80]. Criteria to permit MRI up to 3 T are further defined by recent studies, including MRI-guided neurostimulator placement to increase placement accuracy and reduced intervention time [81,82,83].
Note should be made that a study has revealed poor correlation of heating at different SAR across different scanners even within those of the same manufacturer, and therefore the results of the previous studies should not be applied to other MRI systems [75]. Importantly, several reports of injury during MRI of patients with neurostimulation systems exist in the literature, and experts advise judicious use of MRI, only when clinically warranted, following the specific guidelines of the manufacturer, using send/receive head RF coils, and limiting the SAR to 0.4 W/kg [78, 84,85,86]. Note that limiting SAR in combination with send/receive RF coils is not currently approved for all MRI scanners in conjunction with deep brain stimulators.
Intra-aortic Balloon Pumps
An animal study assessed the recovery of left ventricular function after myocardial infarction with and without balloon counterpulsation via MRI. During this study, the intra-aortic balloon pump was paused during the scan. Although MRI safety was not the primary outcome assessed; no untoward side effects of MRI in this setting were reported [86]. More studies are needed to assess the safety of MRI in the setting of intra-aortic balloon counterpulsation prior to human studies with MRI.
Ventricular Assist Devices
Ventricular assist devices have high metal content, complicated circuitry, and in some cases magnetic field dependence for appropriate function. There is no literature regarding MRI in patients with implanted assist devices. Because of the issues listed and high potential for catastrophic device failure, MRI is an absolute contraindication in patients with current ventricular assist devices.
Orthopedic Implants
Most orthopedic implants have been reported to be non-ferromagnetic or weakly ferromagnetic and therefore are safe for undergoing MRI at 1.5 T [87]. The Perfix interference screw (Instrument Macar, Okemos, MI) used for anterior cruciate ligament reconstruction is the only hardware found to be highly ferromagnetic. However, the strength of the surrounding tissue provides sufficient retentive force to provide for safe imaging of patients with these implants. Some non-ferromagnetic hardware, especially cervical and external fixation systems, show significant heating due to their shape and the formation of conductive loops, especially at 3 T [88].
Shellock tested a variety of orthopedic implants at 3 T and found low torque measurements for most devices except for the bone fusion stimulator. However, this device is also likely safe for imaging given its typical position in the patient with respect to the static magnetic field of MRI and retentive strength of the subcutaneous and granulation tissue [10].
Image Artifacts
The presence of ferromagnetic materials can cause variations in the surrounding magnetic field, resulting in image distortion, signal voids or bright areas, and poor fat suppression, especially at 3 T [89]. Susceptibility artifacts appear to be most pronounced on inversion recovery and steady-state free precession sequences.
In our experience, artifacts on inversion recovery prepared delayed cardiac MRI show high signal intensity and can mimic areas of delayed enhancement, which would otherwise indicate myocardial scar. Correlation of suspect areas on different pulse sequences can help avoid misidentification of artifact. Using imaging planes perpendicular to the plane of the device generator, shortening of the echo time, and using spin echo and fast spin-echo sequences appear to reduce the qualitative extent of artifact.
Risks Associated with the Use of Gadolinium-Based Contrast Agents
Pharmacokinetics of Gadolinium-Based Contrast Agents: Relation with Kidney Function
GBCAs are administrated intravenously with rapid equilibration in the extracellular fluid space which consists of the intravascular and interstitial compartments. Six out of nine currently clinically approved GBCAs (gadopentetate dimeglumine [Magnevist], gadodiamide [Omniscan], gadoversetamide [OptiMARK], gadoterate dimeglumine [Dotarem], gadobutrol [Gadovist], and gadoteridol [Prohance]) only distribute in the extracellular fluid space. These agents are almost exclusively eliminated by the kidneys with a half-life time in the blood of 1.5 h in patients with normal kidney function [90,91,92,93,94].
Gadobenate dimeglumine [Multihance] and gadoxetate disodium [Eovist in the USA, Primovist in Europe] are specifically used for imaging of the hepatobiliary system. They are taken up by hepatocytes and partially cleared by the hepatobiliary system (2–4% for gadobenate dimeglumine and 50% for gadoxetate disodium), with renal excretion for the remainder [95]. Gadofosveset trisodium [Ablavar in the USA, not approved in Europe] is a blood pool agent used for MRI angiography. Gadofosveset reversibly binds serum albumin which limits its distribution mainly to the intravascular space. Like other GBCAs it is mainly renally excreted [96].
Given the central role of renal excretion, the clearance rate of GBCAs is strongly correlated with kidney function. Chronic kidney disease (CKD) is divided into five stages based on the glomerular filtration rate (GFR), and GBCA half-life increases exponentially with more severe stages of CKD [97]:
Stage | GFR (mL/min/1.73m2) | |
|---|---|---|
Stage 1 | Normal kidney function but urine or other abnormalities point to kidney disease | ≥90 |
Stage 2 | Mildly reduced kidney function, urine, or other abnormalities point to kidney disease | 60–89 |
Stage 3 | Moderately reduced kidney function | 30–59 |
Stage 4 | Severely reduced kidney function | 15–29 |
Stage 5 | Very severe or end-stage kidney failure/dialysis | <15 (or dialysis) |
Delayed elimination of GBCAs in patients with renal insufficiency has important consequences. One possible side effect is the dissociation of the GBCA molecule in a free gadolinium ion (Gd3+) and ligand. Free gadolinium had poor solubility, precipitates in the interstitial tissue, and is extremely toxic. Another side effect is the slow diffusion of the contrast agent into different deep compartments. Several reports have shown enhancement of the cerebrospinal fluid space on MRI 1–2 days after GBCA administration in patients with renal failure [98, 99]. Possible health risks associated with free gadolinium diffusion include nephrogenic systemic fibrosis (NSF) and recent observations of gadolinium deposition in the brain which are discussed below.
