
Abstract
The recent revolution in cancer genetics offers the promise of using genetic information to individualize patient treatment. In pancreatic cancer, numerous studies have described a genetic landscape characterized by a set of commonly mutated genes aggregated into core molecular pathways accompanied by numerous but infrequently mutated genes. Studies have also demonstrated significant intratumoral heterogeneity. Resistance against chemotherapeutic agents has also been attributed to difficulty of drug delivery through a rich stromal microenvironment. For these reasons, therapeutic development against pancreatic cancer has been challenging, and a number of promising agents have failed clinical trial testing. Personalized models have been studied as a tool for testing candidate drugs to select the most efficacious treatment. The patient-derived xenograft (PDX) is a well-established preclinical tool to improve the drug screening and development. The PDX model requires adequate tissue for transplantation, and failure is common. A recently described, innovative three-dimensional organoid culture platform can be exploited for genomic and functional studies at the level of the individual patient for personalized treatment approach. Organoid technology may fill the gap between cancer genetics and patient trials and allow personalized therapy design. Combination of genome-based medicine and individualized model-based drug screening may fulfill the promise of precision medicine for pancreatic cancer.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Precision medicine
- Three-dimensional organoid culture
- Patient-derived xenograft (PDX)
- Genomic-based medicine
Introduction
The field of oncology is rapidly evolving from treating large, unselected populations to targeting small numbers of patients using deep evaluation of molecular features and selection of the most appropriate treatment. President Obama announced the launching of a Precision Medicine Initiative in his 2015 State of the Union Address, and he requested 215 million dollars to fund this endeavor in the fiscal year 2016. The time is right to pursue this strategy, using the individual patient’s genetic information to guide individualized therapy. The significant revolution in cancer genetics is allowing, for the first time, the gathering of enormous amounts of genomic information, including the assessment of complete cancer genomes, to aid in clinical decision-making. From this approach, numerous potential targets have emerged for individual patients that may potentially be linked to clinical response.
Genomic-based treatment has already provided examples of remarkable success stories. The development of Imatinib to treat CML and GIST, BRAF inhibitors to treat melanoma, HER2 antibodies to treat HER2 positive breast cancer, and EGFR inhibitors and ALK inhibitors to treat nonsmall cell lung cancer are just some examples that have dramatically changed the treatment paradigms and improved the survival of patients.
Targeted therapy development continues to evolve rapidly, and this approach has intuitively expanded to precision medicine. NCI-Molecular Analysis for Therapy Choice (NCI-MATCH) (ClinicalTrials.gov number, NCT02465060) is a clinical trial to treat cancer patients according to their molecular abnormalities using DNA sequencing from biopsy specimens. The drugs included in this trial are approved by US Food and Drug Administration (FDA) for another cancer indication or are being tested in clinical trials and have shown a promising result against solid tumors such as breast, colon, lung, prostate, or lymphoma with specific mutations. The AURORA clinical trial is expected to establish detailed molecular profiling of metastatic breast cancer for deeper understanding of the molecular biology, promising to lead to personalized cancer medicine (ClinicalTrials.gov number, NCT02102165).
Pancreatic ductal adenocarcinoma (PDA) remains one of the most deadly cancers worldwide, with 5-year survival below 7%. Surgical resection, the only potentially curative treatment for PDA, is performed in only 15 to 20% of PDA patients, as most cases are diagnosed at a late stage when surgery is not possible. Recent advances in chemotherapy, such as development of the FOLFIRINOX (5-fluorouracil, leucovorin, irinotecan, and oxaliplatin) regimen, and gemcitabine and nanoalbumin-bound paclitaxel, have extended the survival of PDA patients. Although other types of cancer patients are treated based on specific markers, there are no effective markers for targeted therapy in PDA.
The epidermal growth factor receptor (EGFR) inhibitor, erlotinib, the only FDA approved targeted agent for treating PDA, only marginally extends overall survival in combination with gemcitabine. Poly-ADP-ribose polymerase (PARP) inhibitors have shown promising preliminary results [1]. This agent was first reported for BRCA1/2 positive breast cancer and ovarian cancer.
Recent expression analysis has identified four molecular subtypes of PDA [2]. This and other integrated molecular analyses are expected to give insights with therapeutic relevance. One hypothesizes that treatments could be individualized based on a patient’s molecular subtype. For example, immune modulators could be tested in patients with an immunogenic subtype. In terms of precision treatment, categorizing some specific patients according to active, available drugs is a logical way forward. Recent clinical trials have shown the efficacy of PARP inhibitor for patients with BRCA1/2 or PALB2 mutations [3, 4]. The frequency of BRCA1/2 deficiency is 5–8% in the general population and 12–15% in certain groups such as Ashkenazi Jewish patients with a family history of breast cancer. Patients with BRCA deficiency driven tumors have increased sensitivity to platinum agents. In addition to platinum agents, BRCA deficient cancers have shown high sensitivity to PARP inhibitors. Recent sequencing data suggest that mutations in BRCA pathway component genes and surrogate measures of defects in DNA maintenance (genomic instability and the BRCA mutational signature) have potential implications for therapeutic selection for PDA in the absence of BRCA or PALB2 mutations [5].
Personalized medicine for PDA patients will be based on an enhanced understanding of biological features of PDA, advancement of technology, and treatment development. Advances in technology currently allows for faster and less expensive whole-genome, exome, and transcriptome analyses compared with traditional Sanger-based methods, enabling routine and rapid characterization of genetic and pathway alterations. Some trials are already underway to test this concept. In the IMPaCT (The Individualized Molecular Pancreatic Cancer Therapy) trial [6], HER2 amplification, KRAS wild-type, and mutations in DNA damage repair pathways (BRCA1, BRCA2, PALB2, ATM) are assessed for guiding treatment. Another approach utilizes the patient derived xenograft (PDX) mouse model, a so-called avatar model. The PDX represents a valuable preclinical tool for studying human cancer biology and patient response to treatments, which suggest the potential for precision medicine. Due to the short survival seen in PDA, participants of clinical trials are often unable to be treated according to their molecular analysis due to their worsening conditions or progression of their disease. For precision medicine to be effective in PDA, developing rapid analyses is a prerequisite.
Genetic Screening and Genomic-Based Treatment
Based on rigorous molecular pathology studies and genomic analyses, the generally accepted model of carcinogenesis describes a stepwise progression from normal pancreatic epithelia to pancreatic intraepithelial neoplasia (PanIN) and finally to frank adenocarcinoma with accumulation of accompanying signature mutations. Recent genomic analyses of PDA have revealed a complex mutational landscape [2, 5, 7]. More than 90% of PDA carry activating KRAS mutations. Mutations in KRAS are seen in all stages of PanIN. Inactivation of tumor suppressor genes such as TP53, Smad4, and p16 are seen with progressive PanIN development and occur at rates of more than 50%. The prevalence of recurrently mutated genes then drops to ~10% which aggregate into core molecular pathways including KRAS, WNT, NOTCH, DNA damage repair, RNA processing, cell cycle regulation, TGF-β signaling, SWI-SNF, chromatin regulation, and axonal guidance. For a number of reasons, including inter- and intra tumor heterogeneity, and an inability to target commonly mutated genes, development of targeted and effective therapeutics remains challenging.
Jones et al. [8] reported a core set of 12 cellular signaling pathways altered in PDA, including apoptosis (100%), DNA damage control (83%), regulation of G1/S phase transition (100%), hedgehog signaling (100%), homophilic cell adhesion (79%), integrin signaling (67%), c-Jun N-terminal kinase signaling (96%), KRAS signaling (100%), regulation of invasion (92%), small GTPase–dependent signaling (other than KRAS) (79%), TGF-β signaling (100%), and Wnt/Notch signaling (100%). Jones and colleagues determined the sequences of 23,219 transcripts, representing 20,661 protein-coding genes and found that PDA contains an average of 63 genetic alterations, the majority of which are point mutations. They collected 24 PDA DNA samples from 10 PDXs and 14 cell lines from 17 patients with surgically resected and 7 patients who underwent a rapid autopsy. Normal tissues were obtained from tumor-negative duodenum, liver, or spleen. These 12 pathways are genetically altered in the great majority of pancreatic cancers. However, the pathway components that are altered in any individual tumor vary widely and the specific genes altered in each tumor are largely different. In addition, it is difficult to determine whether each identified mutation plays a functional role in the pathway or process identified.
