
Abstract
β-thalassemia and sickle cell disease (SCD) are prototypical Mendelian single gene disorders, both caused by mutations affecting the adult β-globin gene. Despite the apparent genetic simplicity, both disorders display a remarkable spectrum of phenotypic severity and share two major genetic modifiers—α-globin genotype and innate ability to produce fetal hemoglobin (HbF, α2γ2).
This article provides an overview of the genetic basis for SCD and β-thalassemia, and genetic modifiers identified through phenotype correlation studies. Identification of the genetic variants modifying HbF production in combination with α-globin genotype provide some prediction of disease severity for β-thalassemia and SCD but generation of a personalized genetic risk score to inform prognosis and guide management requires a larger panel of genetic modifiers yet to be discovered.
Nonetheless, genetic studies have been successful in characterizing some of the key variants and pathways involved in HbF regulation, providing new therapeutic targets for HbF reactivation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Introduction
The inherited disorders of hemoglobin (Hb) production are the most common human monogenic disorders, among which those affecting the adult β globin gene (HBB) —β-thalassemia and sickle cell disease (SCD)—are the most clinically significant [1, 2]. β-thalassemia is caused by a spectrum of mutations that results in a quantitative reduction of β globin chains that are structurally normal [3], while SCD is caused by a single nucleotide substitution (GAG to GTG) in the sixth codon of HBB gene, substituting valine for glutamic acid in adult β globin (βGlu6Val), resulting in an abnormal Hb variant, HbS (α2β2 S) [1, 4]. This change allows HbS to polymerize when deoxygenated, a primary event indispensable in the molecular pathogenesis of SCD.
Both β-thalassemia and SCD occurs widely in a broad belt of tropical and sub-tropical regions including the Mediterranean, parts of North and sub-Saharan Africa, the Middle East, Indian subcontinent and Southeast Asia, with some variations in frequencies in the two diseases. For example, SCD is predominant in sub-Saharan Africa but it is also prevalent in small pockets in the Mediterranean region, Middle East and the Indian sub-continent. It appears that heterozygotes for both thalassemia [5] and the βS gene [6] are protected from the severe effects of falciparum malaria, and natural selection has increased and maintained their gene frequencies in these malarious regions. In these prevalent regions, gene frequencies for β-thalassemia range between 2 and 30%; in sub-Saharan Africa, the βS gene frequency is uneven but rarely reaches 20% [7]. However, owing to population movements in recent years, both β-thalassemia and SCD are no longer confined to these high-incidence regions, but have become an important part of clinical practice posing an important public health problem in many countries, including North America and Europe [1].
The β-Globin Gene (HBB) and Normal Expression
Βeta-globin is encoded by a structural gene found in a cluster with the other β-like genes on chromosome 11 (11p 15.15) [8]. The cluster contains five functional genes, ε (HBE), Gγ (HBG2), Aγ (HBG1), δ (HBD), and β (HBB), which are arranged along the chromosome in the order of their developmental expression to produce different Hb tetramers: embryonic (Hb Gower-1 (ζ2ε2), Hb Gower-2 (α2ε2), and Hb Portland (ζ2β2)), fetal (α2γ2), and adult (HbA, α2β2 and HbA2, α2δ2) [8]. Expression of the globin genes is dependent on local promoter sequences as well as the upstream β-globin locus control region (β-LCR) which consists of five Dnase 1 hypersensitive (HS) sites (designated HS1 to HS5) distributed between 6 and 20 kb 5′ of HBE gene [9,10,11]. There is one HS site at approximately 20 kb downstream of HBB gene. All these regulatory regions bind a number of key erythroid-specific transcription factors, notably GATA-1, GATA-2, NF-E2, KLF1 (also known as EKLF), and SCL as well as various co-factors (e.g., FOG, p300), and factors that are more ubiquitous in their tissue distribution, such as Sp1 [8, 12, 13].
The β-like globin genes are each expressed at distinct stages of development through a process referred to as hemoglobin switching (embryonic → fetal → adult). Transcription of HBE in the embryonic yolk sac during the initial period of pregnancy switches during the second month of gestation to transcription of the γ-globin genes, and then around the time of birth, to that of the adult β-globin gene . At 6 months after birth, fetal Hb (HbF, α2γ2) which comprises less than 5% of the total Hb, continues to fall reaching the adult level of <1% at 2 years of age, when adult Hb becomes the major Hb [8]. It is at this stage that mutations affecting the adult HBB gene, i.e. β-thalassemia and SCD become manifested [14,15,16].
The tissue- and developmental-specific expression of the individual globin genes, i.e. hemoglobin switching, relies on a timely and direct physical interactions between the globin promoters and the β-LCR, the interaction being mediated through binding of erythroid-specific and ubiquitous transcription factors [17]. Tissue-specific expression may be explained by the presence of binding sites for the erythroid-specific transcription factors [12]. The binding of hemopoietic-specific factors activates the LCR, which renders the entire β-globin gene cluster transcriptionally active. Transcription factors which bind to enhancer and local promoter sequences within each gene, work in tandem to regulate the expression of the individual genes in the clusters. Some of the transcription factors are developmental stage specific and may be involved in the (still poorly understood) differential expression of embryonic, fetal and adult globin genes. A dual mechanism has been proposed for the developmental expression: (a) gene competition for the upstream β-LCR , conferring advantage for the gene closest to the LCR [18], and (b) autonomous silencing (transcriptional repression) of the preceding gene [19, 20]. The ability to compete for the β-LCR and autonomous silencing depends on the change in the transcriptional factor environment—in the abundance and repertoire of various transcription factors—favouring promoter-LCR interaction. While the ε and γ-globin genes are autonomously silenced at the appropriate developmental stage, expression of the adult β-globin gene depends on lack of competition from the upstream γ-gene for the LCR sequences. Concordant with this mechanism, when the γ-gene is upregulated by point mutations in their promoter causing a non-deletion hereditary persistence of fetal hemoglobin (HPFH), expression of the downstream cis β-gene is downregulated [21]. Further, mutations which affect the β globin promoter, which removes competition for the β-LCR , are associated with higher than expected increases in γ (HbF) and δ (HbA2, α2δ2) expression [22,23,24]. A thorough understanding of the switch from fetal to adult Hb expression may provide insights on strategies of delaying the switch to allow persistent expression of the fetal globin genes for treating both SCD and β-thalassemia.
So far, the best-defined example of a developmental stage-specific regulatory factor is the erythroid Krüppel-like factor (EKLF , also known as KLF1) without which the β genes cannot be fully activated in the definitive cells [25,26,27]. Not only is KLF1 expression restricted mainly to erythroid cells [28] but it is also a highly promoter-specific activator, binding with high affinity to the β-globin CACCC box [24]. Its greater affinity for the β-globin than the γ-globin promoter accelerates the silencing of the γ-genes [29]. A genetic network regulating the switch from γ- to β-globin expression has emerged involving the interaction of KLF1, BCL11A and MYB with each other, and other transcription factors (e.g., GATA-1) and co-repressor complexes that involve chromatin modeling and epigenetic modifiers [30, 31]. BCL11A, previously known as an oncogene involved in leukemogenesis, was ‘discovered’ as an important genetic locus regulating HbF through genome-wide association studies (GWAS) [32, 33]. Downstream functional studies in cell lines, primary human erythroid cells and transgenic mice, have shown that BCL11A is a repressor of γ-globin expression [34,35,36]. KLF1 is a direct activator of BCL11A [37, 38]. KLF1 is key in the switch from γ-globin to β-globin expression; it not only activates the β-globin gene directly, providing a competitive edge, but also silences the γ-globin genes indirectly via activation of BCL11A. KLF1 may also play a role in the silencing of the embryonic globin genes. KLF1 has emerged as a major erythroid transcription factor with pleiotropic roles underlying many of the previously uncharacterized anemias (e.g., congenital dyserythropoietic anemia type IV) [39,40,41]. KLF1 variants have also been associated with variable increases in HbF and HbA2 levels, as a primary phenotype [39,40,41]. A recent case report indicated that complete absence of KLF1 causes hydrops fetalis and nonspherocytic hemolytic anemia [42].
Genetics of β-Thalassemia
Almost 300 β-thalassemia alleles have now been described (http://globin.cse.psu.edu) but only about forty account for 90% or more of the β-thalassemias worldwide. This is because in the areas where β-thalassemia is prevalent, only a few mutations are common, possibly reflecting local selection due to malaria. Each of these populations thus has its own spectrum of β-thalassemia alleles.
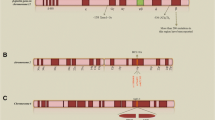
Downregulation of the β-globin gene can be caused by a whole spectrum of molecular lesions ranging from point changes to small deletions limited to the β-gene or extensive deletions of the whole β globin cluster or βLCR (Fig. 2.1) [3]. Deletions causing β-thalassemia, however, are rare; the vast majority (~250 of the 300) of mutations causing β-thalassemia are non-deletional.

Mutations causing β-thalassemia. A summary of the mechanisms downregulating β-globin gene expression. The upper panel depicts the β-globin gene (HBB) cluster with the upstream β-locus control region (βLCR). The vast majority are point mutations affecting the structural HBB gene. Deletions downregulating HBB are rare and are either restricted to the gene or extensive, involving the βLCR with or without HBB. Dashed lines represent variation in the amounts of flanking DNA removed by the deletions
Functionally, the β-thalassemia alleles can be considered as β0 where no β-globin is produced, or β+ in which some β-globin is produced, but less than normal. A range of severity is encountered within the β+ thalassemia group; the less severe forms are sometimes designated β++ to reflect the minimal deficit in β chain production. Some β++ alleles are so mild that they are ‘silent’; carriers do not display any evident hematological phenotypes; their red cell indices and HbA2 levels are within normal limits, the only abnormality being an imbalanced α:non-α chain synthesis [43]. These β-thalassemia alleles have usually been ‘discovered’ in individuals with thalassemia intermedia who have inherited a silent β-thalassemia allele in compound heterozygosity with a severe β-thalassemia allele. In this case, one parent has typical β-thalassemia trait, and the other (with the β++ thalassemia) is apparently normal.
Non-deletion β-Thalassemia
These non-deletional mutations, i.e. single base substitutions, small insertions or deletions of one to a few bases are located within the gene or its immediate flanking sequences. They downregulate the β-globin gene via almost every known stage of gene expression, from transcription to RNA processing and translation of β-globin mRNA. Approximately half of the non-deletional mutations completely inactivate the β-gene with no β globin production resulting in β0 thalassemia.