To prevent these adverse health effects, renal function should be assessed in all patients before GBCA administration. The creatinine clearance rate is the golden standard to assess kidney function before MRI and shows good correlation with the blood half-life of GBCAs. However, its assessment requires blood sampling and is time-consuming. An alternative approach is the use of the “Choyke questionnaire,” originally developed in 1998 to prevent iodinated contrast agent-induced nephropathy [100, 101]. A completely negative response to this six-question patient survey (history of (1) preexisting renal disease, (2) proteinuria, (3) prior kidney surgery, (4) hypertension, (5) gout, or (6) diabetes) can identify patients with normal creatinine levels with a sensitivity of 94%.
Immediate Adverse Reactions
The first GBCA was approved for clinical use in 1988 (gadopentetate dimeglumine, Magnevist, Bayer Healthcare Pharmaceuticals, Wayne, NJ), and >200 million doses have been administered worldwide since then. GBCAs are well tolerated by the vast majority of patients, and the frequency of immediate adverse reactions in GBCAs is lower than in iodinated contrast agents [102, 103]. Contrary to iodinated contrast agents, the risk of reaction is not related to the osmolality of GBCAs as the low doses used make the osmolar load very small. The frequency of all acute adverse effects after clinical GBCA injection (0.1–0.2 mmol/kg) ranges from 0.07% to 2.4%. Most of these adverse effects are mild and non-allergic, including nausea, dizziness, headache, and local paresthesia. Allergic reactions are extremely rare with an estimated frequency of 0.004–0.079%. Urticaria and rash are the most common allergic reactions, while severe, life-threatening anaphylactic reactions are exceedingly rare (0.001–0.01%).
Risk factors for allergic reactions to GBCAs are female sex, asthma, and various allergies, including previous allergic reaction to iodinated contrast agents. The recurrence rate of hypersensitivity reactions in patients with previous reaction to GBCAs is estimated at 30%, and appropriate premedication should be considered in patients with a previous allergic reaction [104]. The treatment of immediate adverse reactions to GBBAs is similar to that for adverse reactions to iodinated contrast agents. Guidelines for treatment of adverse reactions to iodinated and gadolinium-based contrast agents can be found on the websites of the American College of Radiology and the European Society of Urogenital Radiology (www.ACR.com and www.ESUR.org).
Late Adverse Reactions
Nephrogenic Systemic Fibrosis
Nephrogenic systemic fibrosis (NSF) is a potentially fatal disorder first identified in 1997 and reported in 2000 [105]. NSF is characterized by progressive tissue fibrosis, predominately involving the skin but also affecting internal organs such as the heart, lungs, liver, and muscles. In 2006, an association between gadodiamide (Omniscan, GE Healthcare, Piscataway, NJ) and NSF was proposed [106].
Free gadolinium ions (Gd3+) have been shown to stimulate the expression of profibrotic cytokines and growth factors in vitro and in vivo [107]. Therefore, the dissociation of free gadolinium ions from the chelate GBCA molecule is generally accepted to be the primary etiology of NSF. The stability of GBCAs is dependent on their molecular structure with two major determining factors: linear versus macrocyclic and ionic versus nonionic structure. Macrocyclic agents are more stable than linear ones, while ionic agents are more stable than nonionic ones. Thus, ionic macrocyclic GBCAs are least likely to release free gadolinium which may induce NSF, followed by nonionic macrocyclic, ionic linear, and nonionic linear GBCAs, respectively. In an in vitro stability study in human serum, nonionic linear GBCAs showed 20% dissociation, while ionic linear GBCAs demonstrated only 1.1–1.9% gadolinium release. All three macrocyclic GBCAs showed no detectable free gadolinium dissociation [108]. Consequently, the least stable GBCA (gadodiamide) is associated with the most cases of NSF in the current literature. Reduced kidney function is another important risk factor for NSF, as the slower renal excretion of GBCAs in these patients allows more time for gadolinium dissociation [109, 110].
The American College of Radiology 2013 Manual on Contrast Media classifies GBCAs in three groups based on the association with NSF [111]:
-
Group 1 agents (gadodiamide, gadopentetate dimeglumine, and gadoversetamide) are the least stable and are associated with the majority of NSF cases.
-
Group 2 agents (gadobenate dimeglumine, gadobutrol, gadoterate meglumine, and gadoteridol) are associated with few, if any, unconfounded cases of NSF. (Gadoterate meglumine is a relatively newer contrast agent; it is a macrocyclic agent and will likely also be classified as a Group 2 agent.)
-
Group 3 agents (gadofosveset trisodium and gadoxetate disodium) only recently appeared on the market with only limited data for these agents, though to date, no, if any, cases of NSF have been associated with these agents.
GBCAs can be used at label doses, only if the added diagnostic information is essential and not obtainable with non-contrast-enhanced MRI. The use of group 1 agents in patients with GFR ≤ 40 mL/min/1.73m2 is not recommended by the American College of Radiology. The US Food and Drug Administration (FDA) has indicated these group 1 agents are contraindicated for GFR ≤ 30 mL/min/1.73m2. Both the American College of Radiology and the European Society for Urogenital Radiology have published guidelines for the use of GBCAs in patients at risk for NSF [111, 112]. Cautious use of GBCAs in patients with reduced kidney function and a shift to more stable agents have resulted in no confirmed cases of NSF since 2009.