Biankin et al. [7] performed exome sequencing and copy number analysis of early (stage I and II) PDA. Biankin and colleagues identified substantial heterogeneity with 2016 nonsilent mutations and 1628 copy-number variations from the analysis of informative 99 tumor samples. They defined 16 significantly mutated genes, reaffirming known mutations (KRAS, TP53, CDKN2A, SMAD4, MLL3, TGFBR2, ARID1A, and SF3B1), and uncovered novel mutated genes including additional genes involved in chromatin modification (EPC1 and ARID2), DNA damage repair (ATM), and other mechanisms (ZIM2, MAP 2 K4, NALCN, SLC16A4, and MAGEA6). Pathway-based analysis of recurrently mutated genes identified mechanisms known to be important in cancer: G1/S checkpoint machinery, apoptosis, regulation of angiogenesis, and TGF-β signaling. They identified frequent and diverse somatic aberrations in genes described traditionally as embryonic regulators of axon guidance, particularly SLIT/ROBO signaling which suggested the potential involvement of axon guidance genes in pancreatic carcinogenesis.
Bailey et al. [2] reported that mutated genes aggregated into 10 molecular mechanisms, including activating mutations of KRAS in 92%; disruption of G1/S checkpoint machinery (TP53, CDKN2A, and TP53BP2) in 78%; TGF-β signaling (SMAD4, SMAD3, TGFBR1, TFGBR2, ACVR1B, and ACVR2A) in 47%; histone modification (KDM6A, SETD2, and ASCOM complex members MLL2 and MLL3) in 24%; the SWI/SNF complex (ARID1A, PBRM1, and SMARCA4) in 14%; the BRCA pathway (BRCA1, BRCA2, ATM, and PALB2: 5% germline, 12% somatic); WNT signaling defects through RNF43 mutation (5%); and RNA processing genes, SF3B1, U2AF1, and RBM10 (16%).
Genomic instability is a characteristic feature of almost all human cancers. Germline mutations in DNA mismatch repair (MMR) genes have been reported in hereditary cancers. With regard to the molecular basis of genomic instability in sporadic cancers, recent genome-wide studies by the use of Sanger sequencing reported that mutations in DNA repair genes and mitotic checkpoint genes were infrequent. Wang et al. sequenced the exomes of 15 human PDA-derived cell lines and their matched normal samples and identified a total of 1517 somatic mutations. Among them, 56 genes were recurrently mutated in two or more cell lines and showed dramatically increased rate of both indels and substitutions involved in all nine core signaling pathways. They revealed that MLH1 expression levels appear to be correlated with the mutation rates. Among the MMR proteins, the loss of MLH1 is the most common cause of MSI [9].
Epigenome
While a significant effort has been made to understand the somatic genetic alterations acquired in PDA, research into epigenetic mechanisms has expanded our understanding of altered gene expression in PDA. Research has focused on several well-characterized epigenetic mechanisms, including DNA methylation, histone modification, and microRNAs. It is increasingly understood that multiple epigenetic mechanisms are indeed crucial in the development and progression of PDA. In addition to genetic changes, epigenetic alterations add another layer of complexity and contribute to the heterogeneity of PDA.
Studies on chromatin dynamics alone are unveiling the existence of robust machineries that can mediate epigenetic changes in pancreatic cells. These findings highlight the need to further our insight into how epigenetic mechanisms are able to independently and cooperatively influence gene regulation and thereby PDA development.
Furthermore, it is important to emphasize one of the characteristics of epigenetic mechanisms of gene regulation – their reversibility. This feature provides a unique target for the introduction of specific therapeutic interventions for PDA.
Nones et al. reported a large-scale methylation and expression profiling study of 167 PDA compared with 29 adjacent nonmalignant pancreas. A total of 11,634 CpG sites associated with 3522 genes and pathway analysis revealed an enrichment of aberrantly methylated genes involved in core signaling pathways including TGF-β, WNT, integrin signaling, cell adhesion, stellate cell activation, and axon guidance. Notably, they revealed epigenetic suppression of SLIT-ROBO signaling and upregulation of MET and ITGA2 expression, which is correlated with poor outcome. Biankin et al. identified genomic aberration of ROBO1 in 11% and SLIT in 10% of PDA samples. Nones et al. suggested that hypermethylation of SLIT-ROBO is a more widespread mechanism of inactivation of this pathway. From the 58 tumors 48% showed hypermethylation of all four genes (ROBO1, ROBO3, SKIT2, and SLIT3). Tumor suppressor genes with a low incidence of mutations may be inactivated by epigenetic mechanisms more frequently. DNA methylation cooperating with other genetic mechanisms alter key signaling pathways critical to cancer development [10].
Chromatin regulators such as HDACs and BET proteins are currently being analyzed as potential strategies for PDAC patients [11, 12].
Transcriptomic PDA Subtypes
Treatment outcomes are improved by targeting drugs according to tumor subtypes in other cancers. Identification of therapeutic molecular subtypes in PDA has been challenging. Collisson et al., for the first time, demonstrated three gene expression subtypes using a 62-gene signature (PDAssigner; [13]) applied to laser capture–microdissected epithelial PDA tumors. They designated these subtypes as classical, quasimesenchymal (QM), and exocrine-like. Classical PDA [14] is characterized by high adhesion-associated ribosomal and epithelial gene expression, and elevated GATA6 expression, which is essential for pancreatic development [13]. QM-PDA showed high expression of mesenchymal-associated genes. Exocrine-like PDA shows high expression of tumor cell–derived digestive genes. However, in 19 human and 15 mouse PDA cell lines, only the classical and the QM-PDA subtypes were identified, suggesting that currently used PDA cell lines inadequately represent the heterogeneity of human PDA. They showed that classical PDA lines are relatively more dependent on Kras and more sensitive to erlotinib than QM-PDA lines. Conversely, QM-PDA lines are more sensitive to gemcitabine than classical PDA. However, the drug sensitivity of the exocrine-like subtype has yet to be determined.
The presence of the exocrine-like subtype was validated by Noll and colleagues [15], by deriving matched exocrine-like PDA patient-derived xenograft tumors and cell lines. In addition, they showed that the exocrine-like PDA subtype is resistant to small-molecule drugs dasatinib, erlotinib, and paclitaxel and that this resistance is mediated by a cell-autonomous CYP (cytochrome P450) 3A5-dependent drug detoxification mechanism. CYP3A5 also contributes to acquired drug resistance in other subtypes of PDA and in other malignancies.
They identified the subtype by two surrogate markers, HNF1A for exocrine-like PDA and KRT81 for QM-PDA. Classical PDA was defined as double negative of these markers. HNF1A+ cases are more differentiated whereas KRT81+ cases are less differentiated. Exocrine-like PDAs were found to have the best survival rates.
Moffitt et al. [14] identified two tumor subtypes as classical and basal-like and two stromal subtypes as normal and activated by digitally separating tumor, stromal and normal gene expression. The Collisson classical and QM subtypes appeared to be a mixed collection of genes from the Moffitt basal-like and stromal subtypes. Although the basal-like tumor subtype, which is molecularly similar to basal tumors in bladder and breast cancers, demonstrated worse outcomes, basal-like tumors showed better response to adjuvant therapy. The activated stromal subtype showed worse prognosis than normal stromal subtype. The KRAS mutation encoding G12D was associated with basal-like subtype, and the KRAS-G12 V allele was higher in African Americans. In addition, Collisson and colleagues demonstrated high inter-patient tumor heterogeneity and low heterogeneity between primary and metastatic sites.
Bailey et al. [2] demonstrated four subtypes of PDA using RNA-sequencing data from 96 bulk tumors with high epithelial content. They named these subtypes squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine (ADEX). These four subtypes were associated with specific histological characteristics. Squamous showed adenosquamous carcinoma, pancreatic progenitor and immunogenic showed mucinous noncystic (colloid) adenocarcinoma and carcinoma arising from IPMN, and ADEX showed acinar cell carcinoma. Three of four subtypes overlap with the Collisson subtypes with the exception of immunogenic subtype. The Collisson QM, classical, and exocrine-like subtypes correspond to the Bailey squamous, pancreatic progenitor, and ADEX subtypes, respectively. The immunogenic class shares many of the characteristics of the pancreatic progenitor class but is uniquely associated with a significant immune cells infiltration.