Transcriptional Mutations
Transcriptional mutants involve the conserved DNA sequences that form the β-globin promoter (from 100 bp upstream to the site of the initiation of transcription, including the functionally important CACCC, CCAAT and ATAA boxes) and the stretch of 50 nucleotides in the 5′UTR. Generally, these transcriptional mutants result in a mild to minimal reduction of β globin output i.e. β+ or β++ thalassemia alleles, and occasionally they are ‘silent’. A silent β-thalassemia allele which has been observed fairly frequently in the Mediterranean region is the −101 C→T mutation where it interacts with a variety of more severe β-thalassemia mutations to produce milder forms of β-thalassemia [44]. Other ‘silent’ mutations include those in the 5′ UTR; the extremely mild phenotype is exemplified in a homozygote for the +1 A→C mutation who has the hematologic values of a thalassemia carrier, heterozygotes are ‘silent’ [45].
Within this group of transcriptional mutants, ethnic variation in phenotype has been observed. Black individuals homozygous for the −29 A→G mutation have an extremely mild disease [46], while a Chinese individual homozygous for the same mutation had severe anemia and was transfusion-dependent [47]. The cause of this striking difference in phenotype is not known but likely to be related to the different chromosomal backgrounds on which the apparently identical mutations have arisen. One difference is the C-T polymorphism at position –158 upstream of the Gγ globin gene (Xmn1-Gγ site) present in the β chromosome carrying the −29 A→G mutation in Blacks but absent in that of the Chinese. The XmnI-Gγ site, considered to be a quantitative trait locus for HbF, is associated with increased HbF production under conditions of erythropoietic stress (see later on ‘Update on the genetic control on HbF).
Mutations Affecting RNA Processing
A wide variety of mutations interfere with processing of the primary mRNA transcript. Those that affect the invariant dinucleotide GT or AG sequences at exon-intron splice junctions prevent normal splicing altogether, and cause β0 thalassemia. Mutations involving the consensus sequences adjacent to the GT or AG dinucleotides, allow normal splicing to varying degrees and produce a β-thalassemia phenotype that ranges from mild to severe. For example, mutations at position 5 IVS1 G→C, T or A, considerably reduce splicing at the mutated donor site compared with the normal β allele [21]. On the other hand, the substitution of C for T in the adjacent nucleotide, intron 1 position 6, only mildly affects normal RNA splicing. Although the IVS1-6 T-C mutation is generally associated with milder β-thalassemia, studies have shown differential severities for apparently identical mutations; again this is presumably related to the chromosomal background on which the mutations have arisen [48].
Both exons and introns also contain ‘cryptic’ splice sites which are sequences very similar to the consensus sequence for a splice site but are not normally used. Mutations can occur in these sites creating a sequence that resembles more closely the normal splice site, such that during RNA processing the newly created site is utilized preferentially, leading to aberrant splicing; incorrectly spliced mRNA is not functional because spliced intronic sequences generate a frameshift and a premature termination code. Such mutations have been identified in both introns 1 and 2, and exon 1 of HBB gene. The associated phenotype may be either β+ or β0 thalassemia, depending on the proportion of normal and abnormal mRNA species generated. One such mutation is the (GAG → AAG) mutation in codon 26 in exon 1 [49], that results in the HbE variant. The single base substitution leads to a minor use of the alternative pathway; as the major β mRNA that codes for the variant is normally spliced, HbE has a mild β+ thalassemia phenotype. Clinical phenotypes of compound HbE/β-thalassemia heterozygotes resemble those of homozygous β-thalassemia ranging from severe anemia and transfusion-dependency to non-transfusion dependent states (i.e. thalassemia intermedia) depending on the non-HbE β-thalassemia allele and other genetic factors [50].
Other RNA processing mutants affect the polyadenylation signal (AATAA) and the 3′ UTR. These are generally mild β+ thalassemia alleles [3].
Translational Mutations
About half of the β thalassemia alleles completely inactivate the gene mostly by generating premature stop codons (PTCs) , either by single base substitution to a nonsense codon , or through a frameshift mutation. As part of the surveillance mechanism that is active in quality control of the processed mRNA, mRNA harboring a PTC are destroyed and not transported to the cytoplasm in a phenomenon called (nonsense mediated RNA decay or NMD) to prevent the accumulation of mutant mRNAs coding for truncated peptides [51]. However, some in-phase PTCs that occur later in the β sequence, in 3′ half of exon 2 and in exon 3, escape NMD and are associated with substantial amounts of mutant β-mRNA leading to a synthesis of β chain variants that are highly unstable and non-functional with a dominant negative effect (see Dominantly inherited β-thalassemia) [52]. Other mutations of RNA translation involve the initiation (ATG) codon. Nine of these have been described; apart from an insertion of 45 bp all are single base substitutions and again they result in β0 thalassemia [21].
Deletions Causing β-Thalassemia
β-thalassemia is rarely caused by deletions , 15 restricted to the HBB gene itself have been described, of which two remove the 3′ end of the gene but leave the 5′ end intact [3]. The 0.6 kb deletion at the 3′ end of the β-gene is relatively common, but restricted to the Sind populations of India and Pakistan where it accounts for about one-third of the β-thalassemia alleles [53]. The other deletion which removes 7.7 kb 3′ from the second intron of HBB, was described in compound heterozygosity with βS gene in a woman with SCD from Cape Verde islands [54]. The other thirteen deletions differ widely in size (from 290 bp to >67 kb) but remove in common a region in the β promoter (from position −125 to +78 relative to the mRNA CAP site) which includes the CACCC, CCAAT, and TATA elements. They are extremely rare, but of particular clinical interest because they are associated with an unusually high levels of HbA2 and HbF in heterozygotes. These deletions result in β0 thalassemia, yet the increase in HbF is adequate to compensate for the complete absence of HbA in homozygotes for these deletions [55,56,57]. It has been proposed that the mechanism underlying the elevated levels of HbA2 and HbF is related to deletion of the β promoter removing competition for the upstream β-LCR and limiting transcription factors, resulting in an increased interaction of the LCR with the γ- and δ-genes in cis, thus enhancing their expression. This mechanism may also explain the unusually high HbA2 levels that accompany the point mutations in the β promoter region [23].
Dominantly Inherited β-Thalassemia
In contrast to the common β-thalassemia alleles that are inherited typically as Mendelian recessives, some forms of β-thalassemia are dominantly inherited , in that inheritance of a single copy of the β-thalassemia allele results in clinical disease. Carriers have moderate to severe anemia, splenomegaly and the hallmarks of heterozygous β-thalassemia—elevated HbA2 and imbalanced globin chain synthesis [58]. More than thirty dominantly inherited β-thalassemia alleles have now been described; they include a spectrum of molecular lesions—single base missense mutations and minor insertions / deletions that result in truncated or elongated β-globin variants with abnormal carboxy-terminal ends [59]. The underlying denominators of these variants are the production of highly unstable and non-functional β chain variants that are not able to form viable tetramers with α globin. These precipitate in the erythroid precursors and together with the redundant α chains, overload the proteolytic mechanism causing premature death of these cells, and accentuating the ineffective erythropoiesis. Unlike the recessive forms of β-thalassemia that are prevalent in the malarious regions, the dominantly inherited β-thalassemia variants are rare, and found in dispersed geographical regions where the gene frequency for β-thalassemia is very low. Furthermore, many of these variants are unique to the families described, and occur as de-novo mutations.
Unusual Causes of β-Thalassemia
The unusual causes of β-thalassemia are extremely rare and are mentioned here, not just for the sake of completeness but also to illustrate the numerous molecular mechanisms of downregulating the β-globin gene. Transposable elements may occasionally disrupt human genes and result in their activation. The insertion of such an element, a retrotransposon of the LI family into intron 2 of the β-globin gene has been reported to cause β+ thalassemia [60].
Almost all variants downregulating HBB are physically linked to the gene and behave as alleles of the β-globin locus (i.e. they are cis-acting). Rarely, mutations in other genes distinct from the β-globin complex can downregulate β-globin expression. Such trans-acting mutations have been described affecting the XPD protein that is part of the general transcription factor TF11H [61], and the erythroid-specific GATA-1 [62]. Somatic deletion of the β globin gene contributed to thalassemia intermedia in three unrelated families of French and Italian origins [63, 64]. The affected individuals with thalassemia intermedia were constitutionally heterozygous for β0 thalassemia but subsequent investigations revealed a somatic deletion of chromosome 11p15, including the β globin gene complex, in trans to the mutation in a subpopulation of erythroid cells. This results in a somatic mosaic—10–20% of the cells were heterozygous with one normal copy of the β-globin gene, and the rest hemizygous, i.e. without any normal β-globin gene. The sum total of β-globin product is ~25% less than the asymptomatic β0 carrier; these observations offer great promise for potential gene therapy as it shows that expression of a single β-gene in a proportion of red blood cells appears to be sufficient to produce a non-transfusion dependent state. Late presentation of β-thalassemia and transfusion dependency has been reported in a Chinese patient [65], and a Portuguese woman at the age of 15 years [66]. In both cases, the phenotype was caused by uniparental isodisomy of the paternal chromosome 11p15.5 that encompassed the β-thalassemia allele.
Genetics of Sickle Cell Disease
SCD describes a clinical syndrome caused by the presence of HbS (HbS GGlu6Val) [4]. The main genotypes that contribute to SCD include homozygosity for the βS allele (HbSS, specifically referred to as sickle cell anemia), followed by compound heterozygous states of HbS with HbC (HbSC disease). The third genotypic group includes compound heterozygotes of HbS with β-thalassemia, HbS β+ thalassemia or HbS β0 thalassemia depending on whether there is some, or no HbA, respectively. In African-descended populations, HbSS typically accounts for 65–70%, and HbSC 30–35% of the cases of SCD , with most of the remainder having HbSβ-thalassemia (Table 2.1(A)). Other genotypes of SCD have been described, including compound heterozygotes of HbS with HbD, HbO-Arab, but these are rare (Table 2.1(B)). In all affected populations, the βS gene is caused by the same molecular defect (β codon 6 GAG to GTG), found on a genetic background of four different βS haplotypes: three distinct African haplotypes—Senegal, Benin and Central African Republic (or Bantu), and a fourth Indo-European β-haplotype (Arab-Indian) that is linked to the βS gene in Saudi Arabian and Asian Indian patients [67]. The evidence suggests multiple independent origins of the βS mutation although gene conversion on regionally specific β-haplotypes cannot be excluded.