Multiple treatment modalities have been attempted in NSF patients with limited benefit, including physical therapy, ultraviolet A phototherapy, imatinib mesylate, sirolimus, and extracorporeal photopheresis. None of these therapies have been as effective as spontaneous restoration of kidney function or renal transplantation in NSF symptom reduction [113,114,115,116,117]. For patients who are eligible, early transplantation should be considered to halt or reverse the progression of NSF.
Gadofosveset has a specific label indication in the United States for MR angiography for evaluation of aortoiliac occlusive disease in adults with known or suspected peripheral vascular disease. MRI of the heart is considered off-label use of gadolinium contrast agents. The use of these agents is supported by a large number of literature studies, often at doses that exceed that used of other parts of the body [118].
Gadolinium Deposition in the Brain
The first reports of increased signal intensity in the dentate nucleus and globus pallidus on unenhanced T1 brain images in patients who received multiple doses of linear GBCAs were published in 2014 [119]. Autopsy studies provided pathologic confirmation that the T1 shortening is the result of gadolinium deposition with a dose-response relationship with higher gadolinium deposition in patients who received more doses of linear GBCAs [120, 121]. Interestingly, there was no association with renal function nor administration of macrocyclic GBCAs. Similar to NSF, it is presumed that dissociation of gadolinium from the chelate molecule leads to gadolinium deposition in the brain.
To date, the available information does not identify any adverse health effects of brain gadolinium deposition (http://www.fda.gov/downloads/Drugs/DrugSafety/UCM455390.pdf). However, there is sufficient evidence to critically review GBCA administration policy. In our institution, a set of recommendations has been adopted: (1) strictly follow FDA label indications and dosing schemes for GBCA administration; (2) only use GBCAs when clinically indicated or when specified in a research protocol; (3) consider using a macrocyclic GBCA rather than a linear agent; and (4) for patients with document adverse reactions to macrocyclic agents, linear agents can be used [122]. The choice of GBCA should be individualized at each institution, based on specific indications and experience. Further research is warranted to evaluate the hazardous effects, if any, of brain gadolinium deposition.
Cardiac MRI and Pregnancy
Present data have not conclusively documented any adverse effects of MRI on the developing human fetus. Nevertheless, it is recommended to screen all females of reproductive age for pregnancy before MRI. According to the 2013 ACR guidance document on MR safe practices, MRI can be performed during pregnancy if the diagnostic information cannot be obtained by other nonionizing methods (e.g., ultrasonography), the diagnosis will affect the patient care or fetus during the pregnancy, and the referring physician believes that it is not feasible to wait until the patient is no longer pregnant [123]. The ACR does not give special consideration to the first trimester of pregnancy, though many institutions still discourage MRI during the first trimester as some animal studies have shown deleterious effects during this phase of pregnancy [65, 124].
GBCAs should not routinely be administered to pregnant patients. To date, there is no evidence of teratogenesis or mutagenesis after fetal exposure to GBCAs during pregnancy [125, 126]. However, at least some of the GBCAs readily pass through the placental barrier. After fetal renal clearance, these agents may remain in the amniotic fluid, potentially allowing for release of toxic-free gadolinium ions. Even though a recent study suggests that the fetal concentration of GBCAs may be lower than previously thought, the risk of GBCA administration remains unknown, and GBCAs should only be administered if their usage is absolutely necessary and the potential benefits outweigh the risks to the fetus [127]. The dose of the GBCA should be as low as possible with a preference for a stable (macrocyclic) agent.
Premedication is indicated in a pregnant patient with a previous allergic reaction to GBCAs. Most schemes consist of an H1 antihistamine (diphenhydramine) and a steroid (prednisone or methylprednisolone). Diphenhydramine is classified in FDA category B (animal reproduction studies have failed to demonstrate a risk to the fetus, and there are no adequate and well-controlled studies in pregnant women), while prednisone and methylprednisolone are considered FDA category C (animal reproduction studies have shown an adverse effect on the fetus, and there are no adequate and well-controlled studies in humans, but potential benefits may warrant the use of the drug in pregnant women despite potential risks). Steroids may lead to fetal adrenal suppression, and methylprednisolone slightly increases risk for cleft lip in the fetus if used before 10 weeks of gestation [128].
Excretion of GBCA in breast milk is minimal, and available data suggest that interruption of breastfeeding is not necessary after maternal GBCA administration [111, 129]. However, if the patient is concerned, breastfeeding may be interrupted for 12–24 h to allow for excretion of the contrast agent.
References
Prasad SK, Pennell DJ. Safety of cardiovascular magnetic resonance in patients with cardiovascular implants and devices. Heart. 2004;90(11):1241–4.
Manner I, Alanen A, Komu M, Savunen T, Kantonen I, Ekfors T. MR imaging in the presence of small circular metallic implants: assessment of thermal injuries. Acta Radiol. 1996;37(4):551–4.
Okamura Y, Yamada Y, Mochizuki Y, et al. [Evaluation of coronary artery bypass grafts with magnetic resonance imaging]. [Zasshi][Journal] Nihon Kyobu Geka Gakkai. 1997;45(6):801–805.
Hartnell GG, Spence L, Hughes LA, Cohen MC, Saouaf R, Buff B. Safety of MR imaging in patients who have retained metallic materials after cardiac surgery. AJR Am J Roentgenol. 1997;168(5):1157–9.