Proteomics
Proteomics research offers the promise of discovering biomarkers for improvement of early diagnosis and prediction of response to therapy. Several candidate protein biomarkers have been investigated to date. Unfortunately, many of these biomarkers are not specific for PDA or in situ lesions, as they are detected in patients with pancreatitis and other conditions such as smokers. Examples include carbohydrate antigen (CA) 19–9 [16], carcinoembryonic antigen (CEA) and peanutagglutinin (PNA)-binding glycoproteins [17], human telomerase reverse transcriptase (hTert) [18], and matrix metalloproteinase-2 (MMP-2) [19]. More recent attempts to leverage circulating tumor cells and circulating free DNA have yielded similar results [20].
Different sources of pancreatic biomarkers have been evaluated, including blood serum and plasma, duodenal and pancreatic juice, and PDA tissue [21]. Various protein expression detection techniques have been developed, of which the mass spectrometry–based approach is perhaps the most promising. Comprehensive studies to catalog PDA specific proteins have been performed previously, including those by our group [21, 22, 23]. The clinical applicability of these studies was limited by the low concentrations of PDA specific proteins in peripheral blood. Current work is focused on developing and applying novel labeling techniques to improve sensitivity, multiplexing, and quantitative accuracy [24, 25].
A recent study reported proteomic and phosphoproteomic analysis of PDA tissue samples and normal tissue via a LC-MS/MS workflow. The investigators identified new candidate markers such as HIPK1 and MLCK from 2101 proteins identified [26]. They also demonstrated proteins involved in cell migration (Rho guanine nucleotide exchange factors and MRCKa) and formation of focal adhesion by phosphoproteomic analysis. They ascertained phosphorylation sites of known drug targets and suggested Fyn, ERK2, AKT1, and HDAC are potential targets for PDA treatment.
Humphrey et al. reported phosphotyrosine profiling of ATCC PDA cell lines and PDX cell lines they established by immunoaffinity-coupled high-resolution mass spectrometry [27]. They revealed three subtypes of ATCC cell lines, which are associated with cell-cell adhesion and epithelial-mesenchymal transition, mRNA metabolism, and receptor tyrosine kinase (RTK) signaling, respectively. One subtype of PDX cell lines is associated with RTK signaling and showed sensitivity to EGFR inhibitor, erlotinib. These results suggest that a phosphosignature may provide a predictive biomarker for response to targeted therapies.
Metabolomics
Targeting cancer metabolism requires personalized diagnostics for clinical success. Daemena et al. [28] identified three highly distinct metabolic subtypes through broad metabolite profiling of 38 PDA cell lines. One subtype was defined by reduced proliferative capacity, whereas the other two subtypes (glycolytic and lipogenic) showed distinct metabolite levels associated with glycolysis, lipogenesis, and redox pathways, which were confirmed transcriptionally. The glycolytic and lipogenic subtypes showed striking differences in use of glucose and glutamine and showed differential sensitivity to inhibitors of aerobic glycolysis, glutaminolysis, lipid synthesis, and redox balance. In PDA clinical samples, the lipogenic subtype is associated with the Collisson classical subtype, whereas the glycolytic subtype is associated with the Collisson QM-PDA subtype. These findings suggest the utility of broad metabolite profiling to predict sensitivity of tumors to a variety of metabolic inhibitors.
Metabolism in Pancreatic Cancer: Clues from Metabolomics
PDA patients demonstrate many metabolic alterations including signs of muscle wasting, cachexia, fatigue, and changes in lipid and glucose metabolism. These changes cause alterations in levels and distributions of metabolites and recent technological advances have allowed for metabolomic profiling of a variety of relevant biological samples such as serum, tissue, and urine, with the potential for impacting diagnosis, prognosis and therapy. Detecting metabolic markers have been of intense focus in PDA. Many screens have been performed and these studies point to an important role of several metabolites and metabolic pathways.
It is generally understood that the development of tumors requires not only the ability to proliferate uncontrollably but also altered metabolic programs to sustain this rapid expansion. While there are changes common to multiple cancer types such as upregulated glucose uptake and lactate production, known as the Warburg effect, the metabolic profiles of individual tumors and tumors at different stages of development also possess unique features due to the heterogeneous nature of cancers. PDA tumors take up increased amounts of glucose to fuel biosynthetic processes, display elevated glutaminolysis to maintain redox balance, and scavenge fatty acids as well as amino acids from extracellular space to synthesize macromolecules such as lipids and proteins. These metabolic adaptations are the results of oncogenic signaling active in PDA and tumor microenvironment modulation, which collectively meet the cell’s demand to accumulate biomass and proliferate.
Transcriptomic analysis leveraging a doxycycline inducible KrasG12D expressing genetically engineered mouse model (GEMM) and targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) metabolomics revealed that KrasG12D is essential for glucose utilization through stimulation of glucose uptake and channeling of glucose intermediates into the hexosamine biosynthesis pathway for protein glycosylation and pentose phosphate pathways (PPP) for ribose production [29]. This functional validation of several KrasG12D-regulated metabolic enzymes provides candidate therapeutic targets and associated biomarkers for the PDA oncogenic signature.
Kottakis et al. [30] provides evidence for a broader role of metabolic and epigenetic crosstalk in cancer pathogenesis, revealing that LKB1 mutant PDA cells have a marked dependency on pathways linking glycolysis, serine metabolism, and DNA methylation. Their study provides evidence that coupled metabolic and epigenetic states have a more general role in cancer pathogenesis and suggest that LKB1 status is a genetic marker for DNA methyltransferase inhibitor responsiveness.
Recently, studies have focused on communication between tumor and stromal cells, which support tumor cell survival, growth, and proliferation. Notably, this crosstalk includes release of metabolites. Many studies have focused on the role of stromal cells as nutrient suppliers for PDA. Macropinocytosis-mediated internalization of extracellular proteins and their subsequent intracellular degradation was demonstrated as a mechanism for amino acid supply in Ras-transformed cancer cells. These findings suggest the inhibition of macropinocytosis as a promising strategy for therapeutic targeting in a subset of cancers.
Zhao et al. [31] show that fibroblasts smuggle essential nutrients to cancer cells via exosomes, and disable oxygen-based energy production in cancer cells. Oxygen-based energy release was dramatically reduced in the exosome-absorbing cells, and glucose-based energy release increased. They found that contents of the exosomes contain proteins, fatty acids, and other important molecules, which are used by PDA to proliferate. These findings suggest that preventing exosomes from smuggling resources to starving cancer cells might be an effective strategy to treat cancers. Stroma-tumor crosstalk remains under investigations, and this phenomenon reinforces the complexity of PDA. These studies provide new hints regarding the origin of metabolites and approaches to deprive tumors of their benefits.
RNA-sequencing of the PSC transcriptome revealed that, during activation, PSCs decrease expression of genes implicated in lipid storage and lipid metabolism and also increased expression of genes with tumor-supporting potential including cytokines, growth factors, ECM components, and signaling molecules such as Wnt. The transcriptomes of PSCs isolated from patients with PDA identified a PSC “cancer signature” [32]. These analyses also revealed that PSCs express high levels of the vitamin D receptor (VDR), which is maintained in the cancer-associated PSCs. Transcriptome analysis of preactivated and activated PSCs grown in the presence or absence of VDR ligand showed that the vitamin D receptor (VDR) acts as a master genomic suppressor of the PSC activation state. VDR ligand reduces fibrosis and inflammation in a murine pancreatitis model and enhances the efficacy of a coadministered chemotoxic agent. These results highlight a potentially widely applicable strategy to modulate stroma-associated pathologies including inflammation, fibrosis, and cancer.