Genetic Modifiers of β-Hemoglobinopathies
SCD and β-thalassemia are caused by mutations affecting a single gene, and yet, despite the apparent genetic simplicity, both disorders display remarkable diversity in the severity of their disease [4, 68, 69]. The mutations are detectable by DNA analysis, and although the information provides a basis for genetic counselling, predicting disease severity remains difficult. Identification of the genetic modifiers could provide more precise estimates of severity of disease, contributing to the ethos of precision medicine. Further, defining the molecular mechanisms linking the genetic factors could reveal new targets for therapeutic intervention.
Historically, the genetic modifiers in both β-thalassemia and SCD have been derived from an understanding of their pathophysiology, and subsequently validated by family and case control studies. Two important modifiers—co-inheritance of α thalassemia and variants associated with increased synthesis of HbF in adults have emerged from such clinical genetic studies. The modifying effects of HbF and α-thalassemia on SCD and β-thalassemia at the molecular level could not be more different, and yet, elucidation of these genetic modifiers has not been too difficult as these loci have a major clinical effect and the genetic variants are common, and thus would contribute substantially to disease burden. However, these two modifiers do not explain all the clinical heterogeneity. Recent advances in technology and reducing costs have prompted numerous genome-wide association studies (GWAS) in an attempt to derive some of the genetic modifiers in such complex traits [70].
GWASs involve an unbiased scan of the whole human genome and, by design, are more likely to reveal unsuspected interactions [71]. A case in point is the application of GWAS in the highly successful discovery of BCL11A (an oncogene that hitherto, was not known to have a role in erythropoiesis) as a quantitative trait locus (QTL) controlling HbF [32].
Update on the Genetic Control of Fetal Hemoglobin (HbF)
The production of HbF is not completely switched off at birth; all adults continue to produce residual amounts of HbF, with over 20-fold variation [72]. Twin studies have shown that the common HbF variation in adults is predominantly genetically controlled; 89% of the quantitative variation is heritable but the genetic etiology is complex with no clear Mendelian inheritance patterns [73]. Xmn1-HBG2 (rs782144) within the β-globin gene cluster on chromosome 11p, HBS1L-MYB intergenic region, (HMIP) on chromosome 6q23, and BCL11A on chromosome 2p16 are considered to be quantitative trait loci (QTLs) for the common HbF variation in adults [74,75,76].
Xmn1-HBG2 (rs782144) was the first known QTL for HbF and long implicated by clinical genetic studies [77]. SCD individuals in whom the βS gene is on the Senegal or Arab-Indian βS haplotype have the highest HbF levels and a mild clinical course, while SCD individuals with the βS gene on a Bantu haplotype have the lowest HbF levels and the most severe clinical course [78, 79]. The differences in clinical severity was ascribed to the difference in HbF levels implicating the Xmn1-HBG2 site which is linked to the Senegal and Arab-Indian βS haplotype but not to the Bantu haplotype [77]. Recent high resolution genotyping, however, suggests that rs782144 is not likely to be the variant itself, but in tight linkage disequilibrium with causal element(s) that remain to be discovered in the β-globin cluster.
Variants in the HBB, HMIP and BCL11A loci account for 10–50% of the variation in HbF levels in adults, healthy or with sickle cell anemia or β-thalassemia, depending on the population studied [32, 80,81,82,83,84,85,86]. The remaining variation (‘missing heritability’) is likely to be accounted for by many loci with relatively small effects, and/or rare variants with significant quantitative effects on γ-globin gene expression that are typically missed by GWAS population studies. An example of the latter is the KLF1 gene [25, 40].
KLF1 , discovered by Jim Bieker in 1993 [25] re-emerged as a key transcription factor controlling HbF through genetic studies in a Maltese family with β-thalassemia and HPFH that segregated independently of the HBB locus [37]. Linkage studies identified a locus on chromosome 19p13 which encompassed KLF1 and expression profiling of erythroid progenitor cells confirmed KLF1 as the γ-globin gene modifier in this family. Family members with HPFH were heterozygous for the nonsense K288X mutation in KLF1 that disrupted the DNA-binding domain of KLF1, a key erythroid gene regulator. Numerous reports of different mutations in KLF1 associated with increases in HbF soon followed (see review by Borg et al. 2011) [40]. The HbF increases occurred as a primary phenotype or in association with red blood cell disorders such as congenital dyserythropoietic anemia [39, 41], congenital nonspherocytic hemolytic anemia due to pyruvate kinase deficiency [87] and sickle cell anemia [40]. Several GWASs of HbF however, including ones in sickle cell anemia patients of African descent, failed to identify common variants [82, 86]. It would seem that KLF1 does not play an important part in regulation of HbF in SCD. On the contrary, KLF1 variants were over represented in Southern China where β-thalassemia is prevalent compared to North China. Further, KLF1 variants were also over-represented in patients with thalassemia intermedia when compared to thalassemia major [88].
KLF1 is a direct activator of BCL11A (see below) and is also essential for the activation of HBB expression [38, 89]. Collectively, studies suggest that KLF1 is key in the switch from HBG to HBB expression; it not only activates HBB directly, providing a competitive edge, but also silences the gamma-globin genes indirectly via activation of BCL11A , and may play a role in the silencing of embryonic globin gene expression [87]. In the light of these findings, KLF1 has now emerged as a major erythroid transcription factor with pleiotropic roles underlying many of the previously uncharacterized anemias [30].
Functional studies in primary human erythroid progenitor cells and transgenic mice demonstrated that BCL11A acts as a repressor of gamma-globin gene expression that is effected by SNPs in intron 2 of this gene [34]. Fine-mapping demonstrated that these HbF-associated variants, in particular rs1427407, localized to an enhancer that is erythroid-specific and not functional in lymphoid cells [90]. BCL11A does not interact with the γ-globin promoter but occupies discrete regions in the HBB complex [91]. Functional studies in primary human erythroid progenitors and transgenic mice demonstrated that BCL11A represses γ-globin and the silencing effect involves re-configuration of the HBB locus through interaction with GATA-1 and SOX6 that binds the proximal γ-globin promoters [92, 93].
High resolution genetic mapping and resequencing refined the 6q QTL to a group of variants in tight linkage disequilibrium (LD) in a 24-kb block between the HBS1L and MYB gene, referred to as HMIP-2 [94]. The causal single nucleotide polymorphisms (SNPs) are likely to reside in two clusters within the block, at −84 and −71 kb respectively, upstream of MYB [95, 96]. Functional studies in transgenic mice and primary human erythroid cells provide overwhelming evidence that the SNPs at these two regions disrupt binding of key erythroid enhancers affecting long-range interactions with MYB and MYB expression, providing a functional explanation for the genetic association of the 6q HBS1L-MYB intergenic region with HbF and F cell levels [95, 97, 98].
A three-base pair (3-bp) deletion in HMIP-2 is one functional element in the MYB enhancers accounting for increased HbF expression in individuals who have the sentinel SNP rs9399137 that was found to be common in European and Asian populations, although less frequent in African-derived populations [99]. The DNA fragment encompassing the 3-bp deletion had enhancer-like activity that was augmented by the introduction of the 3-bp deletion.
The MYB transcription factor is a key regulator of hematopoiesis and erythropoiesis, and modulates HbF expression via two mechanisms: (1) indirectly through alteration of the kinetics of erythroid differentiation: low MYB levels accelerate erythroid differentiation leading to release of early erythroid progenitor cells that are still synthesizing predominantly HbF [8], and (2) directly via activation of KLF1 and other repressors (e.g., nuclear receptors TR2/TR4) of gamma-globin genes [98, 100, 101].
Modulation of MYB expression also provides a functional explanation for the pleiotropic effect of the HMIP-2 SNPs with other erythroid traits such as red cell count, MCV, MCH, HbA2 levels, and also with platelet and monocyte counts [102,103,104,105]. MYB expression is also reduced by GATA-1 [106], and miRNA-15a and -16-1 [107]. Elevated levels of the latter have been proposed as the mechanism for the elevated HbF levels in infants with trisomy 13. A delayed HbF to HbA switch, along with persistently elevated HbF levels, is one of the unique features in infants with trisomy 13 [108]. One study has provided compelling evidence that the elevated HbF levels relate to the increased expression of microRNAs 15a and 16-1 produced from the triplicated chromosome 13. The increased HbF effect is mediated, at least in part, through down-modulation of MYB via targeting of its 3′ UTR by microRNAs 15a and 16-1 [107].
The HBS1L-MYB intergenic enhancers do not appear to affect expression of HBS1L, the other flanking gene [95]. Further, one study also excluded HBS1L as having a role in the regulation of HbF and erythropoiesis. In whole-exome sequencing of rare uncharacterized disorders, mutations in the HBS1L gene leading to a loss of function in the gene were identified in a female child [109]. The child had normal blood counts and normal HbF levels. Thus, HMIP-2 is likely to affect HbF and hemopoietic traits via regulation of MYB. MYB was also causally implicated by fine-mapping which identified rare missense MYB variants associated with HbF production [110].
The emerging network of HbF regulation also includes SOX6, chromatin-modeling factor FOP and the NURD complex, the orphan nuclear receptors TR2 / TR4 (part of DRED) and the protein arginine methyltransferase PRMT5, involving DNA methylation and HDACs 1 and 2 epigenetic modifiers. Regulators of the key TFs, such as microRNA-15a and 16-1 in controlling MYB, could also have a potential role in regulating HbF levels [111].
Genetic Modifiers of Sickle Cell Disease
Two case reports [112, 113] and a pilot twin study [114] show that despite identical β and α globin genotypes and similarities in growth, hematological and biochemical parameters, the identical twins have quite different prevalence and severity of painful crises and some of the sickle complications. Although sample size of the twin study is small, nevertheless, it does suggest that environmental factors may be of greater importance in determining clinical expression and complications of sickle disease.
SCD should be considered as both a qualitative and quantitative genetic disorder. While presence of HbS is fundamental to the pathobiology, the likelihood of HbS polymerization and sickling is also highly dependent on the concentration of intra-erythrocytic HbS, as well as the presence of non-S Hb [115]. Thus, individuals with HbSS (SCD-SS) or HbSβ0 thalassemia, where the intra-cellular Hb is almost HbS tend to have the most severe disease followed by HbSC and HbSβ+ thalassemia (see Table 2.1).
HbA (α2β2) or HbA2 (α2δ2) do not participate in HbS polymerization, and since the β+ thalassemia alleles in Africans are of the milder type with minimal deficit in β-globin production, HbSβ+ thalassemia in Africans tends to be very mild. In contrast, individuals with HbSβ+ thalassemia in the Mediterranean, have SCD almost as severe as that of HbSS [116]. Subjects with sickle cell trait (HbAS) with HbF of 35–40% do not normally suffer from symptoms of SCD. Under exceptional circumstances however, such as intense physical activity and dehydration, the consequent increased intracellular HbS concentration can induce vaso-occlusive pain [117].