Murphy KJ, Cohan RH, Ellis JH. MR imaging in patients with epicardial pacemaker wires. AJR Am J Roentgenol. 1999;172(3):727–8.
Roguin A, Zviman MM, Meininger GR, et al. Modern pacemaker and implantable cardioverter/defibrillator systems can be magnetic resonance imaging safe in vitro and in vivo assessment of safety and function at 1.5 T. Circulation. 2004;110(5):475–82.
Soulen RL, Budinger TF, Higgins CB. Magnetic resonance imaging of prosthetic heart valves. Radiology. 1985;154(3):705–7.
Edwards M, Taylor KM, Shellock FG. Prosthetic heart valves: evaluation of magnetic field interactions, heating, and artifacts at 1.5 T. J Magn Reson Imaging. 2000;12(2):363–9.
Shellock FG. Prosthetic heart valves and annuloplasty rings: assessment of magnetic field interactions, heating, and artifacts at 1.5 Tesla. J Cardiovasc Magn Reson. 2001;3(4):317–24.
Shellock FG. Biomedical implants and devices: assessment of magnetic field interactions with a 3.0-Tesla MR system. J Magn Reson Imaging. 2002;16(6):721–32.
Edwards M-B, Draper ERC, Hand JW, Taylor KM, Young IR. Mechanical testing of human cardiac tissue: some implications for MRI safety. J Cardiovasc Magn Reson. 2005;7(5):835–40.
Shellock FG. Magnetic resonance safety update 2002: implants and devices. J Magn Reson Imaging. 2002;16(5):485–96.
Condon B, Hadley DM. Potential MR hazard to patients with metallic heart valves: the Lenz effect. J Magn Reson Imaging. 2000;12(1):171–6.
van Gorp MJ, van der Graaf Y, de Mol BAJM, et al. Björk-Shiley convexoconcave valves: susceptibility artifacts at brain MR imaging and mechanical valve fractures 1. Radiology. 2004;230(3):709–14.
Ho JC, Shellock FG. Magnetic properties of Ni–Co–Cr-base Elgiloy. J Mater Sci Mater Med. 1999;10(9):555–60.
Edwards M, Ordidge RJ, Hand JW, Taylor KM, Young IR. Assessment of magnetic field (4.7 T) induced forces on prosthetic heart valves and annuloplasty rings. J Magn Reson Imaging. 2005;22(2):311–7.
Sherif MA, Abdel-Wahab M, Beurich H-W, et al. Haemodynamic evaluation of aortic regurgitation after transcatheter aortic valve implantation using cardiovascular magnetic resonance. EuroIntervention J Eur Collab with Work Gr Interv Cardiol Eur Soc Cardiol. 2011;7(1):57–63.
Strohm O, Kivelitz D, Gross W, et al. Safety of implantable coronary stents during H-magnetic resonance imaging at 1.0 and 1.5 T. J Cardiovasc Magn Reson. 1999;1(3):239–45.
Scott NA, Pettigrew RI. Absence of movement of coronary scents after placement in a magnetic resonance imaging field. Am J Cardiol. 1994;73(12):900–1.
Hug J, Nagel E, Bornstedt A, Schnackenburg B, Oswald H, Fleck E. Coronary arterial stents: safety and artifacts during MR imaging 1. Radiology. 2000;216(3):781–7.
Gerber TC, Fasseas P, Lennon RJ, et al. Clinical safety of magnetic resonance imaging early after coronary artery stent placement. J Am Coll Cardiol. 2003;42(7):1295–8.
Kaya MG, Okyay K, Yazici H, et al. Long-term clinical effects of magnetic resonance imaging in patients with coronary artery stent implantation. Coron Artery Dis. 2009;20(2):138–42.
Porto I, Selvanayagam J, Ashar V, Neubauer S, Banning AP. Safety of magnetic resonance imaging one to three days after bare metal and drug-eluting stent implantation. Am J Cardiol. 2005;96(3):366–8.
Shellock FG, Forder JR. Drug eluting coronary stent: in vitro evaluation of magnet resonance safety at 3 tesla. J Cardiovasc Magn Reson. 2005;7(2):415–9.
Busch M, Vollmann W, Bertsch T, et al. On the heating of inductively coupled resonators (stents) during MRI examinations. Magn Reson Med. 2005;54(4):775–82.
Engellau L, Olsrud J, Brockstedt S, et al. MR evaluation ex vivo and in vivo of a covered stent-graft for abdominal aortic aneurysms: ferromagnetism, heating, artifacts, and velocity mapping. J Magn Reson Imaging. 2000;12(1):112–21.
Ahmed S, Shellock FG. Magnetic resonance imaging safety: implications for cardiovascular patients. J Cardiovasc Magn Reson. 2001;3(3):171–82.
Stables RH, Mohiaddin R, Panting J, Pennell DJ, Pepper J, Sigwart U. Exclusion of an aneurysmal segment of the thoracic aorta with covered stents. Circulation. 2000;101(15):1888–9.
Marshall MW, Teitelbaum GP, Kim HS, Deveikis J. Ferromagnetism and magnetic resonance artifacts of platinum embolization microcoils. Cardiovasc Intervent Radiol. 1991;14(3):163–6.
Okahara M, Kiyosue H, Hori Y, Yamashita M, Nagatomi H, Mori H. Three-dimensional time-of-flight MR angiography for evaluation of intracranial aneurysms after endosaccular packing with Guglielmi detachable coils: comparison with 3D digital subtraction angiography. Eur Radiol. 2004;14(7):1162–8.