To identify the marker for early diagnosis of PDAC, a number of studies have been performed in serum and, tissue and urine. In a study using gas chromatography mass spectrometry (GC/MS) on serum samples from patients with pancreatic cancer, Kobayashi et al. [33] investigated a diagnostic model based on four serum metabolites (xylitol, 1;5-anhydro-d-glucitol, histidine, and inositol) and found the profile to outperform both CA 19–9 and CEA for diagnosis.
Recently, Mayers et al. [34] reported that branched-chain amino acid (BCAA) serum levels are elevated 2–5 years before the onset of carcinogenesis in PDA, suggesting that BCAA elevation is an independent risk factor for PDA. Metabolic changes alter systemic amino acid profiles together with changes in plasma BCAA concentrations in the precancerous phase or extremely early stages of PDA. However, BCAA levels return to normal levels within the 2 years before confirmation of cancer. In addition, the results of a mouse study indicated that the period of BCAA elevation was bell-shaped and only temporary. Fukutake et al. [35] indicated novel plasma free amino acids (PFAA) profiles from a large cohort of PDA patients. Concentrations of 19 PFAAs were measured by liquid chromatography–mass spectrometry. Plasma serine concentrations were especially elevated, while tryptophan and histidine concentrations were diminished in PDA patients compared with healthy control subjects. The PFAA profiles of PDA patients with stage 0–IIB disease, the resectable stage subgroup, were similar to those of all other PDA patients. This study identified characteristics of PDA phases, and the PFAA index is a promising biomarker for screening and diagnosis of PDA.
Zhang et al. found specific alterations in free fatty acid (FFA) metabolites, which were decreased in cancer patients [36]. Alterations in the lipid metabolism network included key lipolytic enzymes. Gene expression of these lipases was significantly decreased in pancreatic tumors as compared with nontumor tissues, leading to a reduction in FFA. These results may open new therapeutic options for targeting PDA.
Urinary metabolomics was explored using nuclear magnetic resonance (NMR) spectroscopy to investigate metabolomics profiles in the urine of PDA patients. A distinct urinary metabolomics signature was found in urine of patients with newly diagnosed PDA [37], which reliably could separate patients with PDA and controls with benign disease. Of particular interest was the finding that the increased urinary metabolomic profile decreased after surgical R0 resection.
While metabolomics studies using different technology platforms and samples from various tissue types can provide further insight into cancer biology, the current challenge with these results is confirming validity and reproducibility. Markers and panels appear to change across studies and technological platforms, thus making it difficult to find any one panel with a superior diagnostic, predictive, or prognostic value over the other. Metabolomic profiles of PDA patients have been reported in several previous studies, among which, several amino acid profiles were similar, although there were some obvious discrepancies. First, previous studies included relatively small numbers of subjects compared with the recent studies, which included the largest number of subjects to date. Second, differences may have occurred because of variations in sample preparation conditions and analytical methods. Third, metabolite profiles exhibit diurnal fluctuations and are largely dependent on recent meals. Furthermore, leaving collected blood samples at room temperature is known to alter plasma amino acid concentrations. Furthermore, genetic, racial, and geographical elements may also be factors impacting metabolic profiles, all issues which should be clarified in future research.
Tumors are often highly heterogeneous, with distinct areas dependent on different signaling pathways. Tumor cells adapt and reprogram their metabolism to cope with different environmental conditions. All this makes metabolomic mapping quite difficult. With the hypoxic versus normoxic mosaic, PDA perfectly reflects the idea that different metabolic environments may be found within a single tumor mass, an area worthy of further study. As with other fields of study, tumor metabolism likely results from disturbances in several pathways and will require more sophisticated approaches going forward.
High-Risk Patients
Up to 10% of PDA occur in families with at least two affected first-degree relatives and these are designated familial pancreatic cancers (FPC). FPC is associated with a 2.3- to 32-fold increased risk of PDA development.
The International Cancer of the Patients Screening (CAPS) Consortium has recently reported a suggested guideline for screening, surveillance, and management of high-risk individuals with an inherited predisposition to PDA [38]. A consensus for a screening program to detect and treat T1N0M0 margin-negative PC and high grade dysplastic precursor lesions (pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasm) was reached that the following groups should be offered screening (only to individuals who are surgical candidate): (1) first-degree relatives (FDRs) of the cancer patients from a familial pancreatic cancer cohort with at least two affected (FDRs); (2) patients with Peutz-Jeghers syndrome; and (3) p16, BRCA2 and hereditary nonpolyposis colorectal cancer mutation carriers with at least a single affected FDR. The initial screening should include EUS and/or MRI. However, consensus was not reached on the beginning and the end age of screening/surveillance and the interval of the examination. Their conclusions also included requirements for further studies, and the clinical management should occur at high-volume centers with multidisciplinary teams.
Recent advances in sequencing technology revealed PALB2 and ATM as FPC susceptibility genes, together explaining 3% to 5% of FPC cases. A further 8% to 15% of FPC patients have been reported to harbor other susceptibility genes, including BRCA1, BRCA2, CDKN2A, MLH1, MSH2, MSH6, PMS2, PRSS1, STK11, and TP53. Recent whole genome sequencing demonstrated deleterious variants in the candidate genes BUB1B, CPA1, FANCC, and FANCG as more frequent in FPC patients, many of which are associated with DNA repair or chromosomal stability. CPA1 gene variants have been shown to predispose to chronic pancreatitis, which is strongly associated with an increased risk of PDAC [39].
For FPC patients harboring BRCA1, BRCA2, or PALB2, targeting DNA repair with poly (ADP-ribose) polymerase 1 (PARP-1) inhibitors, platinum compounds, or mitomycin C showed therapeutic benefits [5].
Precision Medicine Clinical Trial
Although we have made great progress in understanding of PDA biology, translating these advances to effective, precision medicine remains a daunting challenge. Both the promise and challenge are illustrated in the IMPaCT (Individualized Molecular Pancreatic Cancer Therapy) trial [6]. In this study, HER2 amplification, KRAS wild-type, and mutations in DNA damage repair pathways (BRCA1, BRCA2, PALB2, ATM) were screened in 76 samples derived from 93 patients.
In this trial, some challenges are illustrated. Of the 22 eligible patients identified for targeted therapy, none were able to receive treatment on protocol because of declining performance status or death. Median time from consent to molecular targeted analysis was 21.5 days. Delays occurred at external testing facilities (n = 6) and the requirement for a repeat biopsy (n = 1). These delays resulting from molecular analysis before treatment initiation are critical in PDA because of the rapid progression of this disease. Von Hoff and colleagues [40] showed that 17.9% (19/106) of participants were unable to be treated according to molecular analyses in a separate molecularly guided study due to worsening physical condition or progression of disease.
Allowing treatment to commence during analysis has not overcome the time lag and perhaps using molecular analysis performed during first-line therapy to guide second-line therapy may be a more practical approach. Randomization in certain studies can also be a deterrent to patient participation.
A paucity of material for molecular analysis remains a major problem. While FNA samples are mainly used for diagnostic material for metastatic PDA patients, the material that remains for molecular analysis is frequently unsuitable. These samples yield low amounts of DNA which is of poor quality for sequencing. Furthermore, as PDA tissue is of low cellularity, limiting eligibility to biopsy samples with cellularity as high as the cancer genome atlas (>60%) would exclude many patients.
Using surrogate biospecimens to perform molecular analysis is a promising approach to overcome some of these obstacles, for example, circulating tumor cells [41] or cell-free DNA. Innovative in vitro approaches, such as expansion of small numbers of tumors cells in three-dimensional organoid culture , can generate adequate numbers of tumor cells, for molecular analysis. Significant efforts are under way to explore these approaches for clinical applicability. Cancer knowledge networks also need to be built to store the results of molecular analysis and medical data of patients, which can then be shared in comprehensive ways among scientists, health care workers, and patients.
Preclinical Models
Cancer Cell Lines
Cancer cell lines have been important tools for drug development. Studies from The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) have established comprehensive catalogs of cancer genes involved in tumorigenesis.
Large-scale drug sensitivity screens in cancer cell lines have been performed to identify potential active drugs. The National Cancer Institute Developmental Therapeutics Program has studied and developed more than 100,000 chemical compounds using 60 human cancer cell lines (NCI-60) since 1990, and this panel of cell lines continue to be used for in vitro drug screening and development.