Other genetic factors that influence the primary event of HbS polymerization include (HbF, α2γ2) and the co-inheritance of α thalassemia (Table 2.2).
Impact of HbF in SCD
HbF inhibits the propensity for HbS polymerization: the hybrid tetramers (α2γβs), not only inhibits HbS polymerization, but intra-erythrocytic HbF presence also dilutes the concentration of HbS [118]. The clinical phenotype of SCD becomes evident within 6 months to 2 years of age as HbF levels decline.
Its impact at the primary level of disease pathology predicts HbF levels to have a global beneficial effect. Indeed, HbF levels are a major predictor of survival in SCD [119], and low levels of HbF have been associated with increased risk of brain infarcts in young children [120]. At the sub-phenotype level, apart from the clear benefit of high HbF levels with acute pain and leg ulceration, there are disparities and less conclusive evidence in its effects on complications such as stroke, renal impairment, retinopathy and priapism [121, 122]. The failure of HbF to uniformly modulate all complications of SCD may be related to the different pathophysiology of the different complications and perhaps also to the small sample sizes in genetic studies and even smaller numbers of end complications, and to ascertainment of phenotypes.
HbF levels vary considerably from 1% to as high as 25% in individuals with SCA and behave as a quantitative genetic trait as in healthy individuals. As discussed earlier, part of this variation resides in regions linked to the HBB complex and is associated, at least in part, with the βS haplotype (Senegal, Arab-Indian, Bantu and Benin) on which the βS gene is found. However, a variance of HbF levels is encountered within each βS haplotype, evidence for the importance of unlinked HbF QTLs such as HMIP-2 on 6q and BCL11A on 2p. Their effect on HbF levels varies with the frequency of the HbF boosting (minor) alleles on the three QTLs (β -globin cluster, HMIP-2 and BCL11A) in different population groups; in patients of African American patients with SCD, the three loci account for 16%-20% of the variation in HbF levels with a corresponding reduction in acute pain rate [80].
A recent GWAS in Tanzanian patients with SCD also confirmed association SNPs at BCL11A and HMIP-2 with HbF but no other HbF QTLs were ‘discovered’ [86]. Similarly, in patients from Cameroon, while these SNPs had a significant impact on HbF levels and rates of hospitalization, they explained less of the HbF variance than that observed in African American patients [123]. The Xmn1-HBG (rs782144) is virtually absent in Cameroon patients with SCD [123].
Impact of α-Thalassemia on SCD Phenotype
About one third of SCD patients of African descent have co-existing α-thalassemia due to the common deletional variant (−α3.7/) [124]. The majority is heterozygous (αα/α-) with 3–5% homozygous for the deletion (α−/α−) [125]. Co-existing α-thalassemia reduces intracellular Hb concentration, decreases the propensity for HbS polymerization, and decrease hemolysis. While the co-existing α-thalassemias have a protective effect against complications associated with severe hemolysis such as priapism, leg ulceration and albuminuria, the increased hematocrit and blood viscosity may account for the increase in other complications associated with microvascular occlusion such as increased acute pain, acute chest syndrome, osteonecrosis and retinopathy [122].
Co-inheritance of α-thalassemia improved hematological indices and was associated with lower rates of hospitalization in Cameroon patients with SCA [126]. In the same study, Rumaney et al also showed that in Cameroon, the incidence of α-thalassemia trait in controls (HbAA) was significantly lower than that in patients with SCA (9.1 vs. 30.4%); the authors proposed that the difference in incidence could be explained by a protective effect of α thalassemia on overall survival in SCA patients. Co-inheritance of α-thalassemia was also associated with lower hospitalization rate in SCA patients.
Coexisting α-thalassemia reduces bilirubin with a quantitative effect that is independent to that of the UGT1A1 promoter polymorphism [125]. In Jamaicans, the absence of α thalassemia and higher HbF levels predicts a benign disease, although a subsequent study reports that α-thalassemia did not promote survival in elderly Jamaicans with SCD-SS [127]. Coinheritance of α-thalassemia blunts the response to hydroxyurea therapy in SCD, that may be explained by its effect on HbF levels and MCV, two key parameters associated with hydroxyurea response [128]. It is quite likely that α-thalassemia carriers could have a poorer response to channel blockers that aim to reduce sickling through preservation of cell hydration [129].
Secondary Modifiers of Sub-phenotypes and Complications
Association studies of candidate genes implicated in the pathophysiology of vasculopathy, such as those encoding factors modifying inflammation , oxidant injury, nitric oxide biology, vaso-regulation and cell adhesion, have been used extensively to identify variants affecting sickle-related complications, such as stroke, priapism, leg ulcers, avascular necrosis, renal disease, acute chest syndrome, gallstones and susceptibility to infection (see following reviews [121, 122, 130]). The majority of these reported associations have not been replicated or validated, and are likely to be false positives. Of the numerous association studies reported, the most robust is the association between serum bilirubin levels and predisposition to gallstones with the 6/7 or 7/7 (TA) repeats in the UGT1A1 promoter [125, 131]. The influence of UGT1A1 polymorphism became more evident in patients while on hydroxycarbamide therapy; children with 6/6 UGT1A1 genotype achieved normal bilirubin levels while children with 6/7 or 7/7 UGT1A1 genotypes did not [132].
Numerous genetic and clinical association studies on cerebrovascular complications in SCD have been carried out [133]. Of the 38 published SNPs associated with stroke, the protective effect of α thalassemia on stroke risk and SNPs in four genes (ADYC9, ANXA2, TEK and TGFBR3) could be replicated although only nominally significant association results were obtained [134]. More recently, GWAS in combination with whole exome sequencing have identified mutations in two genes—GOLGB1 and ENPP1—associated with reduced stroke risk in pediatric patients but again, this needs validation in independent studies. In an attempt to overcome the small sample size in end-point complications, a study utilized a compound phenotype that included one or more sickle-related complications [135]. Patients with complications had a higher frequency of the platelet glycoprotein allele HPA-5B. In this small study, most of the complications were osteonecrosis and only four individuals had more than one complication [135]. As traditional methods are often inadequate in association studies of complex traits, methods of evaluating multi-locus data are promising alternatives. A GWAS was applied to SCD based on a disease severity score that was derived from a Bayesian network that integrates 25 different clinical and laboratory variables [136]. Several genes not known to be related in the pathogenesis of SCD were identified including KCN6 (a potassium channel protein) and TNK5 (gene encoding tankyrase-1, a possible telomere length regulator). However it is important to remember that results from such analytical techniques are dependent on the structure of a model which assumes causality and prior probabilities assigned to the different variables. Using a hemolytic score, GWAS identified a SNP (rs7203560) in NPRL3 that was independently associated with hemolysis [137]. rs7203560 is in perfect LD with SNPs within the α-globin gene regulatory elements (HS-48, HS-40 and HS-33), and also in LD with ITFG3 that is associated with MCV and MCH in several GWASs of different population groups. It is proposed that NPRL3 reduces hemolysis through an independent thalassemic effect on the HBA1/HBA2 genes.
Hydroxyurea remains a major treatment option for SCD [138, 139]; its main effect is mediated primarily through induction of HbF. Clinical and laboratory response to HU therapy however, is variable, a main determinant of response appears to be the baseline HbF levels. Numerous association studies on HbF response to hydroxycarbamide have been reported, of which the association with baseline HbF levels and Xmn1-HBG2 seems to be the most robust [140]. Table 2.1 summarizes the genetic modifiers of SCD.
Genetic Modifiers of β-Thalassemia
The central mechanism underlying the pathophysiology of the β-thalassemias relates to the deleterious effects of the excess α-globin chain on erythroid survival and ineffective erythropoiesis [68]. Clinical studies have shown that disease severity correlates well with the degree of imbalance between α and β globin chains and the size of the free α chain pool. Genetic modifiers can impact the phenotypic severity at the primary level by affecting the degree of globin chain imbalance, and at the secondary level by moderating complications of the disease related to the anemia or therapy e.g. iron chelation.
While the severity of β-thalassemia is primarily determined by the degree of β chain deficiency , for any given β-thalassemia allele the severity of the disease can be alleviated by co-inheritance of α-thalassemia or by co-inheritance of factors that increase γ-globin chain production and HbF levels. In the latter case, γ-globin chains combine with the excess α-globin to form HbF; cells that contain a relatively higher percentage of HbF are protected against the deleterious effect of alpha-globin chain precipitation and premature death, and have selective survival. Thus, all individuals with β-thalassemia have variable increases in HbF due to survival of these F cells.
Certain β-thalassemia mutations, notably those that involve small deletions or mutations of the promoter sequence of the HBB gene, are associated with much higher levels of HbF production than mutations affecting other regions of HBB (see deletions causing β-thalassemia) [3]. This might reflect the competition between the HBG and HBB promoters for interaction with the upstream β-LCR and limited transcription factors. Heterozygotes for such types of β-thalassemia mutations have unusually high HbA2, and although the increases in HbF levels are variable, the increase in HbF production in homozygotes is adequate to compensate for the complete absence of HbA. HbF levels are normal or slightly elevated in β-thalassemia heterozygotes. Higher HbF (and HbA2) levels are found with mutations that involve promoter of the HBB gene, but variations in HbF levels also reflect the genetic background of the individual (e.g. −29 promoter mutation in Blacks and Chinese). In homozygous β-thalassemia, the proportion of HbF ranges from 10%in those with the milder alleles to almost 100% in homozygotes or compound heterozygotes with β0 thalassemia. In the most severe cases, the absolute amount of HbF is approximately 3–5 g/dL, produced as a result of extreme erythroid hyperplasia, selective survival of F cells, and some increase in HBG transcription. Non-transfusion dependent β0 thalassemia intermedia with hemoglobin levels of 8–11 g/dL and 100% HbF has been observed. In some cases, the increase in HbF production reflects the type of β-thalassemia allele, but in others co-inheritance of QTLs associated with increased HBG expression might explain their more benign clinical features [114].