Soeda A, Sakai N, Sakai H, et al. Thromboembolic events associated with Guglielmi detachable coil embolization of asymptomatic cerebral aneurysms: evaluation of 66 consecutive cases with use of diffusion-weighted MR imaging. Am J Neuroradiol. 2003;24(1):127–32.
Albayram S, Selcuk H, Kara B, et al. Thromboembolic events associated with balloon-assisted coil embolization: evaluation with diffusion-weighted MR imaging. Am J Neuroradiol. 2004;25(10):1768–77.
Cottier JP, Bleuzen-Couthon A, Gallas S, et al. Follow-up of intracranial aneurysms treated with detachable coils: comparison of plain radiographs, 3D time-of-flight MRA and digital subtraction angiography. Neuroradiology. 2003;45(11):818–24.
Yamada N, Hayashi K, Murao K, Higashi M, Iihara K. Time-of-flight MR angiography targeted to coiled intracranial aneurysms is more sensitive to residual flow than is digital subtraction angiography. Am J Neuroradiol. 2004;25(7):1154–7.
Cronqvist M, Wirestam R, Ramgren B, et al. Diffusion and perfusion MRI in patients with ruptured and unruptured intracranial aneurysms treated by endovascular coiling: complications, procedural results, MR findings and clinical outcome. Neuroradiology. 2005;47(11):855–73.
Karacozoff AM, Shellock FG, Wakhloo AK. A next-generation, flow-diverting implant used to treat brain aneurysms: in vitro evaluation of magnetic field interactions, heating and artifacts at 3-T. Magn Reson Imaging. 2013;31(1):145–9.
Williamson MR, McCowan TC, Walker CW, Ferris EJ. Effect of a 1.5 tesla magnetic field on greenfield filters in vitro and in dogs. Angiology. 1988;39(12):1022–4.
Liebman CE, Messersmith RN, Levin DN, Lu C-T. MR imaging of inferior vena caval filters: safety and artifacts. Am J Roentgenol. 1988;150(5):1174–6.
Honda M, Obuchi M, Sugimoto H. Artifacts of vena cava filters ex vivo on MR angiography. Magn Reson Med Sci. 2003;2(2):71–7.
Teitelbaum GP, Ortega HV, Vinitski S, et al. Low-artifact intravascular devices: MR imaging evaluation. Radiology. 1988;168(3):713–9.
Grassi CJ, Matsumoto AH, Teitelbaum GP. Vena caval occlusion after Simon nitinol filter placement: identification with MR imaging in patients with malignancy. J Vasc Interv Radiol. 1992;3(3):535–9.
Kim D, Edelman RR, Margolin CJ, et al. The Simon nitinol filter: evaluation by MR and ultrasound. Angiology. 1992;43(7):541–8.
Frahm C, Gehl H, Lorch H, et al. MR-guided placement of a temporary vena cava filter: technique and feasibility. J Magn Reson Imaging. 1998;8(1):105–9.
Bücker A, Neuerburg JM, Adam GB, et al. Real-time MR guidance for inferior vena cava filter placement in an animal model. J Vasc Interv Radiol. 2001;12(6):753–6.
Shellock FG, Morisoli SM. Ex vivo evaluation of ferromagnetism and artifacts of cardiac occluders exposed to a 1.5-T MR system. J Magn Reson Imaging. 1994;4(2):213–5.
Rickers C, Jerosch-Herold M, Hu X, et al. Magnetic resonance image-guided transcatheter closure of atrial septal defects. Circulation. 2003;107(1):132–8.
Shellock FG, Valencerina S. Septal repair implants: evaluation of magnetic resonance imaging safety at 3 T. Magn Reson Imaging. 2005;23(10):1021–5.
Shellock FG, Shellock VJ. Vascular access ports and catheters: ex vivo testing of ferromagnetism, heating, and artifacts associated with MR imaging. Magn Reson Imaging. 1996;14(4):443–7.
Titterington B, Shellock FG. Evaluation of MRI issues for an access port with a radiofrequency identification (RFID) tag. Magn Reson Imaging. 2013;31(8):1439–44.
Masaki F, Shuhei Y, Riko K, Yohjiro M. Iatrogenic second-degree burn caused by a catheter encased tubular braid of stainless steel during MRI. Burns. 2007;33(8):1077–9.
Razavi R, Hill DLG, Keevil SF, et al. Cardiac catheterisation guided by MRI in children and adults with congenital heart disease. Lancet. 2003;362(9399):1877–82.
Susil RC, Yeung CJ, Halperin HR, Lardo AC, Atalar E. Multifunctional interventional devices for MRI: a combined electrophysiology/MRI catheter. Magn Reson Med. 2002;47(3):594–600.
Krämer NA, Krüger S, Schmitz S, et al. Preclinical evaluation of a novel fiber compound MR guidewire in vivo. Investig Radiol. 2009;44(7):390–7.
Saikus CE, Lederman RJ. Interventional cardiovascular magnetic resonance imaging: a new opportunity for image-guided interventions. JACC Cardiovasc Imaging. 2009;2(11):1321–31.
Brown DW, Croft JB, Giles WH, Anda RF, Mensah GA. Epidemiology of pacemaker procedures among Medicare enrollees in 1990, 1995, and 2000. Am J Cardiol. 2005;95(3):409–11.
Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346(12):877–83.