Two recent projects, the Genomics of Drug Sensitivity in Cancer (GDSC) and the Cancer Cell Line Encyclopedia (CCLE) have evaluated genetic correlations of drug sensitivity. GDSC assembled 639 human tumor cell lines and 130 drugs for screening. CCLE described gene expression, chromosomal copy number, and massively parallel sequencing data from 947 human cancer cell lines and the drug response of 24 compounds across 479 cell lines.
PDA cell lines continue to play an important role in studying biology and drug development. Phenotype and genotype of many of these cell lines are well established. Cell lines are homogeneous, grow rapidly in culture, and are easy to study.
Collison et al. [13] evaluated 19 human and 15 mouse PDA cell lines and showed these cell lines do not cover all subtypes of PDA found in patients. They compared their data sets from 27 human microdissected tumors to human and mouse cell lines. Cell lines most closely modeled either classical or QM-PDA subtypes. Classical type was more dependent on Kras than QM-PDA as determined by RNAi. Kras targeted therapy, therefore, may be effective against classical type tumors [42]. QM is more sensitive to gemcitabine than classical and classical is more sensitive to erlotinib than QM.
Generating cancer cell lines results in certain alterations in biologic properties, such as genetic alteration, alteration in growth and invasion properties, and loss of specific cell populations. In addition, cell lines are usually established only from more aggressive tumors and hence are not representative of complex tumor heterogeneity.
Garnett et al. screened 639 human cancer cell lines, representing most tissue types and a wide range of genetic diversity of human cancers to uncover new biomarkers of sensitivity and resistance to cancer therapeutics, using 130 drugs under clinical and preclinical investigation. Cell lines were subjected to sequencing of the full coding exons of 64 commonly mutated cancer genes, copy number analysis, and expression profile. In addition to well-established targeted therapies, such as BCR-ABL-positive CML, BRAF-mutant melanoma, and EGFR-mutant lung cancer, they showed sensitivity of EWS-FLI1-positive Ewing’s sarcoma cell lines to PARP -inhibitors [43].
Iorio et al. [44] analyzed somatic mutations, copy number alterations, and hypermethylation across a total of 11,289 tumors from 29 tissue types and reported how these alterations can be mapped onto 1001 human cancer cell lines and correlated with sensitivity to 265 drugs. They demonstrated that a sufficiently large panel of cancer cell lines recapitulates oncogenic alterations in primary tumors. However, many genetic alterations occurring at low to moderate frequencies (2–5%) are only represented by a single cell line or not at all, and coverage by cancer type is variable. They analyzed the most predictive data types in pan-cancer and cancer-specific analyses. In cancer specific analyses, genomic features generated the most predictive models, while in the pan-cancer analyses, baseline gene expression data was less informative.
Cell Line Base Xenograft Model
Mouse models are the most experimentally tractable mammalian systems for advancing basic understanding of cancer biology. The xenograft mouse model has been widely used as a tool for preclinical drug screening. Human cancer cell lines can be transplanted either orthotopically or ectopically (usually subcutaneously) into immunocompromised mouse. T-cell deficient nude athymic, B and T lymphocytes deficient severe combined immunodeficient (SCID) and SCID on nonobese diabetic background (NOD/SCID) are commonly used host mice.
Among mouse models, the subcutaneous xenograft is a convenient and economical approach and allows for convenient tumor size assessment. Xenografts have facilitated analyzing the efficacy of compound testing, and most currently approved therapies have been preceded by xenograft testing. While xenograft screening in the earliest stages of drug development can be informative, the extensive screening by the NCI demonstrates a moderate predictive value for their xenograft models, and a poor correlation between the therapeutic efficacy in xenografts and in humans [45]. For PDA, a low correlation between in vitro testing data and clinical utility was also reported [46] .
Subcutaneous tumors are a homogeneous mass with limited stromal infiltration and rarely metastasize. Orthotopic transplantation, where cancer cells are transplanted into the relevant tissue of origin, is better than subcutaneous transplantation for modeling tumor stromal interactions. As metastatic models, cancer cells can be injected intravenously, commonly in the tail vein to model lung metastases, or intraventricularly to model systemic metastases. To model liver metastases, cancer cells are injected into the portal vein or spleen. These transplantation systems can be adapted to many different cancer types.
There are also several shortcomings for xenograft mouse models. Host (SCID and nude) mice are immune deficient and not useful for testing of immunomodulatory agents. In addition, in these systems the immunodeficient state of the mouse results in the failure to completely recapitulate the complex tumor-stromal interaction and the impact on drug response of the tumor microenvironment. These are important considerations particularly in PDA, which is characterized by an abundant stromal reaction and unique heterogeneity. Xenograft studies typically use only a few human tumor cell lines, the oncogenomic profiles of which represent only isolated combinations of the wide spectrum of genetic and epigenetic mutations that are resident in the tumors found in human patients. The reliance on small numbers of homogeneous cell lines is a fundamental weakness.
Genetically Engineered Mouse Model
By using pancreas-specific conditional activation or knockout of clinically relevant PDA-related genes and signaling pathways, genetically engineered mouse models (GEMM) of PDA have been described and are now a well-established tool. Histologically, PDA GEMMs generally develop differentiated ductal adenocarcinoma with abundant stromal components including a robust desmoplastic reaction. Some GEMMs develop sarcomatoid or undifferentiated tumors, which are rare in human pancreatic cancer. With regard to TGF-beta signaling, SMAD4 gene mutation or deletion is frequently observed in human PDA tumors; however, mice engineered with pancreas specific Kras activation together with Smad4 knockout were reported to develop cystic tumors of the pancreas, a precancerous lesion distinct from PanINs, intraductal papillary mucinous neoplasms, or mucinous cystic neoplasms [47].
An excellent review of a large number of mouse models was performed, and describes several differences between the pathology identified in GEMMs and that seen in human tumors [48]. First, human PDA tends to be moderate or poorly differentiated, whereas many of the GEMMs produced anaplastic carcinomas. Second, most neoplasms in humans show a single direction of differentiation, whereas multilineage differentiation, including acinar differentiation, was often seen in GEMMs. Third, pancreatic intraepithelial neoplasia in humans often, although not always, occurs in the pancreatic duct. By contrast, many of the duct lesions in GEMMs arose in the background of diffuse acinar-ductal metaplasia. Fourth, most human pancreatic carcinomas are solitary, whereas multifocality, not surprisingly, is commonly seen in GEMMs. Finally, intense desmoplasia is a characteristic feature of invasive ductal adenocarcinoma in humans. By contrast, little desmoplasia is seen in some GEMM carcinomas. Each of these models has its own unique strengths and weaknesses in advancing our understanding of pancreatic neoplasia, to identify target-specific biomarkers to assess drug action and discover resistance mechanisms.
PDA GEMMs have been utilized to make important discoveries. One of the earliest studies described how PDA GEMMs appear to recapitulate the tumor microenvironment better than xenograft tumor models. The GEMM also recapitulated chemotherapy resistance, similar to what is seen in the human disease [49]. One of the most commonly used GEMMs for evaluating preclinical therapeutic agents is the PDX-1-Cre; LSL-KrasG12D; LSL-p53R172/− (KPC) model [50]. The KPC model recapitulates the clinical features of PDA including hemorrhagic ascites and cachexia. This model also demonstrates metastases to liver, lung, peritoneum, and lymph nodes and a short median survival of approximately 5 months. Histopathologically, tumors generally demonstrate ductal adenocarcinoma with dense stromal desmoplasia; however, sarcomatoid and anaplastic tumors do also occur. Unlike xenograft models using immunocompromised mouse, GEMMs have an intact immune system and stromal reaction. An intact tumor microenvironment was important for the preclinical study of PEGPH20 [51], a PEGylated human recombinant PH20 hyaluronidase. The glycosaminoglycan hyaluronan (HA) is abundant in PDA stroma and transduces signaling through CD44 to regulate receptor tyrosine kinases and small GTPase activity which play important roles in angiogenesis, epithelial-mesenchymal transition, and chemoresistance [52]. PEGPH20 treatment increases intratumoral delivery of chemotherapeutic agents by digesting HA. These preclinical studies have prompted further clinical development of PEGPH20, which is currently in randomized phase III testing for the treatment of advanced PDA (NCT02715804). Hedgehog pathway inhibition was first reported to inhibit the stromal component in KPC mice, which increased the delivery of gemcitabine to tumors and improved survival in combination with gemcitabine [49]. Unfortunately, in a randomized phase II clinical trial, the hedgehog pathway inhibitor IPI-926 in combination with gemcitabine was ineffective. Using a separate GEMM, Rhim et al. demonstrated that prolonged hedgehog inhibition as a monotherapy led to more aggressive tumor behavior [53]. These results suggest GEMM models are a useful tool to evaluate the efficacy of drugs targeting tumor microenvironment and mechanism of efficacy of chemotherapeutic agents. GEMM models also play an important role in evaluating immune modulating agents. Feig and colleagues reported that KPC models do not respond to antagonism of the immune checkpoints anti-cytotoxic T lymphocyte-associated protein 4 (α-CTLA-4) and α-programmed cell death 1 ligand 1 (α-PD-L1), as is seen in human clinical trials. However, the depletion of cancer-associated fibroblast enabled control of tumor growth using these inhibitors. Treatment with a CXCL12 receptor inhibitor resulted in T cell accumulation in tumors and potentiated anticancer effects of α-PD-L1 [54]. GEMMs can be used to understand the disease biology and drug development, particularly focused on tumor microenvironment and immune response.