Effect of the Primary Modifiers: HbF Quantitative Trait Loci and α-Globin Genotype
While selection of F cells provides an explanation for the increases in HbF in β-thalassemia, the mechanism does not explain the wide variation in the amount produced. Much of this variability is genetically determined, in part from the co-inheritance of one or more of HbF-boosting alleles of the Xmn1-HBG2, HMIP-2 and BCL11A HbF quantitative trait loci (QTLs) [33, 81, 84, 142, 143]. The Xmn1-HBG2 QTL is a common sequence variation in all population groups, present at a frequency of approximately 0.35. Although increases in HbF and F cells associated with Xmn1-HBG2 are minimal or undetectable in healthy adults, clinical studies have shown that under conditions of stress erythropoiesis, as in homozygous β-thalassemia, the presence of Xmn1-HBG2 leads to a much higher HbF response [141]. This could explain why the same mutation on different beta chromosomal backgrounds, some with and others without the Xmn1-HBG2 variant, are associated with different clinical severity. High resolution genotyping studies suggest that Xmn1-HBG2 may not be the causal element but in tight linkage disequilibrium to another, as yet undiscovered, variant(s) on chromosome 11p.
Together Xmn1-HBG2, BCL11A, and HMIP-2, and perhaps other loci , linked and unlinked to the HBB complex, constitute the loosely-defined entity of heterocellular HPFH. These HbF QTLs play an important role in fine-tuning γ-globin production in healthy adults and in response to the stress erythropoiesis of sickle cell anemia and β-thalassemia. The three QTLs are associated with HbF and severity of thalassemia in diverse population groups including Sardinian, French, Chinese, and Thai [33, 84, 143, 144]. More than 95% of Sardinian β-thalassemia patients are homozygous for the same codon 39 β0 thalassemia mutation but have extremely variable clinical severity. Co-inheritance of variants in BCL11A and HMIP-2, and α-thalassemia accounts for 75% of the differences in disease severity [81]. In France, a combination of the beta thalassemia genotype, Xmn1-HBG2 and SNPs in BCL11A and HMIP-2, can predict up to 80% of disease severity [84]. In a cohort of 316 β0 thalassemia patients, delayed or absent transfusion requirements correlated with status of the three HbF QTLs and the α-globin genotype [145]. Using a combination of the HbF QTLs, the type of β-thalassemia mutations, and the α-globin genotype, a predictive score of severity has been proposed [146].
In many populations where β-thalassemia is prevalent, α-thalassemia also occurs at a high frequency and hence it is not uncommon to co-inherit both conditions [15]. Homozygotes or compound heterozygotes for β-thalassemia who co-inherit α-thalassemia will have less redundant α globin and tend to have a less severe anemia. The degree of amelioration depends on the severity of the β-thalassemia alleles and the number of functional α-globin genes. At one extreme, patients with homozygous β-thalassemia who have also co-inherited HbH (equivalent of only one functioning α-globin gene) have thalassemia intermedia [68].
In individuals with one β-thalassemia allele (heterozygotes), co-inheritance of α thalassemia normalizes the hypochromia and microcytosis but the elevated HbA2 remains unchanged. Increased α globin production through co-inheritance of extra α-globin genes (triplicated—ααα/αα or ααα/ααα, quadruplicated—αααα/αα, or duplication of the whole α globin gene cluster—αα/αα/αα) with heterozygous β-thalassemia tips the globin chain imbalance further, converting a typically clinically asymptomatic state to thalassemia intermedia [14, 68, 147]. Again, the severity of anemia depends on the number of extra α globin genes and the severity of the β-thalassemia alleles.
At the primary level of chain imbalance, the proteolytic capacity of the erythroid precursors in catabolising the excess α globin has often been suggested, but this effect has been difficult to define. Alpha hemoglobin stabilizing protein, a molecular chaperone of α globin has also been suggested as another genetic modifier but its impact on disease severity has been inconclusive [148].
Secondary Modifiers of Complications of β-Thalassemia
These modifiers do not affect globin imbalance directly but might moderate the different complications of β-thalassemia that are directly related to the anemia, or to therapy such as iron overload [69, 149, 150]. They include genetic variants which affect bilirubin metabolism, iron metabolism, bone disease and cardiac complications. Jaundice and a predisposition to gallstones, a common complication of β-thalassemia, are associated with a polymorphic variant in the promoter of the UGT1A1 gene. Individuals who are homozygous for 7 [TA]s, also referred to as Gilbert’s syndrome , have higher levels of bilirubin and increased predisposition to gallstones, an observation that has been validated at all levels of β-thalassemia. Several genes involved in iron homeostasis have now been characterized, including those encoding HFE, (HFE), transferrin receptor 2 (TFR2), ferroportin (FPN), hepcidin (HAMP) and hemojuvelin (HJV) [151]. The H63D variant, a common polymorphism in the HFE gene, appears to have a modulating effect on iron absorption. β-thalassemia carriers who are homozygous for HFE H63D variant, have higher serum ferritin levels than carriers without the variant (see Table 2.3 for modifiers of β-thalassemia).
The degree of iron loading, bilirubin levels and bone mass are quantitative traits with a genetic component; variants affect the genes that are involved in the regulation of these traits that contribute to the complications.
References
Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008 Jun;86(6):480-7. PubMed PMID: 18568278.
Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010 Jun 3;115(22):4331-6. PubMed PMID: 20233970. Pubmed Central PMCID: 2881491. Epub 2010/03/18. eng.
Thein SL. The Molecular Basis of beta-Thalassemia. Cold Spring Harb Perspect Med. 2013;3(5):3/5/a011700 [pii] 10.1101/cshperspect.a. PubMed PMID: 23637309. Epub 2013/05/03. eng.
Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010 Dec 11;376(9757):2018-31. PubMed PMID: 21131035. Epub 2010/12/07. eng.
Willcox M. The haemoglobin pattern of sickle cell and haemoglobin C b+-thalassaemia in Liberia. Journal of Medical Genetics. 1983;20:430-2.
Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Williams TN, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104. PubMed PMID: 21045822. Pubmed Central PMCID: 3060623. Epub 2010/11/04. eng.
Piel FB, Patil AP, Howes RE, Nyangiri OA, Gething PW, Dewi M, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013 Jan 12;381(9861):142-51. PubMed PMID: 23103089. Pubmed Central PMCID: 3547249. Epub 2012/10/30. eng.
Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Experimental hematology. 2005 Mar;33(3):259-71. PubMed PMID: 15730849.
Carter D, Chakalova L, Osborne CS, Dai YF, Fraser P. Long-range chromatin regulatory interactions in vivo. Nature genetics. 2002;32(4):623-6. PubMed PMID: 12426570.
Li Q, Peterson KR, Fang X, Stamatoyannopoulos G. Locus control regions. Blood. 2002;100(9):3077-86. PubMed PMID: 12384402.
Palstra RJ, de Laat W, Grosveld F. Beta-globin regulation and long-range interactions. Adv Genet. 2008;61:107-42. PubMed PMID: 18282504.
Cantor AB, Orkin SH. Transcriptional regulation of erythropoiesis: an affair involving multiple partners. Oncogene. 2002 May 13;21(21):3368-76. PubMed PMID: 12032775.
Bauer DE, Orkin SH. Update on fetal hemoglobin gene regulation in hemoglobinopathies. Curr Opin Pediatr. 2011 Feb;23(1):1-8. PubMed PMID: 21157349. Pubmed Central PMCID: 3092400. Epub 2010/12/16. eng.
Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. PubMed PMID: 20492708. Pubmed Central PMCID: 2893117. Epub 2010/05/25. eng.
Weatherall DJ, Clegg JB. The Thalassaemia Syndromes. 4th ed. Oxford: Blackwell Science; 2001.
Serjeant GR. The natural history of sickle cell disease. In: Weatherall DJ, Schechter AN, Nathan DG, editors. Hemoglobin and its diseases. Cold Spring Harbor Perspectives in Medicine. New York: Cold Spring Harbor Laboratory Press; 2013. p. 301-11.
Kiefer CM, Hou C, Little JA, Dean A. Epigenetics of beta-globin gene regulation. Mutat Res. 2008 Dec 1;647(1-2):68-76. PubMed PMID: 18760288. Pubmed Central PMCID: 2617773. Epub 2008/09/02. eng.
Hanscombe O, Whyatt D, Fraser P, Yannoutsos N, Greaves D, Dillon N, et al. Importance of globin gene order for correct developmental expression. Genes and Development. 1991;5:1387-94.
Raich N, Enver T, Nakamoto B, Josephson B, Papayannopoulou T, Stamatoyannopoulos G. Autonomous developmental control of human embryonic globin gene switching in transgenic mice. Science. 1990;250:1147-9.
Dillon N, Grosveld F. Human g-globin genes silenced independently of other genes in the b-globin locus. Nature. 1991;350:252-4.
Thein SL, Wood WG. The molecular basis of β-thalassemia, δβ-thalassemia, and hereditary persistence of fetal hemoglobin. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Second ed. Cambridge, UK: Cambridge University Press; 2009. p. 323-56.
Giardine B, Borg J, Higgs DR, Peterson KR, Philipsen S, Maglott D, et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nature genetics. 2011 Apr;43(4):295-301. PubMed PMID: 21423179. Epub 2011/03/23. eng.
Huisman THJ. Levels of Hb A2 in heterozygotes and homozygotes for beta-thalassemia mutations: Influence of mutations in the CACCC and ATAAA motifs of the beta-globin gene promoter. Acta haematologica. 1997;98:187-94. PubMed PMID: 9401495.
Moi P, Faa V, Marini MG, Asunis I, Ibba G, Cao A, et al. A novel silent beta-thalassemia mutation in the distal CACCC box affects the binding and responsiveness to EKLF. British journal of haematology. 2004 Sep;126(6):881-4. PubMed PMID: 15352994.
Miller IJ, Bieker JJ. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Krüppel family of nuclear proteins. Molecular and cellular biology. 1993;13(5):2776-86.
Perkins AC, Sharpe AH, Orkin SH. Lethal b-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375(6529):318-22.
Nuez B, Michalovich D, Bygrave A, Ploemacher R, Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375(6529):316-8.
Bieker JJ. Erythroid-specific transcription. Current opinion in hematology. 1998;5:145-50.
Bieker JJ. Putting a finger on the switch. Nature genetics. 2010 Sep;42(9):733-4. PubMed PMID: 20802474. Epub 2010/08/31. eng.
Siatecka M, Bieker JJ. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood. 2011 Aug 25;118(8):2044-54. PubMed PMID: 21613252. Pubmed Central PMCID: 3292426. Epub 2011/05/27. eng.
Yien YY, Bieker JJ. EKLF/KLF1, a tissue-restricted integrator of transcriptional control, chromatin remodeling, and lineage determination. Molecular and cellular biology. 2013 Jan;33(1):4-13. PubMed PMID: 23090966. Pubmed Central PMCID: 3536305. Epub 2012/10/24. eng.
Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nature genetics. 2007 Oct;39(10):1197-9. PubMed PMID: 17767159.
Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proceedings of the National Academy of Sciences of the United States of America. 2008 Feb 5;105(5):1620-5. PubMed PMID: 18245381.
Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, et al. Human Fetal Hemoglobin Expression Is Regulated by the Developmental Stage-Specific Repressor BCL11A. Science. 2008 Dec 19;322(5909):1839-42. PubMed PMID: 19056937.
Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, et al. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009 Aug 27;460(7259):1093-7. PubMed PMID: 19657335.
Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, et al. Correction of Sickle Cell Disease in Adult Mice by Interference with Fetal Hemoglobin Silencing. Science. 2011 Nov 18;334(6058):993-6. PubMed PMID: 21998251. Epub 2011/10/15. eng.
Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nature genetics. 2010 Aug 1;42(9):801-5. PubMed PMID: 20676099. Epub 2010/08/03. Eng.
Zhou D, Liu K, Sun CW, Pawlik KM, Townes TM. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nature genetics. 2010 Sep;42(9):742-4. PubMed PMID: 20676097. Epub 2010/08/03. Eng.
Arnaud L, Saison C, Helias V, Lucien N, Steschenko D, Giarratana MC, et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. American journal of human genetics. 2010 Nov 12;87(5):721-7. PubMed PMID: 21055716. Pubmed Central PMCID: 2978953. Epub 2010/11/09. eng.
Borg J, Patrinos GP, Felice AE, Philipsen S. Erythroid phenotypes associated with KLF1 mutations. Haematologica. 2011 May;96(5):635-8. PubMed PMID: 21531944. Pubmed Central PMCID: 3084906. Epub 2011/05/03. eng.
Jaffray JA, Mitchell WB, Gnanapragasam MN, Seshan SV, Guo X, Westhoff CM, et al. Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm. Blood cells, molecules & diseases. 2013 Aug;51(2):71-5. PubMed PMID: 23522491. Epub 2013/03/26. eng.
Magor GW, Tallack MR, Gillinder KR, Bell CC, McCallum N, Williams B, et al. KLF1 null neonates display hydrops fetalis and a deranged erythroid transcriptome. Blood. 2015 Feb 27;ePub ahead of print. PubMed PMID: 25724378.
Gonzalez-Redondo JM, Stoming TA, Kutlar A, Kutlar F, Lanclos KD, Howard EF, et al. A C->T substitution at nt -101 in a conserved DNA sequence of the promoter region of the ß-globin gene is associated with 'silent' ß-thalassemia. Blood. 1989;73:1705-11.
Maragoudaki E, Kanavakis E, Trager-Synodinos J, Vrettou C, Tzetis M, Metxotou-Mavrommati A, et al. Molecular, haematological and clinical studies of the -101 C®T substitution in the β-globin gene promoter in 25 β-thalassaemia intermedia patients and 45 heterozygotes. British journal of haematology. 1999;107(4):699-706. PubMed PMID: 10606872.
Wong C, Dowling CE, Saiki RK, Higuchi RG, Erlich HA, Kazazian HHJ. Characterization of beta-thalassaemia mutations using direct genomic sequencing of amplified single copy DNA. Nature. 1987;330:384-6.
Safaya S, Rieder RF, Dowling CE, Kazazian HHJ, Adams JGI. Homozygous b-thalassemia without anemia. Blood. 1989;73:324-8.
Huang S-Z, Wong C, Antonarakis SE, Ro-Lein T, Lo WHY, Kazazian HHJ. The same TATA box b-thalassemia mutation in Chinese and US Blacks: Another example of independent origins of mutation. Human Genetics. 1986;74:162-4.
Rund D, Oron-Karni V, Filon D, Goldfarb A, Rachmilewitz E, Oppenheim A. Genetic analysis of ß-thalassaemia intermedia in Israel: diversity of mechanisms and unpredictability of phenotype. American journal of hematology. 1997 Jan;54(1):16-22. PubMed PMID: 8980256.
Orkin SH, Kazazian HHJ, Antonarakis SE, Ostrer H, Goff SC, Sexton JP. Abnormal RNA processing due to the exon mutation of bE-globin gene. Nature. 1982;300:768-9.
Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med [Internet]. 2012; 2(8):[a011734 p.]. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22908199.
Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nat Rev Genet. 2012;13(4):246-59. PubMed PMID: 22392217. Epub 2012/03/07. eng.
Hall GW, Thein SL. Nonsense codon mutations in the terminal exon of the ß-globin gene are not associated with a reduction in ß-mRNA accumulation: a mechanism for the phenotype of dominant ß-thalassemia. Blood. 1994;83:2031-7.
Varawalla NY, Old JM, Sarkar R, Venkatesan R, Weatherall DJ. The spectrum of b-thalassaemia mutations on the Indian subcontinent: the basis for prenatal diagnosis. British journal of haematology. 1991;78:242-7.
Andersson BA, Wering ME, Luo HY, Basran RK, Steinberg MH, Smith HP, et al. Sickle cell disease due to compound heterozygosity for Hb S and a novel 7.7-kb beta-globin gene deletion. European journal of haematology. 2007 Jan;78(1):82-5. PubMed PMID: 17038017.
Gilman JG. The 12.6 kilobase DNA deletion in Dutch b0-thalassaemia. British journal of haematology. 1987;67:369-72.
Schokker RC, Went LN, Bok J. A new genetic variant of ß-thalassaemia. Nature. 1966 Jan 1;209:44-6. PubMed PMID: 5925329.
Craig JE, Kelly SJ, Barnetson R, Thein SL. Molecular characterization of a novel 10.3 kb deletion causing beta-thalassaemia with unusually high Hb A2. British journal of haematology. 1992;82(4):735-44.
Thein SL. Structural Variants with a Beta-Thalassemia Phenotype. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge,UK: Cambridge University Press, Cambridge,UK; 2001. p. 342-55.
Thein SL. Is it dominantly inherited beta thalassaemia or just a beta-chain variant that is highly unstable? British journal of haematology. 1999 Oct;107(1):12-21. PubMed PMID: 10520021.
Kimberland ML, Divoky V, Prchal J, Schwahn U, Berger W, Kazazian HH, Jr. Full-length human L1 insertions retain the capacity for high frequency retrotransposition in cultured cells. Hum Mol Genet. 1999 Aug;8(8):1557-60. PubMed PMID: 10401005.
Viprakasit V, Gibbons RJ, Broughton BC, Tolmie JL, Brown D, Lunt P, et al. Mutations in the general transcription factor TFIIH result in beta-thalassaemia in individuals with trichothiodystrophy. Hum Mol Genet. 2001;10(24):2797-802. PubMed PMID: 11734544.
Yu C, Niakan KK, Matsushita M, Stamatoyannopoulos G, Orkin SH, Raskind WH. X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood. 2002;100(6):2040-5. PubMed PMID: 12200364.
Badens C, Mattei MG, Imbert AM, Lapoumérouliee C, Martini N, Michel G, et al. A novel mechanism for thalassaemia intermedia. The Lancet. 2002;359(9301):132-3. PubMed PMID: 11809258.
Galanello R, Perseu L, Perra C, Maccioni L, Barella S, Longinotti M, et al. Somatic deletion of the normal beta-globin gene leading to thalassaemia intermedia in heterozygous beta-thalassaemic patients. British journal of haematology. 2004 Dec;127(5):604-6. PubMed PMID: 15566365.
Chang JG, Tsai WC, Chong IW, Chang CS, Lin CC, Liu TC. Beta-thalassemia major evolution from beta-thalassemia minor is associated with paternal uniparental isodisomy of chromosome 11p15. Haematologica. 2008 Jun;93(6):913-6. PubMed PMID: 18413893.
Bento C, Maia TM, Milosevic JD, Carreira IM, Kralovics R, Ribeiro ML. beta thalassemia major due to acquired uniparental disomy in a previously healthy adolescent. Haematologica. 2013 Jan;98(1):e4-6. PubMed PMID: 22875618. Pubmed Central PMCID: 3533667. Epub 2012/08/10. eng.
Nagel RL, Steinberg MH. Genetics of the βS Gene: Origins, Genetic Epidemiology, and Epistasis in sickle cell Anemia. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge,UK: Cambridge University Press, Cambridge,UK; 2001. p. 711-55.
Thein SL. Pathophysiology of Beta Thalassemia--A Guide to Molecular Therapies. Hematology (Am Soc Hematol Educ Program). 2005:31-7. PubMed PMID: 16304356.
Thein SL. Genetic association studies in β-hemoglobinopathies. Hematology ASH Education Program book 2013. 2013 Dec 2013;Dec 2013(1):354-61. PubMed PMID: 24319204. Eng.
Raychaudhuri S. Mapping rare and common causal alleles for complex human diseases. Cell. 2011 Sep 30;147(1):57-69. PubMed PMID: 21962507. Pubmed Central PMCID: 3198013.
Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008 Nov 7;322(5903):881-8. PubMed PMID: 18988837.
Thein SL, Craig JE. Genetics of Hb F/F cell variance in adults and heterocellular hereditary persistence of fetal hemoglobin. Hemoglobin. 1998;22:401-14.
Garner C, Tatu T, Reittie JE, Littlewood T, Darley J, Cervino S, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000;95(1):342-6. PubMed PMID: 10607722.
Thein SL, Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. British journal of haematology. 2009 May;145(4):455-67. PubMed PMID: 19344402.
Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009 Oct 15;18(R2):R216-23. PubMed PMID: 19808799.
Sankaran VG, Xu J, Orkin SH. Advances in the understanding of haemoglobin switching. British journal of haematology. 2010 Apr;149(2):181-94. PubMed PMID: 20201948.
Labie D, Pagnier J, Lapoumeroulie C, Rouabhi F, Dunda-Belkhodja O, Chardin P, et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proceedings of the National Academy of Sciences, USA. 1985 Apr;82(7):2111-4. PubMed PMID: 2580306. Pubmed Central PMCID: 397502. Epub 1985/04/01.
Nagel RL, Fabry ME, Pagnier J, Zohoun I, Wajcman H, Baudin V, et al. Hematologically and genetically distinct forms of sickle cell anemia in Africa. The Senegal type and the Benin type. New England Journal of Medicine. 1985 Apr 4;312(14):880-4. PubMed PMID: 2579336.
Nagel RL, Rao SK, Dunda-Belkhodja O, Connolly MM, Fabry ME, Georges A, et al. The hematologic characteristics of sickle cell anemia bearing the Bantu haplotype: the relationship between Gg and Hb F level. Blood. 1987;69:1026-30.
Lettre G, Sankaran VG, Bezerra MA, Araujo AS, Uda M, Sanna S, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America. 2008 Aug 19;105(33):11869-74. PubMed PMID: 18667698.
Galanello R, Sanna S, Perseu L, Sollaino MC, Satta S, Lai ME, et al. Amelioration of Sardinian beta-zero thalassemia by genetic modifiers. Blood. 2009 Oct 29;114(18):3935-7. PubMed PMID: 19696200.
Bhatnagar P, Purvis S, Barron-Casella E, Debaun MR, Casella JF, Arking DE, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. Journal of human genetics. 2011 Apr;56(4):316-23. PubMed PMID: 21326311. Epub 2011/02/18. eng.
Makani J, Menzel S, Nkya S, Cox SE, Drasar E, Soka D, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood. 2011 Jan 27;117(4):1390-2. PubMed PMID: 21068433. Epub 2010/11/12. eng.
Badens C, Joly P, Agouti I, Thuret I, Gonnet K, Fattoum S, et al. Variants in genetic modifiers of beta-thalassemia can help to predict the major or intermedia type of the disease. Haematologica. 2011 Nov;96(11):1712-4. PubMed PMID: 21791466. Pubmed Central PMCID: 3208691. Epub 2011/07/28. eng.
Bae HT, Baldwin CT, Sebastiani P, Telen MJ, Ashley-Koch A, Garrett M, et al. Meta-analysis of 2040 sickle cell anemia patients: BCL11A and HBS1L-MYB are the major modifiers of HbF in African Americans. Blood. 2012 Aug 30;120(9):1961-2. PubMed PMID: 22936743. Pubmed Central PMCID: 3433099. Epub 2012/09/01. eng.
Mtatiro SN, Singh T, Rooks H, Mgaya J, Mariki H, Soka D, et al. Genome wide association study of fetal hemoglobin in sickle cell anemia in Tanzania. PloS one. 2014;9(11):e111464. PubMed PMID: 25372704. Pubmed Central PMCID: 4221031.
Viprakasit V, Ekwattanakit S, Riolueang S, Chalaow N, Fisher C, Lower K, et al. Mutations in Kruppel-like factor 1 cause transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Blood. 2014 Jan 17. PubMed PMID: 24443441. Epub 2014/01/21. Eng.
Liu D, Zhang X, Yu L, Cai R, Ma X, Zheng C, et al. KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of beta-thalassemia. Blood. 2014 Jul 31;124(5):803-11. PubMed PMID: 24829204. Pubmed Central PMCID: 4118488.
Esteghamat F, Gillemans N, Bilic I, van den Akker E, Cantu I, van Gent T, et al. Erythropoiesis and globin switching in compound Klf1::Bcl11a mutant mice. Blood. 2013 Mar 28;121(13):2553-62. PubMed PMID: 23361909. Epub 2013/01/31. eng.
Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013 Oct 11;342(6155):253-7. PubMed PMID: 24115442. Epub 2013/10/12. eng.
Jawaid K, Wahlberg K, Thein SL, Best S. Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood cells, molecules & diseases. 2010 Aug 15;45(2):140-6. PubMed PMID: 20542454.
Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, Kim W, et al. Transcriptional silencing of {gamma}-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes & development. 2010 Apr 15;24(8):783-98. PubMed PMID: 20395365.
Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, et al. Corepressor-dependent silencing of fetal hemoglobin expression by BCL11A. Proceedings of the National Academy of Sciences of the United States of America. 2013 Apr 16;110(16):6518-23. PubMed PMID: 23576758. Pubmed Central PMCID: 3631619. Epub 2013/04/12. eng.
Thein SL, Menzel S, Peng X, Best S, Jiang J, Close J, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proceedings of the National Academy of Sciences of the United States of America. 2007 Jul 3;104(27):11346-51. PubMed PMID: 17592125.
Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, van Ijcken W, et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. The Journal of clinical investigation. 2014 Apr;124(4):1699-710. PubMed PMID: 24614105. Pubmed Central PMCID: 3973089.
Menzel S, Rooks H, Zelenika D, Mtatiro SN, Gnanakulasekaran A, Drasar E, et al. Global Genetic Architecture of an Erythroid Quantitative Trait Locus, HMIP-2. Annals of human genetics. 2014 Jul 29. PubMed PMID: 25069958. Pubmed Central PMCID: 4303951.
Stadhouders R, Thongjuea S, Andrieu-Soler C, Palstra RJ, Bryne JC, van den Heuvel A, et al. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. The EMBO journal. 2012 Feb 15;31(4):986-99. PubMed PMID: 22157820. Pubmed Central PMCID: 3280550. Epub 2011/12/14. eng.
Suzuki M, Yamazaki H, Mukai HY, Motohashi H, Shi L, Tanabe O, et al. Disruption of the Hbs1l-Myb locus causes hereditary persistence of fetal hemoglobin in a mouse model. Molecular and cellular biology. 2013 Apr;33(8):1687-95. PubMed PMID: 23428869. Pubmed Central PMCID: 3624254. Epub 2013/02/23. eng.
Farrell JJ, Sherva RM, Chen ZY, Luo HY, Chu BF, Ha SY, et al. A 3-bp deletion in the HBS1L-MYB intergenic region on chromosome 6q23 is associated with HbF expression. Blood. 2011 May 5;117(18):4935-45. PubMed PMID: 21385855. Pubmed Central PMCID: 3100700. Epub 2011/03/10. eng.
Bianchi E, Zini R, Salati S, Tenedini E, Norfo R, Tagliafico E, et al. c-Myb supports erythropoiesis through the transactivation of KLF1 and LMO2 expression. Blood. 2010 Nov 25;116(22):e99-110. PubMed PMID: 20686118. Epub 2010/08/06. Eng.
Tallack MR, Perkins AC. Three fingers on the switch: Kruppel-like factor 1 regulation of gamma-globin to beta-globin gene switching. Current opinion in hematology. 2013 May;20(3):193-200. PubMed PMID: 23474875. Epub 2013/03/12. eng.
Menzel S, Jiang J, Silver N, Gallagher J, Cunningham J, Surdulescu G, et al. The HBS1L-MYB intergenic region on chromosome 6q23.3 influences erythrocyte, platelet, and monocyte counts in humans. Blood. 2007 Nov 15;110(10):3624-6. PubMed PMID: 17712044.
Soranzo N, Spector TD, Mangino M, Kuhnel B, Rendon A, Teumer A, et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nature genetics. 2009 Nov;41(11):1182-90. PubMed PMID: 19820697.
van der Harst P, Zhang W, Mateo Leach I, Rendon A, Verweij N, Sehmi J, et al. Seventy-five genetic loci influencing the human red blood cell. Nature. 2012 Dec 20;492(7429):369-75. PubMed PMID: 23222517. Epub 2012/12/12. eng.
Menzel S, Garner C, Rooks H, Spector TD, Thein SL. HbA2 levels in normal adults are influenced by two distinct genetic mechanisms. British journal of haematology. 2013 Jan;160(1):101-5. PubMed PMID: 23043469. Epub 2012/10/10. eng.
Welch JJ, Watts JA, Vakoc CR, Yao Y, Wang H, Hardison RC, et al. Global regulation of erythroid gene expression by transcription factor GATA-1. Blood. 2004 Nov 15;104(10):3136-47. PubMed PMID: 15297311.
Sankaran VG, Menne TF, Scepanovic D, Vergilio JA, Ji P, Kim J, et al. MicroRNA-15a and -16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proceedings of the National Academy of Sciences of the United States of America. 2011 Jan 25;108(4):1519-24. PubMed PMID: 21205891. Pubmed Central PMCID: 3029749. Epub 2011/01/06. eng.
Huehns ER, Hecht F, Keil JV, Motulsky AG. Developmental hemoglobin anomalies in a chromosomal triplication: D1 trisomy syndrome. Proc Natl Acad Sci USA. 1964 Jan;51:89-97. PubMed PMID: 14104609. Pubmed Central PMCID: 300609. Epub 1964/01/01. eng.
Sankaran VG, Joshi M, Agrawal A, Schmitz-Abe K, Towne MC, Marinakis N, et al. Rare complete loss of function provides insight into a pleiotropic genome-wide association study locus. Blood. 2013 Nov 28;122(23):3845-7. PubMed PMID: 24288412. Epub 2013/11/30. eng.
Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nature genetics. 2010 Nov 7;40(12):1049-51. PubMed PMID: 21057501. Epub 2010/11/09. Eng.
Suzuki M, Yamamoto M, Engel JD. Fetal globin gene repressors as drug targets for molecular therapies to treat the beta-globinopathies. Molecular and cellular biology. 2014 Oct 1;34(19):3560-9. PubMed PMID: 25022757. Pubmed Central PMCID: 4187729.
Amin BR, Bauersachs RM, Meiselman HJ, Mohandas N, Hebbel RP, Bowen PE, et al. Monozygotic twins with sickle cell anemia and discordant clinical courses: clinical and laboratory studies. Hemoglobin. 1991;15(4):247-56. PubMed PMID: 1723971.
Joishy SK, Griner PF, Rowley PT. Sickle b-thalassemia: identical twins differing in severity implicate nongenetic factors influencing course. American journal of hematology. 1976;1(1):23-33. PubMed PMID: 988745.
Weatherall MW, Higgs DR, Weiss H, Weatherall DJ, Serjeant GR. Phenotype/genotype relationships in sickle cell disease: a pilot twin study. Clin Lab Haematol. 2005 Dec;27(6):384-90. PubMed PMID: 16307540.
Noguchi CT, Torchia DA, Schechter AN. Intracellular polymerization of sickle hemoglobin. Effects of cell heterogeneity. The Journal of clinical investigation. 1983 Sep;72(3):846-52. PubMed PMID: 6886006. Pubmed Central PMCID: 1129249.
Serjeant GR, Serjeant BE. Sickle Cell Disease. Third ed. Oxford, UK: Oxford University Press; 2001. 772 p.
Bonham VL, Dover GJ, Brody LC. Screening student athletes for sickle cell trait--a social and clinical experiment. The New England journal of medicine. 2010 Sep 9;363(11):997-9. PubMed PMID: 20825310. Epub 2010/09/10. eng.
Noguchi CT, Rodgers GP, Serjeant G, Schechter AN. Levels of fetal hemoglobin necessary for treatment of sickle cell disease. New England Journal of Medicine. 1988;318(2):96-9. PubMed PMID: 2447498.
Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease: Life expectancy and risk factors for early death. New England Journal of Medicine. 1994;330(23):1639-44.
Wang WC, Pavlakis SG, Helton KJ, McKinstry RC, Casella JF, Adams RJ, et al. MRI abnormalities of the brain in one-year-old children with sickle cell anemia. Pediatric blood & cancer. 2008 Nov;51(5):643-6. PubMed PMID: 18478575.
Thein SL. Genetic modifiers of sickle cell disease. Hemoglobin. 2011;35(5-6):589-606. PubMed PMID: 21967611. Epub 2011/10/05. eng.
Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. American journal of hematology. 2012 Aug;87(8):795-803. PubMed PMID: 22641398. Epub 2012/05/30. eng.
Wonkam A, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Cooper RS, Tayo B, et al. Association of variants at BCL11A and HBS1L-MYB with hemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PloS one. 2014;9(3):e92506. PubMed PMID: 24667352. Pubmed Central PMCID: 3965431.
Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Second ed. Cambridge,UK: Cambridge University Press, Cambridge,UK; 2009.
Vasavda N, Menzel S, Kondaveeti S, Maytham E, Awogbade M, Bannister S, et al. The linear effects of alpha-thalassaemia, the UGT1A1 and HMOX1 polymorphisms on cholelithiasis in sickle cell disease. British journal of haematology. 2007 Jul;138(2):263-70. PubMed PMID: 17593033.
Rumaney MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Kengne AP, Ngogang J, et al. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PloS one. 2014;9(6):e100516. PubMed PMID: 24978191. Pubmed Central PMCID: 4076272.
Serjeant GR, Higgs DR, Hambleton IR. Elderly survivors with homozygous sickle cell disease. The New England journal of medicine. 2007 Feb 8;356(6):642-3. PubMed PMID: 17287491.
Vasavda N, Badiger S, Rees D, Height S, Howard J, Thein SL. The presence of alpha-thalassaemia trait blunts the response to hydroxycarbamide in patients with sickle cell disease. British journal of haematology. 2008 Nov;143(4):589-92. PubMed PMID: 18764867.
Castro OL, Gordeuk VR, Gladwin MT, Steinberg MH. Senicapoc trial results support the existence of different sub-phenotypes of sickle cell disease with possible drug-induced phenotypic shifts. British journal of haematology. 2011 Dec;155(5):636-8. PubMed PMID: 21732927. Epub 2011/07/08. eng.
Fertrin KY, Costa FF. Genomic polymorphisms in sickle cell disease: implications for clinical diversity and treatment. Expert Rev Hematol. 2010 Aug;3(4):443-58. PubMed PMID: 21083035. Epub 2010/11/19. eng.
Milton JN, Sebastiani P, Solovieff N, Hartley SW, Bhatnagar P, Arking DE, et al. A genome-wide association study of total bilirubin and cholelithiasis risk in sickle cell anemia. PloS one. 2012;7(4):e34741. PubMed PMID: 22558097. Pubmed Central PMCID: 3338756. Epub 2012/05/05. eng.
Heeney MM, Howard TA, Zimmerman SA, Ware RE. UGT1A promoter polymorphisms influence bilirubin response to hydroxyurea therapy in sickle cell anemia. J Lab Clin Med. 2003 Apr;141(4):279-82. PubMed PMID: 12677174.
Flanagan JM, Sheehan V, Linder H, Howard TA, Wang YD, Hoppe CC, et al. Genetic mapping and exome sequencing identify 2 mutations associated with stroke protection in pediatric patients with sickle cell anemia. Blood. 2013 Apr 18;121(16):3237-45. PubMed PMID: 23422753. Epub 2013/02/21. eng.
Flanagan JM, Frohlich DM, Howard TA, Schultz WH, Driscoll C, Nagasubramanian R, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011 Jun 16;117(24):6681-4. PubMed PMID: 21515823. Pubmed Central PMCID: 3123027. Epub 2011/04/26. eng.
Castro V, Alberto FL, Costa RN, Lepikson-Neto J, Gualandro SF, Figueiredo MS, et al. Polymorphism of the human platelet antigen-5 system is a risk factor for occlusive vascular complications in patients with sickle cell anemia. Vox sanguinis. 2004 Aug;87(2):118-23. PubMed PMID: 15355504.
Sebastiani P, Solovieff N, Hartley SW, Milton JN, Riva A, Dworkis DA, et al. Genetic modifiers of the severity of sickle cell anemia identified through a genome-wide association study. American journal of hematology. 2010 Jan;85(1):29-35. PubMed PMID: 20029952. Epub 23 Oct 2009.
Milton JN, Rooks H, Drasar E, McCabe EL, Baldwin CT, Melista E, et al. Genetic determinants of haemolysis in sickle cell anaemia. British journal of haematology. 2013 Apr;161(2):270-8. PubMed PMID: 23406172. Epub 2013/02/15. eng.
Ware RE. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood. 2010 Jul 1;115(26):5300-11. PubMed PMID: 20223921. Pubmed Central PMCID: 2902131. Epub 2010/03/13. eng.
Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA : the journal of the American Medical Association. 2014 Sep 10;312(10):1033-48. PubMed PMID: 25203083.
Ware RE, Eggleston B, Redding-Lallinger R, Wang WC, Smith-Whitley K, Daeschner C, et al. Predictors of fetal hemoglobin response in children with sickle cell anemia receiving hydroxyurea therapy. Blood. 2002;99(1):10-4. PubMed PMID: 11756146.
Ho PJ, Hall GW, Luo LY, Weatherall DJ, Thein SL. Beta thalassemia intermedia: is it possible to consistently predict phenotype from genotype? British journal of haematology. 1998;100(1):70-8.
So CC, Song YQ, Tsang ST, Tang LF, Chan AY, Ma ES, et al. The HBS1L-MYB intergenic region on chromosome 6q23 is a quantitative trait locus controlling fetal haemoglobin level in carriers of beta-thalassaemia. J Med Genet. 2008 Nov;45(11):745-51. PubMed PMID: 18697826.
Nuinoon M, Makarasara W, Mushiroda T, Setianingsih I, Wahidiyat PA, Sripichai O, et al. A genome-wide association identified the common genetic variants influence disease severity in β0-thalassemia/hemoglobin E. Hum Genet. 2010 March;127(3):303-14. PubMed PMID: 19924444.
Sedgewick AE, Timofeev N, Sebastiani P, So JC, Ma ES, Chan LC, et al. BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood cells, molecules & diseases. 2008 Nov-Dec;41(3):255-8. PubMed PMID: 18691915.
Danjou F, Anni F, Perseu L, Satta S, Dessi C, Lai ME, et al. Genetic modifiers of beta-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica. 2012 Jul;97(7):989-93. PubMed PMID: 22271886. Pubmed Central PMCID: 3396667.
Danjou F, Francavilla M, Anni F, Satta S, Demartis FR, Perseu L, et al. A genetic score for the prediction of beta-thalassemia severity. Haematologica. 2014 Dec 5. PubMed PMID: 25480500.
Harteveld CL, Higgs DR. α-thalassaemia. Orphanet Journal of Rare Diseases. 2010 May 2010;5(Item 13):21.
Weiss MJ, dos Santos CO. Chaperoning erythropoiesis. Blood. 2009 Mar 5;113(10):2136-44. PubMed PMID: 19109556.
Danjou F, Anni F, Galanello R. Beta-thalassemia: from genotype to phenotype. Haematologica. 2011 Nov;96(11):1573-5. PubMed PMID: 22058279. Pubmed Central PMCID: 3208672. Epub 2011/11/08. eng.
Thein SL. Genetic modifiers of the beta-haemoglobinopathies. British journal of haematology. 2008 May;141(3):357-66. PubMed PMID: 18410570.
Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004 Apr 30;117(3):285-97. PubMed PMID: 15109490.
Nagel RL, Fabry ME, Steinberg MH. The paradox of hemoglobin SC disease. Blood reviews. 2003 Sep;17(3):167-78. PubMed PMID: 12818227.
Masiello D, Heeney MM, Adewoye AH, Eung SH, Luo HY, Steinberg MH, et al. Hemoglobin SE disease: a concise review. American journal of hematology. 2007 Jul;82(7):643-9. PubMed PMID: 17278112.
Moo-Penn W, Bechtel K, Jue D, Chan MS, Hopkins G, Schneider NJ, et al. The presence of hemoglobin S and C Harlem in an individual in the United States. Blood. 1975 Sep;46(3):363-7. PubMed PMID: 1148394.
Monplaisir N, Merault G, Poyart C, Rhoda MD, Craescu C, Vidaud M, et al. Hemoglobin S antilles: A variant with lower solubility than hemoglobin S and producing sickle cell disease in heterozygotes. Proceedings of the National Academy of Sciences of the United States of America. 1986 Dec;83(24):9363-7. PubMed PMID: 3467311.
Witkowska HE, Lubin BH, Beuzard Y, Baruchel S, Esseltine DW, Vichinsky EP, et al. Sickle cell disease in a patient with sickle cell trait and compound heterozygosity for hemoglobin S and hemoglobin Quebec-Chori. The New England journal of medicine. 1991 Oct 17;325(16):1150-4. PubMed PMID: 1891024.
Steinberg MH. Other sickle hemoglobinopathies. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Second ed. Cambridge,UK: Cambridge University Press, Cambridge,UK; 2009. p. 564-86.
Geva A, Clark JJ, Zhang Y, Popowicz A, Manning JM, Neufeld EJ. Hemoglobin Jamaica plain--a sickling hemoglobin with reduced oxygen affinity. The New England journal of medicine. 2004 Oct 7;351(15):1532-8. PubMed PMID: 15470216.
Nagel RL, Daar S, Romero JR, Suzuka SM, Gravell D, Bouhassira E, et al. HbS-Oman heterozygote: A new dominant sickle syndrome. Blood. 1998;92:4375-82.
Swensen JJ, Agarwal AM, Esquilin JM, Swierczek S, Perumbeti A, Hussey D, et al. Sickle cell disease due to uniparental disomy in a child who inherited sickle cell trait. Blood. 2010 Oct 14;116(15):2822-5. Epub July 1, 2010.
Koenig SC, Becirevic E, Hellberg MS, Li MY, So JC, Hankins JS, et al. Sickle cell disease caused by heterozygosity for Hb S and novel LCR deletion: Report of two patients. American journal of hematology. 2009 Sep;84(9):603-6. PubMed PMID: 19650141.
Acknowledgement
I thank Claire Steward for help with preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media LLC
About this chapter

Cite this chapter
Thein, S.L. (2017). Genetic Basis and Genetic Modifiers of β-Thalassemia and Sickle Cell Disease. In: Malik, P., Tisdale, J. (eds) Gene and Cell Therapies for Beta-Globinopathies. Advances in Experimental Medicine and Biology(), vol 1013. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7299-9_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7299-9_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-7297-5
Online ISBN: 978-1-4939-7299-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)