Bardy GH, Lee KL, Mark DB, et al. Amiodarone or an implantable cardioverter–defibrillator for congestive heart failure. N Engl J Med. 2005;352(3):225–37.
Kalin R, Stanton MS. Current clinical issues for MRI scanning of pacemaker and defibrillator patients. Pacing Clin Electrophysiol. 2005;28(4):326–8.
Shellock FG, Tkach JA, Ruggieri PM, Masaryk TJ. Cardiac pacemakers, Icds, and loop recorder: evaluation of translational attraction using conventional (“long-bore”) and “short-bore” 1.5-and 3.0-tesla Mr systems: SAFETY. J Cardiovasc Magn Reson. 2003;5(2):387–97.
Erlebacher JA, Cahill PT, Pannizzo F, Knowles RJR. Effect of magnetic resonance imaging on DDD pacemakers. Am J Cardiol. 1986;57(6):437–40.
Hayes DL, Holmes DR, Gray JE. Effect of 1.5 tesla nuclear magnetic resonance imaging scanner on implanted permanent pacemakers. J Am Coll Cardiol. 1987;10(4):782–6.
Smith JM. Industry viewpoint: Guidant: pacemakers, ICDs, and MRI. Pacing Clin Electrophysiol. 2005;28(4):264.
Stanton MS. Industry viewpoint: medtronic: pacemakers, ICDs, and MRI. Pacing Clin Electrophysiol. 2005;28(4):265.
Levine PA. Industry viewpoint: St. Jude medical: pacemakers, ICDs and MRI. Pacing Clin Electrophysiol. 2005;28(4):266–7.
Shellock FG, Crues JV. MR procedures: biologic effects, safety, and patient care 1. Radiology. 2004;232(3):635–52.
Faris OP, Shein MJ. Government viewpoint: US food & drug administration: pacemakers, ICDs and MRI. Pacing Clin Electrophysiol. 2005;28(4):268–9.
Gimbel J, Johnson D, Levine PA, Wilkoff BL. Safe performance of magnetic resonance imaging on five patients with permanent cardiac pacemakers. Pacing Clin Electrophysiol. 1996;19(6):913–9.
Sommer T, Vahlhaus C, Lauck G, et al. MR imaging and cardiac pacemakers: in vitro evaluation and in vivo studies in 51 patients at 0.5 T 1. Radiology. 2000;215(3):869–79.
Vahlhaus C, Sommer T, Lewalter T, et al. Interference with cardiac pacemakers by magnetic resonance imaging: are there irreversible changes at 0.5 Tesla? Pacing Clin Electrophysiol. 2001;24(4):489–95.
Martin ET, Coman JA, Shellock FG, Pulling CC, Fair R, Jenkins K. Magnetic resonance imaging and cardiac pacemaker safety at 1.5-Tesla. J Am Coll Cardiol. 2004;43(7):1315–24.
Del Ojo J, Moya F, Villalba J, et al. Is magnetic resonance imaging safe in cardiac pacemaker recipients? Pacing Clin Electrophysiol. 2005;28(4):274–8.
Gimbel J, Kanal E, Schwartz KM, Wilkoff BL. Outcome of magnetic resonance imaging (MRI) in selected patients with implantable cardioverter defibrillators (ICDs). Pacing Clin Electrophysiol. 2005;28(4):270–3.
Brignole M, Auricchio A, Baron-Esquivias G, et al. ESC guidelines on cardiac pacing and cardiac resynchronization therapy. Eur Heart J. 2013;2013:eht150.
Nazarian S, Roguin A, Zviman MM, et al. Clinical utility and safety of a protocol for noncardiac and cardiac magnetic resonance imaging of patients with permanent pacemakers and implantable-cardioverter defibrillators at 1.5 tesla. Circulation. 2006;114(12):1277–84.
Baker KB, Tkach JA, Nyenhuis JA, et al. Evaluation of specific absorption rate as a dosimeter of MRI-related implant heating. J Magn Reson Imaging. 2004;20(2):315–20.
Gimbel J. Magnetic resonance imaging of implantable cardiac rhythm devices at 3.0 tesla. Pacing Clin Electrophysiol. 2008;31(7):795–801.
Mollerus M, Albin G, Lipinski M, Lucca J. Ectopy in patients with permanent pacemakers and implantable cardioverter-defibrillators undergoing an MRI scan. Pacing Clin Electrophysiol. 2009;32(6):772–8.
Rezai AR, Phillips M, Baker KB, et al. Neurostimulation system used for deep brain stimulation (DBS): MR safety issues and implications of failing to follow safety recommendations. Investig Radiol. 2004;39(5):300–3.
Finelli DA, Rezai AR, Ruggieri PM, et al. MR imaging-related heating of deep brain stimulation electrodes: in vitro study. Am J Neuroradiol. 2002;23(10):1795–802.
Bhidayasiri R, Bronstein JM, Sinha S, et al. Bilateral neurostimulation systems used for deep brain stimulation: in vitro study of MRI-related heating at 1.5 T and implications for clinical imaging of the brain. Magn Reson Imaging. 2005;23(4):549–55.
Baker KB, Nyenhuis JA, Hrdlicka G, Rezai AR, Tkach JA, Shellock FG. Neurostimulation systems: assessment of magnetic field interactions associated with 1.5-and 3-Tesla MR systems. J Magn Reson Imaging. 2005;21(1):72–7.
Fraix V, Chabardes S, Krainik A, et al. Effects of magnetic resonance imaging in patients with implanted deep brain stimulation systems: clinical article. J Neurosurg. 2010;113(6):1242–5.
Foltynie T, Zrinzo L, Martinez-Torres I, et al. MRI-guided STN DBS in Parkinson’s disease without microelectrode recording: efficacy and safety. J Neurol Neurosurg Psychiatry. 2011;82(4):358–63.
Henderson JM, Tkach J, Phillips M, Baker K, Shellock FG, Rezai AR. Permanent neurological deficit related to magnetic resonance imaging in a patient with implanted deep brain stimulation electrodes for Parkinson’s disease: case report. Neurosurgery. 2005;57(5):E1063.
Rezai AR, Baker KB, Tkach JA, et al. Is magnetic resonance imaging safe for patients with neurostimulation systems used for deep brain stimulation? Neurosurgery. 2005;57(5):1056–62.
Azevedo CF, Amado LC, Kraitchman DL, et al. The effect of intra-aortic balloon counterpulsation on left ventricular functional recovery early after acute myocardial infarction: a randomized experimental magnetic resonance imaging study. Eur Heart J. 2005;26(12):1235–41.
Kumar R, Lerski RA, Gandy S, Clift BA, Abboud RJ. Safety of orthopedic implants in magnetic resonance imaging: an experimental verification. J Orthop Res. 2006;24(9):1799–802.
Liu Y, Chen J, Shellock FG, Kainz W. Computational and experimental studies of an orthopedic implant: MRI-related heating at 1.5-T/64-MHz and 3-T/128-MHz. J Magn Reson Imaging. 2013;37(2):491–7.
Bernstein MA, Huston J, Ward HA. Imaging artifacts at 3.0 T. J Magn Reson Imaging. 2006;24(4):735–46.
Weinmann HJ, Laniado M, Mützel W. Pharmacokinetics of GdDTPA/dimeglumine after intravenous injection into healthy volunteers. Physiol Chem Phys Med NMR. 1983;16(2):167–72.
Van Wagoner M, Worah D. Gadodiamide injection: first human experience with the nonionic magnetic resonance imaging enhancement agent. Investig Radiol. 1993;28:S44–8.
McLachlan SJ, Eaton S, De Simone DN. Pharmacokinetic behavior of gadoteridol injection. Investig Radiol. 1992;27:S16.
Tombach B, Bremer C, Reimer P, et al. Pharmacokinetics of 1M gadobutrol in patients with chronic renal failure. Investig Radiol. 2000;35(1):35.
Baker JF, Kratz LC, Stevens GR, Wible JH Jr. Pharmacokinetics and safety of the MRI contrast agent gadoversetamide injection (OptiMARK) in healthy pediatric subjects. Investig Radiol. 2004;39(6):334–9.
Pascolo L, Cupelli F, Anelli PL, et al. Molecular mechanisms for the hepatic uptake of magnetic resonance imaging contrast agents. Biochem Biophys Res Commun. 1999;257(3):746–52.
McMurry TJ, Parmelee DJ, Sajiki H, et al. The effect of a phosphodiester linking group on albumin binding, blood half-life, and relaxivity of intravascular diethylenetriaminepentaacetato aquo gadolinium (III) MRI contrast agents. J Med Chem. 2002;45(16):3465–74.
Levey AS, Eckardt K-U, Tsukamoto Y, et al. Definition and classification of chronic kidney disease: a position statement from kidney disease: improving global outcomes (KDIGO). Kidney Int. 2005;67(6):2089–100.
Rai AT, Hogg JP. Persistence of gadolinium in CSF: a diagnostic pitfall in patients with end-stage renal disease. Am J Neuroradiol. 2001;22(7):1357–61.
Morris JM, Miller GM. Increased signal in the subarachnoid space on fluid-attenuated inversion recovery imaging associated with the clearance dynamics of gadolinium chelate: a potential diagnostic pitfall. Am J Neuroradiol. 2007;28(10):1964–7.
Choyke PL, Cady J, DePollar SL, Austin H. Determination of serum creatinine prior to iodinated contrast media: is it necessary in all patients? Tech Urol. 1998;4(2):65–9.
Sena BF, Stern JP, Pandharipande PV, et al. Screening patients to assess renal function before administering gadolinium chelates: assessment of the Choyke questionnaire. Am J Roentgenol. 2010;195(2):424–8.
Murphy KPJ, Szopinski KT, Cohan RH, Mermillod B, Ellis JH. Occurrence of adverse reactions to gadolinium-based contrast material and management of patients at increased risk: a survey of the American Society of Neuroradiology Fellowship Directors. Acad Radiol. 1999;6(11):656–64.
Prince MR, Zhang H, Zou Z, Staron RB, Brill PW. Incidence of immediate gadolinium contrast media reactions. Am J Roentgenol. 2011;196(2):W138–43.
Jung J-W, Kang H-R, Kim M-H, et al. Immediate hypersensitivity reaction to gadolinium-based MR contrast media. Radiology. 2012;264(2):414–22.
Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000–1.
Grobner T. Gadolinium–a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006;21(4):1104–8.
Morcos SK. Experimental studies investigating the pathophysiology of nephrogenic systemic fibrosis; what did we learn so far? Eur Radiol. 2011;21(3):496–500.
Frenzel T, Lengsfeld P, Schirmer H, Hütter J, Weinmann H-J. Stability of gadolinium-based magnetic resonance imaging contrast agents in human serum at 37 C. Investig Radiol. 2008;43(12):817–28.
Martin DR, Krishnamoorthy SK, Kalb B, et al. Decreased incidence of NSF in patients on dialysis after changing gadolinium contrast-enhanced MRI protocols. J Magn Reson Imaging. 2010;31(2):440–6.
Kuo PH. Gadolinium-containing MRI contrast agents: important variations on a theme for NSF. J Am Coll Radiol. 2008;5(1):29–35.
American College of Radiology (ACR) Website. ACR manual on contrast media, version 10.1. http://www.acr.org/~/media/ACR/Documents/PDF/QualitySafety/Resources/ContrastManual/2015_Contrast_Media.pdf. Accessed 16 Mar 2016.
Thomsen HS, Morcos SK, Almén T, et al. Nephrogenic systemic fibrosis and gadolinium-based contrast media: updated ESUR contrast medium safety committee guidelines. Eur Radiol. 2013;23(2):307–18.
Tran KT, Prather HB, Cockerell CJ, Jacobe H. UV-A1 therapy for nephrogenic systemic fibrosis. Arch Dermatol. 2009;145(10):1170–4.
Elmholdt TR, Buus NH, Ramsing M, Olesen AB. Antifibrotic effect after low-dose imatinib mesylate treatment in patients with nephrogenic systemic fibrosis: an open-label non-randomized, uncontrolled clinical trial. J Eur Acad Dermatology Venereol. 2013;27(6):779–84.
Ross C, De Rosa N, Marshman G, Astill D. Nephrogenic systemic fibrosis in a gadolinium-naïve patient: successful treatment with oral sirolimus. Australas J Dermatol. 2015;56(3):e59–62.
Mathur K, Morris S, Deighan C, Green R, Douglas KW. Extracorporeal photopheresis improves nephrogenic fibrosing dermopathy/nephrogenic systemic fibrosis: three case reports and review of literature. J Clin Apher. 2008;23(4):144–50.
Panesar M, Banerjee S, Barone GW. Clinical improvement of nephrogenic systemic fibrosis after kidney transplantation. Clin Transpl. 2008;22(6):803–8.
Nacif MS, Arai AE, Lima JA, Bluemke DA. Gadolinium-enhanced cardiovascular magnetic resonance: administered dose in relationship to United States Food and Drug Administration (FDA) guidelines. J Cardiovasc Magn Reson. 2012;14:18.
Kanda T, Ishii K, Kawaguchi H, Kitajima K, Takenaka D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2013;270(3):834–41.
Kanda T, Fukusato T, Matsuda M, et al. Gadolinium-based contrast agent accumulates in the brain even in subjects without severe renal dysfunction: evaluation of autopsy brain specimens with inductively coupled plasma mass spectroscopy. Radiology. 2015;276(1):228–32.
McDonald RJ, McDonald JS, Kallmes DF, et al. Intracranial gadolinium deposition after contrast-enhanced MR imaging. Radiology. 2015;275(3):772–82.
Malayeri AA, Brooks KM, Bryant LH, et al. National Institutes of Health perspective on reports of gadolinium deposition in the brain. J Am Coll Radiol. 2016;13:237.
Kanal E, Barkovich AJ, Bell C, et al. ACR guidance document on MR safe practices: 2013. J Magn Reson Imaging. 2013;37(3):501–30.
Saito K, Suzuki H, Suzuki K. Teratogenic effects of static magnetic field on mouse fetuses. Reprod Toxicol. 2006;22(1):118–24.
Marcos HB, Semelka RC, Worawattanakul S. Normal placenta: gadolinium-enhanced dynamic MR imaging. Radiology. 1997;205(2):493–6.
Shoenut JP, Semelka RC, Silverman R, Yaffe CS, Micflikier AB. MRI in the diagnosis of Crohn’s disease in two pregnant women. J Clin Gastroenterol. 1993;17(3):244–7.
Oh KY, Roberts VHJ, Schabel MC, Grove KL, Woods M, Frias AE. Gadolinium chelate contrast material in pregnancy: fetal biodistribution in the nonhuman primate. Radiology. 2015;276(1):110–8.
Wang PI, Chong ST, Kielar AZ, et al. Imaging of pregnant and lactating patients: part 1, evidence-based review and recommendations. Am J Roentgenol. 2012;198(4):778–84.
Kubik-Huch RA, Gottstein-Aalame NM, Frenzel T, et al. Gadopentetate Dimeglumine excretion into human breast milk during lactation 1. Radiology. 2000;216(2):555–8.
Summary
The diagnostic benefits of cardiovascular MRI are of critical importance in the management of an ever-increasing number of patients with cardiovascular disease. Patients with higher morbidity are referred for MRI, raising specific safety concerns. Although techniques for safe imaging in the setting of certain devices have been developed, the potential for catastrophic complications still exists and dictates a high degree of vigilance for safe imaging. The reader is encouraged to consult websites that provide more specific information regarding individual devices and safe administration of GBCAs (e.g., www.mrisafety.com and www.ACR.org). The final decision to perform cardiac MRI should be made on an individual basis considering the potential benefit of MRI relative to the associated risks.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Science+Business Media, LLC, part of Springer Nature
About this chapter

Cite this chapter
Symons, R., Nazarian, S., Halperin, H.R., Bluemke, D.A. (2019). Safety and Monitoring for Cardiac Magnetic Resonance Imaging. In: Kwong, R., Jerosch-Herold, M., Heydari, B. (eds) Cardiovascular Magnetic Resonance Imaging. Contemporary Cardiology. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-8841-9_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-8841-9_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-8839-6
Online ISBN: 978-1-4939-8841-9
eBook Packages: MedicineMedicine (R0)