It is evident that an understanding of genetic events and signaling pathways is crucial for the development of effective targeted therapies in PDA. GEMMs will continue to play a significant role in the crucial first step of drug discovery and target validation. Pdx1-Cre; LSL-KrasG12D; Ptenflox/flox mouse model which demonstrates elevated mTOR (mammalian target of rapamycin) signaling showed response to mTOR inhibitor [55]. In clinical trial, mTOR inhibitor did not show the efficacy for unselected pancreatic cancer patients. However, patients with mutations in mTOR pathway showed efficacy for mTOR inhibitor [55]. A Ptf1a-Cre; LSL-KrasG12D; Tgfbr2flox/flox mouse model was used to assess the efficacy of the EGFR inhibitor erlotinib in combination with gemcitabine [56]. Systematic studies using 2D cancer cells of cancer genomes and drug efficacy implied the efficacy of EGFR/ERBB2 inhibitors against cancer cells with Smad4 mutation [44].
Recent whole genome sequencing, exome sequencing and RNA sequencing studies revealed some characteristics of PDA, but these subtypes are not predictive for drug sensitivity. GEMMs recapitulate many of the features of human PDA. GEMMs can be useful to evaluate drug response against PDA patients with specific genetic backgrounds. With regard to the discovery of specific biomarkers in cancer patients, it is necessary to collect large numbers of specimens because of interindividual variability, which makes the discovery of biomarkers difficult. However, the use of a GEMM, designed to develop the desired cancer with a predicted latency could allow for identification of candidate biomarkers, which can then be validated in human clinical samples.
By using tetracycline-regulated and CRE-inducible alleles, the timing, duration and tissue compartment of gene expression or inactivation can be further controlled. An alternative method for generating GEMMs uses the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated proteins) gene-editing system. Chiou and colleague reported CRISPR-mediated targeting of liver kinase B1 (LKB1) in combination with Kras expression [57]. In this study, they also reported in vivo gene editing by retrograde injection of adenoviral-Cre and lentiviral-Cre into the pancreas of LSL-KRasG12D; p53flox/flox mice.
GEMMs are an important tool for studying biology and drug development. GEMMs are customizable to perturb any number of genetic alterations, which will hopefully continue to lead to more effective therapies.
Patient Avatars
Patient-Derived Xenograft
For a number of reasons previously discussed, the establishment of cell lines is not an effective strategy for personalized medicine. The principal limitation of conventional 2D cell line–based xenograft models is their poor predictive value with regard to clinical outcome [58]. Generally, PDX models have been reported to retain the principal characteristics of donor tumors both histologically and biologically. An analysis of genetic profiles show good concordance between primary tumors and the models derived from them, except discordance in genes involved in the stromal compartment and immune function, which is due to the replacement of the human stroma by murine elements. Although the gene expression profile of PDX models is similar to the original tumor, cell lines developed from the same specimen demonstrate a different expression profile that is not restored by in vivo subcutaneous propagation in mice in SCLC. In PDA models, similar results have been observed in which the frequency of mutations in genes such as TP53 or RAS closely mirrors the frequency of these mutations in human samples [59, 60].
PDX models are an attractive preclinical tool to improve drug screening and development. PDX models are expected to faithfully model the human patients from whom the tumor is derived, both with regards to cancer biology and response to treatments. Personalized PDX models have been studied as a tool for testing candidate regimens which may be effective for treating the patient’s own tumor [61]. Evaluating the relationship of drug response with genetic information could lead to the discovery of new biomarkers of drug efficacy. These results suggest that PDX models hold promise for precision medicine in PDA.
One study found a good correlation between response in patient derived PDX and clinical response to gemcitabine in PDA patients [62]. Drug response of PDX models has been reported to be stably maintained across generations (up to 10 passages) [59].
Hidalgo et al. found in a pilot study that treatment of PDA patients with drugs selected according to preclinical PDX drug screening was predictive of tumor response, which suggests that response in PDX models correlates with clinical outcome [63]. This work showed that the combination of nab-paclitaxel and gemcitabine is effective in PDX models of PDA, which correlated with the clinical efficacy of this combination. This regimen has subsequently been demonstrated to provide a survival benefit for patients with advanced PDA in a randomized phase III study. Likewise, failure to exert antitumor efficacy in PDX models correlates with negative clinical results. This is illustrated in PDAC for agents such as the SRC inhibitor saracatinib and the mTOR inhibitor sirolimus, for which lack of efficacy in unselected PDX preclinical studies predicted failure of the same strategy in the clinic [61]. Based on these data, PDX models have now become an integral part of the preclinical screening of new anticancer agents.
The concordance between PDX models and human trials with regard to biomarkers of drug susceptibility and drug resistance is an important finding. In PDA, PDX studies with gemcitabine identified expression of the gemcitabine-activating enzyme deoxycytidine kinase as a predictor of drug efficacy [59, 64]. Likewise, PDX models have been used to identify metabolic as well as imaging biomarkers. PDX models are also versatile tools for simulating resistance when exposed to treatment strategies used in the clinical setting and to study strategies for overcome resistance.
In most patients, derivation of a personalized PDX for guiding therapy is not feasible for a combination of reasons such as failure of the tumor to engraft, lack of effective agents, and length of time required for a complete study [62, 63]. For patients whose tumors do not take in mice or those who require a long time to be established and characterized, an alternative to a personalized PDX strategy could be to determine treatment choices based on drug responses in a similar, established PDX. Biopsies of primary tumors or metastases would be molecularly characterized and compared with available PDX collections from the same pathology, for which responses to chemotherapies and targeted agents have been previously determined.
PDX models generally rely on surgical specimens, which provide large quantities of tumor tissue. As most PDA patients are inoperable, it is more useful to generate PDX from smaller samples, such as fine-needle aspiration for personalized therapy. Four to eight months are required to generate PDX models for preclinical treatment study. The success rate of engraftment is about 60% and it is important to establish the best engraftment methods according to the phenotype of cancer. Human cancer stroma included in the cancer specimens are replaced rapidly by mouse stromal cells including fibroblasts, inflammatory cells, blood vessels, and immune cells. PDX models require an immunocompromised mouse host which limits the ability to evaluate immune modulators, such as vaccines, anti-PD-1, and anti-CD40 antibodies.
PDX models may also be used as part of co-clinical trials. In co-clinical trials, a personalized PDX model is developed from a patient enrolled in a clinical trial and treated with the same experimental agents to emulate clinical response by using appropriate endpoints such as response rate or tumor growth delay. The availability of a larger collection of models extensively characterized at the histologic, molecular, and genomic level would enable these larger screens. Biologic and genetic comparisons between sensitive and resistant models can be explored for the prioritization of biomarkers for inclusion in clinical studies.
This strategy permits the assessment of drug response simultaneously in the patient and mouse model, providing an interesting platform to investigate biomarkers of susceptibility and resistance, as well as interrogation of novel combination strategies to overcome emergent resistance pathways. Novel approaches, such as short-term primary cultures or organoids, are being developed and are expected to be used for preclinical screening studies.
Organoid: A Promising New Model
New and innovative culture approaches have been developed which address several obstacles to studying and treating PDA. As previously discussed, samples for genetic screening are frequently unsuitable, of low cellularity, yield low quantities and poor quality DNA for sequencing. 2D cell lines established from human PDA samples are useful; however, the process of cell line establishment results in clonal loss, therefore cell lines do not accurately reflect tumor heterogeneity.
Loss of tumor heterogeneity is a similar weakness of 2D cell line–based xenografts. While studies of PDX models have demonstrated the presence of dense desmoplastic stroma, maintenance of tumor heterogeneity, and good correlation between drug response and human clinical response, transplant success rates are biased towards more aggressive tumors and require a large piece of tumor tissue. PDX models require 4 to 8 months before drug screening can be performed. GEMMs recapitulate the stromal reaction, genetic mutations and progression from normal to PanIN to adenocarcinoma; however, GEMMs lack the genetic and cellular heterogeneity which can only be captured in the human disease.
New 3D culture techniques have been developed in the past decades, providing a new tool with the potential for addressing many of the issues described above.
The first description of this long-term culture system, termed organoids, was reported by Sato et al. [65]. Sato and colleagues used cells derived from the murine small intestine. Several key growth factors appear important for long-term organoid maintenance. For example, supplementation with Wnt ligand supports crypt proliferation, epidermal growth factor (EGF) supports intestinal proliferation, Noggin induces expansion of crypt numbers, inhibition of anoikis is necessary, and finally, laminin-rich Matrigel acts as an extracellular matrix and supports intestinal epithelial growth. At the same time, another long-term culture was established by Ootani et al. [66] for small and large intestine. Successively, long-term 3D culture methods were described for other organs such as stomach, liver, and mammary gland. In addition, long-term 3D culture system was described for malignant tumors derived from breast, colon, and prostate. More recently, normal pancreas and PDA organoid systems have been established.
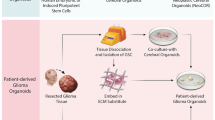
Boj et al. [67] described an organoid culture system for both normal and neoplastic epithelial cells derived from both mice and humans. Pancreatic organoids are embedded in Matrigel, which contains essential components of a basement membrane. The culture media contains Wnt3a, Noggin, EGF, and R-spondin-1, which are key growth factors. For human organoid culture, FGF10, nicotinamide, A83–01, and prostaglandin E2 are additionally required. Pancreatic organoids can be passaged indefinitely except for human normal organoids, which generally can only be cultured for 20–25 passages. PDA organoids can be expanded from a minimal piece of tissue, such as from a fine needle aspiration (Fig. 1).

Representative images of human organoid cultures established from normal tissues (hN), primary tumors (hT), and metastatic tumors (hM). Bar: 500 μm
Expansion of small amounts of tumor or normal tissue to large-scale organoid cultures allows for parallel precision medicine analysis including drug screening, genomic, transcriptomic, metabolomics, and proteomic analyses. Boj et al. performed gene expression analysis comparing mouse normal, PanIN, and tumor organoids and showed similar changes in gene expression patterns comparing mouse PanIN and tumor organoids to normal organoids, as seen with oncogenic Kras activation in KrasG12D mice. These analyses demonstrated the ability of the organoid system to characterize molecular alterations associated with PDA progression. Proteomic analysis of mouse normal, PanIN, and tumor organoids was also performed. Few protein expression changes were seen comparing mouse PanIN and tumor organoids, whereas many more changes were seen comparing mouse normal and PanIN organoids, or mouse normal and tumor organoids. Gene Set Enrichment Analysis (GSEA) of RNA sequencing and proteomic data comparing mouse PanIN to normal organoids revealed up regulated genes and proteins involved in glutathione metabolism and biological oxidations, consistent with previous studies. Similar to the PDX mouse model, organoid transplant mouse models are a promising tool for drug screening and studying biology. Using organoids for in vitro drug screening is possible a couple of months after samples are collected. Organoids can be reliably established from virtually every patient sample. Preliminary studies suggest maintenance of tumor heterogeneity even after several passages.
Interestingly, orthotopic transplantation of organoids develops a full spectrum of lesions associated with disease progression, including early PanIN and late PanIN, invasive ductal adenocarcinoma, and metastasis. This model is a promising tool to study the earliest stage of human cancer to understand fundamental biology and to identify biomarkers of early disease.
Hunag et al. generated pancreatic progenitor cells from pluripotent stem cells in 3D culture and induced differentiation of their organoid progenitor cells into pancreatic exocrine cells which express ductal and acinar markers [68]. They adapted their culture condition for growing human PDA. Among 20 human pancreatic samples, they established 17 tumor organoid lines and showed similar morphological and cytological features to those of the primary tumors they were derived from after 16 days in 3D culture. They transplanted 50,000 cells subcutaneously and tumors grew within 4–7 weeks. Xenograft tumors demonstrated similar histoarchitecture to the primary tumor or origin and also maintained histological heterogeneity. They tested an EZH2 (enhancer of zeste homolog 2) inhibitor against human tumor organoids and suggested the usefulness of organoids as a platform for personalized drug testing, although they were not able to correlate organoid response to patient outcomes.
Walsh et al. established mouse and human organoids for drug testing and optical metabolic imaging (OMI) which probes the fluorescence intensity and lifetime of NAD(P)H and FAD [69]. After mechanical digestion, organoids are embedded in Matrigel and subjected to drug testing and optical metabolic imaging. This method does not allow for passage of organoids but can be useful as a tool to evaluate drug response for personalized medicine. They observed three distinctive morphologies of murine PDAC including spherical organoids (type 1), symmetric organoids (type 2), and fibroblasts. Type 1 and type 2 organoids are positive for epithelial markers. Type 1 organoids show the greatest OMI index and type 2 organoids showed the smallest OMI index. Optical redox index ratio of type 2 organoids was lower than that of type 1 organoids and fibroblast. Organoids were treated with a JAK2 inhibitor, MEK inhibitor, PI3K inhibitor, and combinations to evaluate drug-induced metabolic changes, which revealed heterogeneous metabolic responses among cell populations [69]. Human PDAC organoids demonstrated a broad spectrum of morphologies, which were difficult to classify into subtypes. They showed that the OMI index reduction was detected with gemcitabine treatment and gemcitabine with JAK2 inhibitor treatment.
Li et al. cultured organoids with both epithelial and mesenchymal components from embryonic pancreas using an air-liquid interface culture method with an inner collagen gel-containing transwell with direct air exposure. This system does not require exogenous factor supplementation [70].
Wetering et al. [71] reported the establishment of a “living biobank” from 20 colorectal cancer patients. They demonstrated that the organoid culture platform can be exploited for genomic and functional studies at the level of the individual patient for personalized treatment approach. Organoid technology may fill the gap between cancer genetics and patient trials, complement cell-line- and xenograft-based drug studies, and help to achieve an effective, personalized therapy approach.
Conclusion
Integrated genomic, epigenomic, and transcriptomic analyses are generating biological insights with potential therapeutic relevance in PDA. The recurrently mutated genes aggregate into core molecular pathways including KRAS, Wnt, Notch, DNA damage repair, RNA processing, cell cycle regulation, TGF-β signaling, SWI-SNF, chromatin regulation, and axonal guidance. Genomic-based treatment has resulted in paradigm changing therapies for other cancers, dramatically improving survival and cures. However, this remains an unfulfilled promise in PDA due to apparently untargetable mutations, high resistance to available chemotherapeutic agents, and the difficulty of drug delivery through a rich stromal component. In addition, individual tumors have infrequently mutated genes, result in significant inter- and intratumoral heterogeneity. Due to this diversity, therapeutic development has been challenging. Familial pancreatic cancer patients harboring BRCA or PALB2 may have sensitivity to PARP-1 inhibitors, platinum compounds, or mitomycin C. In the IMPaCT (The Individualized Molecular Pancreatic Cancer Therapy) trial [6], HER2 amplification, KRAS wild-type, and mutations in DNA damage repair pathways (BRCA1, BRCA2, PALB2, ATM) were targeted for treatment. Personalized PDX models have the potential to identify effective drug therapies, however, with significant limitations, including a long lead-time and large amounts of tumor tissue for testing. The three-dimensional organoid culture platform can be exploited for genomic and functional studies at the level of the individual patient for personalized treatment approach. Organoid technology may fill the gap between cancer genetics and patient trials and allow personalized therapy design, although further studies to validate this approach are needed (Fig. 2). A combination of genome-based medicine and individualized model drug screening may prove to be the key tools needed for precision medicine for PDA (Table 1).

The design of precision medicine
References
Kaufman B, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–50.
Bailey P, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52.
Jones S, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217.
Villarroel MC, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10:3–8.
Waddell N, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501.
Chantrill LA, et al. Precision medicine for advanced pancreas cancer: the individualized molecular pancreatic cancer therapy (IMPaCT) trial. Clin Cancer Res. 2015;21:2029–37.
Biankin AV, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405.
Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6.
Wang L, et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012;22:208–19.
Nones K, et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014;135:1110–8.
Garcia PL, et al. The BET bromodomain inhibitor JQ1 suppresses growth of pancreatic ductal adenocarcinoma in patient-derived xenograft models. Oncogene. 2016;35:833–45.
Mazur PK, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015;21:1163–71.
Collisson EA, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500–3. https://doi.org/10.1038/nm.2344.
Moffitt RA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168–78.
Noll EM, et al. CYP3A5 mediates basal and acquired therapy resistance in different subtypes of pancreatic ductal adenocarcinoma. Nat Med. 2016;22:278–87.
Rosty C, Goggins M. Early detection of pancreatic carcinoma. Hematol Oncol Clin North Am. 2002;16:37–52.
Ching CK, Rhodes JM. Enzyme-linked PNA lectin binding assay compared with CA19-9 and CEA radioimmunoassay as a diagnostic blood test for pancreatic cancer. Br J Cancer. 1989;59:949–53.
Uehara H, et al. Diagnosis of pancreatic cancer by detecting telomerase activity in pancreatic juice: comparison with K-ras mutations. Am J Gastroenterol. 1999;94:2513–8.
Yokoyama M, et al. Matrix metalloproteinase-2 in pancreatic juice for diagnosis of pancreatic cancer. Pancreas. 2002;24:344–7.
Bettegowda C, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24.
Chen R, Pan S, Aebersold R, Brentnall TA. Proteomics studies of pancreatic cancer. Proteomics Clin Appl. 2007;1:1582–91.
Yu KH, Rustgi AK, Blair IA. Characterization of proteins in human pancreatic cancer serum using differential gel electrophoresis and tandem mass spectrometry. J Proteome Res. 2005;4:1742–51.
Wehr AY, Furth EE, Sangar V, Blair IA, Yu KH. Analysis of the human pancreatic stellate cell secreted proteome. Pancreas. 2011;40:557–66.
Yu KH, et al. Stable isotope dilution multidimensional liquid chromatography-tandem mass spectrometry for pancreatic cancer serum biomarker discovery. J Proteome Res. 2009;8:1565–76.
Wehr AY, Hwang W-T, Blair IA, Yu KH. Relative quantification of serum proteins from pancreatic ductal adenocarcinoma patients by stable isotope dilution liquid chromatography-mass spectrometry. J Proteome Res. 2012;11:1749–58.
Britton D, et al. Quantification of pancreatic cancer proteome and phosphorylome: indicates molecular events likely contributing to cancer and activity of drug targets. PLoS One. 2014;9:e90948.
Humphrey ES, et al. Resolution of novel pancreatic ductal adenocarcinoma subtypes by global phosphotyrosine profiling. Mol Cell Proteomics. 2016;15:2671–85.
Daemen A, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc Natl Acad Sci USA. 2015;112:E4410–7.
Ying H, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70.
Kottakis F, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539:390–5.
Zhao H, et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife. Sciences. 2016;5:e10250.
Sherman MH, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93.
Kobayashi T, et al. A novel serum metabolomics-based diagnostic approach to pancreatic cancer. Cancer Epidemiol Biomark Prev. 2013;22:571–9.
Mayers JR, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. 2014;20:1193–8.
Fukutake N, et al. A novel multivariate index for pancreatic cancer detection based on the plasma free amino acid profile. PLoS One. 2015;10:e0132223.
Zhang G, et al. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res. 2013;19:4983–93.
Davis VW, Schiller DE, Eurich D, Bathe OF, Sawyer MB. Pancreatic ductal adenocarcinoma is associated with a distinct urinary metabolomic signature. Ann Surg Oncol. 2013;20(Suppl 3):S415–23.
Canto MI, et al. International cancer of the pancreas screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62:339–47.
Witt H, et al. Variants in CPA1 are strongly associated with early onset chronic pancreatitis. Nat Genet. 2013;45:1216–20.
Von Hoff DD, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010;28:4877–83.
Yu KH, et al. Pharmacogenomic modeling of circulating tumor and invasive cells for prediction of chemotherapy response and resistance in pancreatic cancer. Clin Cancer Res. 2014;20:5281–9.
Singh A, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500.
Garnett MJ, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5.
Iorio F, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–54.
Voskoglou-Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res. 2003;9:4227–39.
Abaan OD, et al. The exomes of the NCI-60 panel: a genomic resource for cancer biology and systems pharmacology. Cancer Res. 2013;73:4372–82.
Bardeesy N, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20:3130–46.
Hruban RH, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: consensus report and recommendations. Cancer Res. 2006;66:95–106.
Olive KP, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61.
Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83.
Jacobetz MA, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–20.
Toole BP, Slomiany MG. Hyaluronan: a constitutive regulator of chemoresistance and malignancy in cancer cells. Semin Cancer Biol. 2008;18:244–50.
Rhim AD, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47.
Feig C, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–7.
Morran DC, et al. Targeting mTOR dependency in pancreatic cancer. Gut. 2014;63:1481–9.
Miyabayashi K, et al. Erlotinib prolongs survival in pancreatic cancer by blocking gemcitabine-induced MAPK signals. Cancer Res. 2013;73:2221–34.
Chiou S-H, et al. Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev. 2015;29:1576–85.
Johnson JI, et al. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. Br J Cancer. 2001;84:1424–31.
Rubio-Viqueira B, et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res. 2006;12:4652–61.
Bertotti A, et al. A molecularly annotated platform of patient-derived xenografts (‘xenopatients’) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–23.
Garrido-Laguna I, et al. Integrated preclinical and clinical development of mTOR inhibitors in pancreatic cancer. Br J Cancer. 2010;103:649–55.
Garrido-Laguna I, et al. Tumor engraftment in nude mice and enrichment in stroma-related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin Cancer Res. 2011;17:5793–800.
Hidalgo M, et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther. 2011;10:1311–6.
Sebastiani V. Immunohistochemical and genetic evaluation of deoxycytidine kinase in pancreatic cancer: relationship to molecular mechanisms of gemcitabine resistance and survival. Clin Cancer Res. 2006;12:2492–7.
Sato T, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5.
Ootani A, et al. Sustained in vitro intestinal epithelial culture within a Wnt-dependent stem cell niche. Nat Med. 2009;15:701–6.
Boj SF, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–38.
Huang L, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med. 2015;21:1364–71. https://doi.org/10.1038/nm.3973.
Walsh AJ, Castellanos JA, Nagathihalli NS, Merchant NB, Skala MC. Optical imaging of drug-induced metabolism changes in murine and human pancreatic cancer organoids reveals heterogeneous drug response. Pancreas. 2016;45:863–9.
Li X, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 2014;20:769–77.
van de Wetering M, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–45.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media, LLC, part of Springer Nature
About this entry

Cite this entry
Miyabayashi, K., Tuveson, D.A., Yu, K.H. (2018). Multiparameter Modalities for the Study of Patients in the Setting of Individualized Medicine. In: Neoptolemos, J., Urrutia, R., Abbruzzese, J., Büchler, M. (eds) Pancreatic Cancer. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-7193-0_65
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7193-0_65
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-7191-6
Online ISBN: 978-1-4939-7193-0
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences