
Abstract
Rapid eye movement (REM) sleep behavior disorder (RBD) is a parasomnia characterized by repeated episodes of dream enactment behavior and REM sleep without atonia (RSWA) during polysomnography. RSWA is characterized by increased phasic or tonic muscle activity seen on polysomnographic electromyogram channels. RSWA is a requisite diagnostic feature of RBD, but may also be seen in patients without clinical symptoms or signs of dream enactment as an incidental finding in neurologically normal individuals, especially in patients receiving antidepressant therapy. RBD may be idiopathic or symptomatic. Patients with idiopathic RBD often later develop other neurological features including parkinsonism, orthostatic hypotension, anosmia, or cognitive impairment. RSWA without clinical dream eneactment, as well as clinically overt RBD, also often occurs concomitantly with synucleinopathy neurodegenerative disorders, which include idiopathic Parkinson disease, Lewy body dementia, and multiple system atrophy. This chapter reviews the epidemiology of RBD, clinical, and polysomnographic diagnostic standards for both RBD and RSWA, previously reported associations of RSWA and RBD with neurodegenerative disorders and other potential causes, pathophysiology of brain structures and networks mediating dysregulation of REM sleep muscle atonia and clinical dream enactment, and considerations for the effective and safe management of RBD.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- REM sleep behavior disorder
- REM sleep without atonia
- Parasomnia
- Synucleinopathy
- Parkinsonism
- Neurodegeneration
- Braak staging
- Melatonin
- Clonazepam
- Treatment
- Neuropsychological testing
Introduction
REM sleep behavior disorder (RBD) was first described in humans in 1986 after a series of patients reported curious nocturnal behaviors that resulted in injury to patients or their bedpartners [1]. Due to the loss of normal REM sleep muscle atonia, RBD patients often “act out their dreams,” most commonly expressing violent complex movements that often mirror dream content [1–10]. RBD patients are primarily divided into two groups: idiopathic RBD, with no obvious cause, and symptomatic RBD, which is primarily associated with synucleinopathy neurodegenerative disorders, including Parkinson’s disease (PD), Lewy body dementia (DLB), and multiple system atrophy (MSA) [3–12]. However, RBD is also common in patients with narcolepsy and in patients receiving antidepressant treatment and may be seen rarely in those with brainstem lesions in dorsal pons and medulla [10, 13–26]. In addition, RBD has also been associated with the use or withdrawal of drugs or alcohol, high chocolate intake [27], and migraine headaches [27–31]. However, because up to 82 % of idiopathic RBD patients develop parkinsonism or dementia over longitudinal follow-up, growing evidence suggests that idiopathic RBD may be a prodromal feature of a neurodegenerative disease, often preceding other characteristic more overt neurological manifestations by several years to decades [3, 5, 7, 8, 12, 32–37]. In addition, recent neuropathology data suggest that up to 94 % of patients with RBD, and 98 % of RBD patients confirmed by polysomnography (PSG), have synucleinopathy neurodegeneration at autopsy, furthering the presumption that RBD may represent the forme fruste of neurodegeneration in many patients [37].
Diagnosis and Classification of RBD
The minimal diagnostic criteria according to the International Classification of Sleep Disorders (ICSD) 2 include: (A) presence of REM sleep without atonia on PSG; (B) sleep-related injurious or potentially injurious disruptive behaviors by history, and/or abnormal REM sleep behaviors during PSG; (C) absence of epileptiform activity during REM sleep (unless RBD can be clearly distinguished from any concurrent REM sleep-related seizure disorder); and (D) sleep disturbance is not better explained by another disorder [38]. However, an evolving diagnostic standard for probable RBD (pRBD) for patients having dream enactment behaviors but who lack PSG evidence for RSWA (due to either unavailability of PSG or failure to record REM sleep) is included in ICSD 3, given the resource intensive nature of confirmatory PSG [38a].
The core clinical feature of RBD is a history of witnessed dream enactment by the patient’s bed partner, with or without recall of dream mentation by the patient himself or herself [1, 5, 11, 34, 39]. Patients are often able to vividly recall their dreams for weeks or longer, and when enacted dreams are recalled, patients typically report that their dream mentation contains a theme of being chased, or defense against an attack by animals or people [11, 40]. However, less aggressive themes such as playing sports or performing household chores are also common [41, 42]. Collateral history obtained from the patient’s bed partner is crucial in diagnosing RBD patients, since NREM parasomnias like sleep walking or sleep terrors also often report frightening dream content. However, dreams of patients with sleep walking or sleep terrors more often involve natural disasters with a “flight” response, as opposed to the “fight” response reported by patients with RBD [5, 11, 39, 42, 43].
RBD patients often have coherent vocalizations such as shouting, screaming, laughing, crying, and swearing, most often matching recalled dream content described by the patient [4–6, 11]. Excessive, repetitive phasic muscle jerking during REM sleep is the most frequent RBD clinical phenomena captured during PSG; however, complex motor behaviors such as punching, kicking, and running can lead the patient to leave the bed, potentially resulting in injuries [5, 11, 44]. Due to the violent nature of dreams, injuries prior to treatment are frequent and have been reported in approximately 55 % of patients [41]. While most often minor injuries such as bruises occur, more serious injuries such as fractures, lacerations requiring suturing, and subdural hematomas occur in as many as 12 % of patients [1, 5, 11, 41].
While PSG is required for a formal diagnosis of RBD, PSG is expensive and requires expertise for accurate interpretation, and is not readily accessible in many locations. Given the resource intensive nature of PSG, several validated screening measures for pRBD are available [39, 43, 45–49]. The RBD-HK consists of 13 questions regarding frequency and severity of dream enactment behavior. Out of 100 possible points, a cutoff score of 18 yields positive and negative predictive values of over 80 % [43]. The REM Sleep Behavior Disorder Screening Questionnaire (RBDSQ), a 10-item patient self-rating questionnaire (maximum total score of 13 points) covering core clinical features of RBD, demonstrated that RBD patients scored an average of 9.5, compared to 4.6 in controls [45]. This survey has been adapted for use in Japanese patients, with a score of 5 yielding a sensitive, specific, and reliable diagnosis of RBD [47]. The Mayo Sleep Questionnaire (MSQ) is another validated diagnostic questionnaire tool for RBD screening in older patients with or without cognitive impairment and/or parkinsonism [39, 48]. While other surveys utilize patient-reported symptoms, one of the main strengths of the MSQ is completion by bed partners, providing valuable collateral history verifying dream enactment symptoms, which may be more reliable for patients who are cognitively impaired and in those who have little recall of their dreams. In addition, the MSQ is 100 % sensitive and 95 % specific for diagnosis of RBD, indicating its utility for screening for RBD when PSG is unavailable [48]. The RBD1Q is a multicenter validated screening questionnaire consisting of a yes or no answer to a single question about dream enactment (which is almost identical to the core RBD question on the MSQ), with a sensitivity of 93.8 % and specificity of 87.2 % for a diagnosis of RBD in patients with Parkinson’s disease [46]. These questionnaires may be valuable for future epidemiological studies given their brevity, efficiency, and accuracy for diagnosis of RBD.
While surveys have been shown to be useful measures for RBD screening, exclusion of RBD-mimics such as nightmares, sleep walking, sleep terrors, nocturnal seizures, obstructive sleep apnea with atypical arousals from REM sleep, post-traumatic stress disorder, nocturnal panic disorder, psychogenic dissociative states, and delirium may be difficult by reliance on questionnaire instruments alone [5, 6, 42, 50, 51]. Given that violent behaviors during sleep in the general population are often associated with vivid dreams in sleep terrors and sleepwalking, the “gold standard” for clinical RBD diagnosis should remain a combination of clinical diagnostic interview with a thorough history, neurological examination, and PSG to confidently diagnose and differentiate RBD from other sleep disorders and parasomnias, and to confirm the necessary diagnostic feature of REM sleep without atonia [1, 5, 11, 38].
Diagnosis of RSWA
PSG diagnosis of RSWA requires the presence of abnormally elevated muscle tone during REM sleep, most often observed in the mentalis and tibialis muscles, but also in arm EMG leads [9, 38, 52, 53]. While most clinical laboratories primarily use mentalis, anterior tibialis, and forearm muscles to identify RSWA, some studies have suggested that an expanded EMG montage including the biceps brachii, flexor digitorum superficialis, and abductor pollicis brevis muscles is the most sensitive for RSWA determination [52, 53]. RSWA is classified as short phasic bursts, with muscle tone reaching at least two to four times larger than the amplitude of the lowest background EMG during REM or NREM sleep, or tonic segments with twice the background amplitude, usually lasting longer than 10–15 s in duration [52–54]. RSWA may also be found incidentally on PSG recording without dream enactment behavior. However, the significance of incidental RSWA for future development of dream enactment or neurodegeneration remains to be elucidated [5, 55]. In addition, RSWA without dream enactment is not uncommon in PD, suggesting that lesions in separate specific structures may be necessary to cause RSWA and DEB [55]. Since a small amount of phasic or tonic muscle activity can also be found during REM sleep in normal individuals [56], the AASM has established a standard for abnormal or potentially clinically relevant RSWA, defined as at least 5 mini-epochs of 3-s duration containing abnormally excessive phasic muscle activity within a single 30-s epoch of REM sleep, or excessive tonic muscle activity in the chin EMG channel lasting over 15 s in duration [38].
Quantitative Methods of RSWA Analysis
In addition to AASM criteria, several manual and automated methods of RSWA quantification have been developed, resulting in definitive cutoff values for a RBD diagnosis [9, 51–53, 57–60]. Many of the manual methods employ variations of the AASM standard, including the percentage of 3-s mini-epochs containing phasic muscle activity, as well as the percentage of 30-s epochs containing tonic activity [53]. In addition, 3-s mini-epochs containing the presence of either scorable phasic or tonic activity have been defined as “any” muscle activity, resulting in percentage of “any” muscle activity [61]. Various phasic, tonic, and “any” percent muscle activity cutoff values have been established for a diagnosis of RBD with 100 % specificity. For the common montage of mentalis and muscles in the submentalis region combined with anterior tibialis muscles, cutoffs of 46.4 % and 44.2 % have been reported for “any” and phasic percent muscle activity. In addition, a submentalis tonic percent muscle activity cutoff of 9.6 % has been reported [61].
Recently, similar methods have been extended to define appropriate density cutoffs for RBD patients with comorbid obstructive sleep apnea, as is commonly seen in clinical populations. Diagnostic RSWA percent muscle activity (phasic, “any”) cutoffs were as follows: submentalis (SM) (15.5, 21.6 %); anterior tibialis (AT) (30.2, 30.2 %); and combined SM/AT (37.9, 43.4 %) [54]. A tonic percent muscle activity cutoff of 1.2 % was 100 % sensitive and specific, while an automated chin REM atonia index of 0.88 is appropriate in this patient population [54]. This manual methods also showed that in addition to standardly determined RSWA percent muscle activity measures, RBD patients have longer phasic muscle burst duration when compared with controls, and that measurement and consideration of phasic muscle burst durations further increases the specificity and sensitivity for diagnosis of RBD when combined with cutoff values for phasic and “any” percent muscle activity [54]. However, whether phasic muscle burst duration measurement may identify which patients with “incidental” RSWA are at greater risk for the development of RBD and future synucleinopathy is yet to be determined [54].
In addition to manual scoring methods, automated methods of chin RSWA analysis have been developed. The REM atonia index (RAI) is a computer generated value from 0 to 1, with a score of 0 indicating absolute absence of REM muscle atonia, and a score of 1 indicating completely preserved REM muscle atonia [58, 62, 63]. RAI of <0.9 has been found to be highly sensitive and specific for a diagnosis of RBD [54, 62]. Another automated method of chin tone analysis found that the number of both short duration (<0.2 s) and long duration (>0.2 s) muscle bursts was significantly greater in RBD patients than controls [60].
While both automated and manual methods of RSWA analysis have been shown to be accurate in the diagnosis of RBD, both have serious limitations. Manual methods are extremely time intensive, and many clinical sleep laboratories do not use expanded recording montages used to determine RSWA cutoff values for RBD diagnosis in some studies. Automated methods, while less time and labor intensive, have yet to be developed for limb muscles, limiting application toward RBD diagnosis in patients with RSWA primarily limited to the limb muscles [64].
Epidemiology
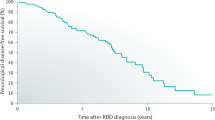
RBD most commonly presents after the age of 50, with 70–90 % of RBD cases occurring in men [34, 37, 65–68]. The prevalence of RBD has traditionally been estimated to be between 0.38 and 0.5 % in large population-based studies, [4, 11] but RBD patients represent up to 4.8 % of patients presenting to sleep disorders clinics, with the most common complaint being self or bedpartner injury due to dream enactment behaviors [67, 69]. More recent data suggest that pRBD (based on the MSQ) may be more common than previously believed, occurring in over 6 % in a community-dwelling subjects aged 70–89 years [70], and a very recent study from a large Korean cohort demonstrated a frequency of RBD of 4.8 % [71]. In addition, data on over 19,000 individuals found that violent behaviors during sleep are present in 1.6 % of the general population and occur more often in male individuals [11]. However, violent behaviors were also associated with NREM parasomnias such as sleep terrors, sleep walking, and confusional arousals, so not all violent behaviors during sleep can be attributed to RBD [11]. While the majority of RBD patients are older men, DEB can begin at any age, and may, in fact, occur as commonly in women as men, although RBD may be under diagnosed in women due to less violent dream enactment behaviors [72–74]. In patients with RBD diagnosed before the age of 50, and in those with comorbid narcolepsy, prevalence is nearly equal between genders [28, 75]. In younger patients with RBD, the most frequent causes are antidepressant use, drug or alcohol withdrawal, underlying narcolepsy, brainstem lesions, or congenital or neurodevelopmental disorders [28–30, 72–74]. In addition, evidence for coexistent autoimmunity is found in 20 % of women with RBD [28, 29]. Psychiatric diagnosis and antidepressant usage have also been reported to increase the likelihood of RBD as much as 10-fold [30]. In addition, limited evidence suggests that there may be a genetic association of RBD with human leukocyte antigen DQw1 [76]. However, this link has not been fully explored. RBD has also recently been shown to be four times more likely in primary relatives (i.e., parents or siblings) of patients with iRBD when compared to primary relatives of individuals without RBD suggesting that there may be familial link in RBD [65]. In addition, history of head injury, smoking, pesticide exposure, lower level of education, and occupation as a farmer have been reported as potential risk factors for RBD. However, further research in this area is needed [77] (Fig. 49.1).

Muscle tone during REM sleep in a normal subject, and in patients with REM sleep behavior disorder. The figure shows representative examples of 30 s polysomnogram epochs recorded during REM sleep. Muscle tone in the chin (submentalis), legs (linked tibialis), and arms (linked extensor digitorum communis) are shown in the sixth, seventh, and eighth channels of each example epoch, respectively. a demonstrates a normal level of REM sleep muscle atonia, while b–d demonstrate clear excessive phasic and tonic muscle tone, b shows predominantly excessive phasic muscle activity, especially in the leg, where there is also subtly increased tonic muscle activity. c demonstrates predominate excess phasic burst activity in the chin, arm, and leg muscle channels, accompanied by probable dream enactment manifested by vocalization; d shows excessive phasic and tonic muscle activity in the chin and leg muscle channels. Each of the examples seen in b–d is consistent with REM sleep without atonia, the neurophysiologic substrate of REM sleep behavior disorder
The Association of RBD with Neurodegenerative Synucleinopathy Disorders
RBD can result from several synucleinopathy phenotypes, as shown in Table 49.1. Both idiopathic and symptomatic RBD have been shown to be especially strongly associated with the synucleinopathies, a group of neurodegenerative disorders of unknown etiology related to intracellular accumulation of the protein alpha-synuclein, including Parkinson disease (PD), Lewy body dementia (DLB), multiple system atrophy (MSA), and pure autonomic failure (PAF) [5, 8, 12, 33, 37, 38, 67, 78]. Mild cognitive impairment (MCI), especially the non-amnestic subtype, has also been associated with RBD [5]. RBD is strongly associated with parkinsonism and/or cognitive impairment, which affects more than 82 % of RBD patients [36, 79]. Idiopathic RBD patients have an approximate 12–45 % risk of developing parkinsonism or dementia within five years of RBD symptom onset, which rises to 60 % within fifteen years, and to approximately 84 % risk 20 years after RBD symptom onset [5, 8, 79, 80].
There is growing evidence in iRBD patients of frequent presence of “soft,” subtle neurological and neuropsychologic abnormalities (also known as non motor signs) consistent with the early stages of a developing synucleinopathy [5, 81–83]. Patients with iRBD show abnormalities in smell and color vision, similar to that seen in patients with PD [81–84]. Some useful batteries for assessment of vision and olfactory function include the Farnsworth-Munsell-100-Hue test (FM 100) for color vision, and the University of Pennsylvania Smell Identification test (UPSIT) and Brief University of Pennsylvania Smell Identification test (B-SIT) to test olfaction [81, 83]. These clinical assessments may help clinicians clarify which RBD patients are most likely to develop parkinsonism, and poor performance on the FM-100 and UPSIT tests appears to be good predictors for the development of impending neurodegenerative disease in patients with iRBD [81, 82, 84]. RBD patients without impaired olfaction have an 86 % chance of remaining disease free for 5 years, contrasting with a 35.4 % chance in those with abnormal olfaction [12]. Abnormal olfaction in RBD appears similar to olfactory deficits in previously diagnosed PD patients, indicating that progressive neurodegeneration in RBD leads to PD [81, 83]. In addition, RBD patients with impaired color vision have an 18–26 % chance of 5-year disease-free survival [12, 83].
Patients with idiopathic RBD and PD with RBD have difficulties with object identification compared to controls and patients with PD lacking RBD [82]. Neuropsychological performance can also be used as a potential predictor of progressive neurodegeneration in iRBD patients [78, 85]. Impairments in attention, executive function, decision-making abilities, verbal memory, and verbal learning are most common in RBD patients, similar to deficits seen in synucleinopathies [86, 87]. Because iRBD is likely an early manifestation of synucleinopathy, serial neuropsychometric testing may help determine the timeline and progression of neurodegeneration in iRBD patients. Similarly, quantitative electroencephalography (EEG) has demonstrated temporo-occipital slowing in patients with iRBD, with higher theta power similar to that seen in patients with DLB and PDD [86, 88]. Of note, mild cognitive impairment appears to be the driver of EEG slowing in iRBD patients and likely heralds impending MCI [89, 90].
Cognitive impairment in iRBD likely varies due to the time point at which patients are sampled. Patients with iRBD are a heterogeneous group and are considered “idiopathic” because they do not yet express clinical symptoms of neurodegenerative disease, but that does not mean patients in the reported studies are at the same point in the disease progression. Because we do not sample patients at a uniform time point in their supposed disease process, and we do not fully understand why patients progress at different rates, it is difficult to draw conclusions about accompanying cognitive, motor, and autonomic dysfunction that would apply to all iRBD patients [91]. RBD patients that have progressed further toward overt clinical manifestations of neurodegenerative disease would very likely have greater impairments in cognition; therefore, differences seen between controls and iRBD patients may be driven by those patients closer to the clinical expressions of synucleinopathies. To our knowledge, no studies have yet examined cognition in the very early stages of dream enactment behavior with subsequent longitudinal, serial follow-up to determine whether or not there is a discernible common pattern of temporal progression of cognitive decline in iRBD. Future studies examining early RBD are of significant importance in the hopes of better understanding the underlying neurodegenerative process in iRBD.
Autonomic dysfunction may be the earliest predictor of impending neurodegeneration in iRBD. Orthostatic blood pressure drop may present as early as 20 years prior to development of overt neurological signs of synucleinopathy [92]. Urinary incontinence (13 years), constipation (15 years), and erectile dysfunction (11.5 years) also begin long before other neurological symptoms [92]. In addition, there is increased cardiac denervation in patients with iRBD and PD, further indicating that RBD may be an early expression of PD in many cases. However, the amount of cardiac denervation does not appear predictive of neurodegenerative disease development [84, 93, 94]. Reduced 123I-MIBG uptake may begin as early as Braak Stage 1, indicating that cardiac scintigraphy may be useful in early identification of PD development. Since cardiac denervation is present in the earliest stages of PD and RBD, but the amount of denervation does not predict neurodegeneration, RBD may represent early PD in a subset of patients [94]. Notably, while 123I-MIBG scintigraphy may be significantly decreased in RBD and PD, iRBD patients have been shown to have even greater reduction of MIBG uptake when compared to PD patients without RBD and neurologically normal controls, possibly indicating that PD-RBD may be a specific PD phenotype [95, 96]. Recently, plasma urate levels have also been shown to be a possible predictive factor of synucleinopathy neurodegeneration, with higher uric acid levels reportedly associated with a decreased risk for PD development [97] and a longer duration of RBD without conversion to PD, suggesting that plasma urate may modulate the progression of neurodegeneration in RBD. However, significantly more research is required to draw definitive conclusions [97].
Imaging in RBD
Similarly, advances in objective neuroimaging tests have begun to allow the non-invasive identification of patients at risk of development of a neurodegenerative disorder [5, 7, 98]. These tests include dopamine transporter uptake 123I-FP-CIT SPECT (DaTscan) and transcranial sonography (TCS), which have shown to be effective in detecting subclinical changes in the substantia nigra that may reflect evolving synucleinopathy; and 99mTc–ethylene cysteinate dimer SPECT (ECD-SPECT), a measure of regional cerberal blood flow (rCBF) which has been shown to predict future development of PD and DLB [98]. These tests may be useful in the identification of patients who could participate in future clinical trials of neuroprotective therapies designed to slow progression or arrest neurodegeneration in patients with iRBD [7]. Abnormalities on TCS (i.e., substantia nigra hyperechogenicity) and DaTscan (i.e., 123I-FP-CIT binding deficiencies in the striatum) in iRBD patients appear predictive of those who subsequently develop more classic features of a synucleinopathy, whereas patients with no abnormalities on neuroimaging remained disease free during follow-up [7]. The sensitivity of combined TCS and DaTscan scans was 100 % for predicting development of neurodegenerative disease within 2.5 years, so DaTscan and TCS may be useful in diagnosing neurodegenerative disease before clinical symptoms of parkinsonism, cognitive decline, or dysautonomia appear [7].
ECD-SPECT scan showed increased hippocampal rCBF in all iRBD patients who developed PD or DLB when compared to iRBD patients who remained disease free [98]. Notably, patients who developed PD displayed increased perfusion in the hippocampus, pons, and anterior cingulate cortex, while patients who developed DLB had increased rCBF confined to the hippocampus. Increased rCBF was also associated with several clinical markers of neurodegeneration. UPDRS-III was positively correlated with rCBF in the right hippocampus, and olfaction was correlated with increased rCBF in the medial frontal gyrus, while impairments in color vision were correlated with increased perfusion in the pons and left hippocampus [98].
Cortical afferent inhibition during transcranial magnetic stimulation (TMS) study was shown to be decreased in iRBD patients when compared to controls and correlates strongly with poor performance in episodic verbal memory and executive function tasks, suggesting that cholinergic dysfunction may underlie cognitive impairment in iRBD [99]. In addition, voxel-based MRI morphometry (VBM) has recently shown gray matter loss in the left and right anterior cerebellum, pontine tegmentum, and left parahippocampal gyrus in iRBD patients, a pattern similar to neurodegeneration in DLB and MSA [100]. These findings indicate the utility of SPECT, TCS, TMS, and MRI scans to predict future neurodegeneration, and possibly synucleinopathy subtype in patients with iRBD, which may enable more accurate prognostication and individualized diagnostic and therapeutic approaches.
Finally, the degree of RSWA appears predictive for future PD development. Patients with a more significant degree of elevation in baseline mentalis tonic density were more likely to develop PD in one study [33]. RSWA increases over time in patients with iRBD, suggesting a progressive neurodegenerative process leading to the destruction of brainstem regions responsible for the control of REM sleep toward clinical manifestations of parkinsonism and dementia [101, 102]. However, RBD symptom duration may not be the correct measure to determine group comparability, and patients with higher RSWA densities may have been studied further along in their disease course toward the eventual expression of PD, so additional prospective studies are necessary to determine whether the degree of RSWA is a marker for progression to PD.
RBD and RSWA Association with Specific Synucleinopathy Disorders: Parkinson Disease, Lewy Body Dementia, and Multiple System Atrophy
RBD and Parkinson Disease
RBD is common in Parkinson disease (PD) with as many as 55 % of PD patients reporting RBD symptoms [12, 32, 103–105]. RBD symptoms can develop prior to the development of clinical PD symptoms or during any of the six stages of PD [67]. Depression has been shown to be a risk factor for future development of PD [106]. In addition, depression and antidepressant use have also been associated with RBD [28, 30, 51, 107]. PD patients with RBD are more likely to have a comorbid psychiatric disorder compared to PD patients without RBD, and iRBD patients with comorbid depression have an increased risk for development of PD compared to RBD patients without comorbid depression [96, 108].
There is some evidence that PD-RBD may be a specific PD subtype. PD patients with RBD are less likely to be of the tremor predominant phenotype, less levodopa responsive, have higher Hoehn–Yahr scores, more frequent hallucinations, a greater degree of autonomic dysfunction, and most important, an increased risk for dementia [95, 96, 105, 109]. PD-RBD patients have a 45 % risk of development of dementia within four years compared to a 0 % risk in PD patients without RBD [95]. Of note, only PD patients with clinical RBD symptoms were at an increased risk for future development of dementia, when compared with PD patients with RSWA on PSG but no history of dream enactment and PD patients with normal REM sleep [109]. This suggests that PD-RBD may be a more widespread disease process compared to PD patients without RBD.
PD-RBD patients may have a gender disparity for the type of dream enactment behaviors similar to that seen in iRBD [110]. Men with PD-RBD report more aggressive and violent dreams with a higher percentage of violent movements during sleep. However, women with PD-RBD report more disturbed sleep with less violent movements during sleep [110]. Clinical RBD symptoms may remit in PD patients and appear less injurious when compared with iRBD patients [41, 111, 112], possibly due to decreased functionality as PD progresses. However, motor activity appears improved during dream enactment behavior compared with awake motor activty, so decreased functioning may not be fully responsible for clinical RBD remission and the lesser injury potential due to RBD behaviors in PD patients [113, 114]. The smoothing of movements during dream enactment represents sleep-related intact corticofugal and corticospinal neural circuitry that bypasses the basal ganglia, where functioning is impaired by synucleinopathy during wakefulness [114].
RBD and Lewy Body Dementia
Cognitive impairment is frequent in RBD and many patients develop dementia, primarily DLB or PDD [67, 84, 115–117]. While DLB and PDD are similar, important differences exist in the time course for the development of cognitive symptoms relative to development of motor symptoms. If motor symptoms are present for >1 year before cognitive decline, patients are considered to have PDD. If motor symptoms are present for <1 year prior to cognitive decline, or if they develop anytime after the onset of cognitive decline, patients have DLB [118]. Dream enactment is common in DLB, and PSG-confirmed RBD is present in as many as 83 % of DLB patients [119]. In fact, DLB and RBD are so closely associated that including RBD as a core feature of DLB increases the likelihood of DLB diagnosis sixfold [117]. Visuospatial impairment is one of the hallmarks of DLB, but can also be present in PDD. In addition, executive functioning is also affected in patients with DLB and PDD [84, 115, 116]. Idiopathic RBD patients show visuospatial and visuoconstructive dysfunction similar to patients with DLB, indicating that RBD may be a predictor for the development of dementia [82, 84]. In addition, many RBD patients eventually express clinical and/or pathologic features of Lewy body dementia/disease [5, 78, 107, 116, 117, 120, 121]. DLB patients with RBD have a shorter duration of dementia, earlier onset of parkinsonism and visual hallucinations, and lower neuritic plaque scores than DLB patients without RBD, indicating that DLB-RBD is a specific subtype of DLB which may have more widespread neurodegeneration than DLB without RBD [121]. Further insights clarifying the association between dementia and RBD, especially determining which patients are at highest risk for deteriorating cognitive functioning, are needed.
RBD and Multiple System Atrophy
Of the synucleinopathies, multiple system atrophy (MSA) appears to be the most strongly associated with RBD, with 68–100 % of patients having RBD or RSWA [85, 122, 113, 123]. Why MSA is more strongly associated with RBD than PD or DLB remains unclear. RBD symptoms in MSA appear to be less severe, even though RSWA is increased, when compared with PD-RBD and iRBD patients, a possible consequence of the more widespread neurodegeneration in MSA and its impact on functionality [124]. A diagnosis of RBD may help differentiate MSA from pure autonomic failure (PAF); in one case series, four patients with MSA had dream enactment behaviors, while all six patients diagnosed with PAF had no sleep disturbances [123]. However, other studies have shown RBD also occurs in PAF, indicating that further research is required on the association of PAF with RBD [122]. RBD has been found to be equally common in both MSA-P and MSA-C subtypes, suggesting that RBD is not a reliable finding to differentiate between subtypes of MSA [125].
RBD and Tauopathies
There have been reported cases of RBD in Alzheimer’s disease, but the pervading theory is that concomitant Lewy body pathology instead may more likely explain the RBD symptoms [126]. REM sleep without atonia and RBD have been variably reported in patients with progressive supranuclear palsy (PSP) and rarely in patients with corticobasal degeneration [127–130]. Evidence is contradictory on whether the amount of RSWA in PSP is similar to the amount seen in Parkinson disease [128, 130, 131]. Regardless, RBD rarely presents years in advance of PSP features, instead tending to occur concurrently with the motor dysfunction [5, 126, 130]. RBD has not been found in other primary tauopathies. Yet rare examples of autopsy-proven Alzheimer’s disease and PSP with RBD have been documented [5]. While it is possible that RBD can occur with a variety of the neurodegenerative proteinopathies, the frequency of RBD associated with synucleinopathies is far greater than in non-synucleinopathy disorders [5].
Narcolepsy and RBD
Since narcolepsy is a boundary state disorder, RSWA and RBD frequently co-occur in up to 60 % of cases [57]. Unlike RBD associated with synucleinopathy neurodegenerative disorders, RBD occurs equally in female as in male narcolepsy patients and has less complex and violent dream enactment behaviors which begin at an earlier age [57, 132]. RSWA without dream enactment occurs more frequently in patients with narcolepsy–cataplexy compared to both idiopathic hypersomnia patients and neurologically normal controls, correlating with the degree of hypocretin cell loss [57]. RSWA may be a useful diagnostic marker when unsure of a diagnosis between narcolepsy and idiopathic hypersomnia [133]. Antidepressants or stimulants may induce clinical RBD symptoms in narcolepsy patients, so sleep neurology clinicians must regularly inquire about dream enactment symptom occurrence and frequency in narcolepsy patients during follow-up care.
RBD Associated with Brainstem Lesions and Autoimmunity
Lesions of the brain stem caused by tumors, paraneoplastic, and autoimmune neurological disorders such as anti-Ma2-associated encephalitis or Morvan syndrome have been associated with clinical RBD symptoms and RSWA [13, 15, 16, 18, 19, 22–25, 128, 134, 135]. In addition, autoimmune disorders have been reported to occur in up to 20 % of women with RBD [29]. Brain stem lesions in patients with subsequent development of RBD symptoms have been helpful in localizing the structures responsible for control of REM sleep muscle tone in humans. One recent case identified RBD due to a discrete lesion in the right dorsomedial pons, the presumed location of the human sublateral dorsal nucleus/subcoeruleus that is believed to play a major role in generation of REM sleep muscle atonia [136]. Other lesional cases of RBD have also impugned the dorsal pontine tegmentum [26, 135]. Further case reports of brain stem lesions may allow insights into RBD pathophysiology. RBD or RSWA has also been found in up to 13 % of patients following traumatic brain injuries, indicating damage to brainstem structures responsible for REM sleep muscle control [137].
RBD Associated with Psychiatric Disorders and Antidepressants
There is growing evidence that RBD is common in psychiatric populations [28, 30, 51, 138]. Depression and antidepressant use are common, with as many as 60 % of RBD patients using antidepressant medications [30, 66]. In addition, RBD patients report significantly greater feelings of anxiety and depression when compared with matched control patients [107]. However, it remains unclear whether psychiatric RBD is a specific RBD subtype with a different pathophysiologic mechanism or simply an early manifestation of synucleinopathy neurodegeneration unveiled by antidepressant medications. A recent longitudinal study comparing iRBD patients taking antidepressant medications compared with antidepressant naïve iRBD patients showed a decreased rate of conversion to symptomatic RBD in the antidepressant users [139]. These findings, along with similar RSWA profiles between iRBD patients taking antidepressants and those not taking antidepressants, support the contention that psychiatric RBD is not a distinct RBD subtype, but simply iRBD that has been unveiled by antidepressant medication use [107, 139]. Notably, patients with depression have shown to be at an increased risk for developing PD, indicating that depression may be an early maker of PD [140, 141]. In addition, iRBD patients with depression appear to be at a significantly greater risk of future development of PD when compared to iRBD patients without depression [108]. PD patients with depression have been shown to have decreased norepinephrine binding in the locus coeruleus compared to PD patients without depression, providing evidence for a possible link between RBD and depression [142]. However, while there appears to be some relationship between psychiatric disease, RBD, and PD, idiopathic and symptomatic RBD patients have significantly greater tonic RSWA elevation when compared to patients with psychiatric RBD on antidepressant medications, indicating that there may be mechanistic differences between these RBD types [107]. Further research in this area is needed to understand these relationships.
Selective serotonin reuptake inhibitors (SSRI) and tricyclic antidepressants have been shown to exacerbate RBD symptoms and increase REM sleep muscle tone in patients without clinical dream enactment [29, 30, 138]. However, it remains unclear whether neurochemical effects mediated by antidepressants cause a reversible state of RSWA and RBD, or these drugs simply unveil RSWA and RBD in predisposed individuals [6, 29, 30]. Patients without a history of dream enactment on antidepressant medications have been reported to have increased RSWA when compared to normal volunteers not on antidepressants [107, 143]. In addition, limited evidence suggests that patients who previously received antidepressant medication use but who were not using antidepressants at the time of polysomnography had normal RSWA indices, suggesting that antidepressant medications do not cause a permanent alteration in REM sleep muscle tone [107]. Clinically, it is important to consider the impact of antidepressant effects in patients with RBD. Gradual cessation of SSRI medications or changing to an alternate antidepressant medication such as bupropion, which may have lesser propensity to aggravate RBD, may help decrease the frequency of dream enactment behaviors [144].
Pathophysiology of RBD
The mechanisms for RSWA and RBD remain poorly understood. Animal studies involving the rat and cat have informed much of our understanding of REM sleep atonia control. RBD-type behaviors were first demonstrated by selective lesioning of the cat locus coeruleus in 1965 [145]. Further study of sleep in the cat and rat has shown that the regulation of REM sleep is complex, and primarily located within the pons and medulla, especially the noradrenergic locus coeruleus (LC) and the cholinergic pedunculopontine nucleus (PPN) and laterodorsal tegmental nucleus (LDTN). Glutamatergic projections from the medullary magnocellular reticular formation (MCRF) also play a role in motor inhibition. In addition, the glycinergic/GABAeric nucleus raphe magnus, and the ventral and alpha gigantocellular nuclei have been implicated in the suppression of REM sleep muscle tone [146]. The hypothalamus, thalamus, substantia nigra, basal forebrain, and frontal cortex also participate in REM sleep regulation [145, 147].
Brainstem regions thought to be involved in the pathophysiology of RSWA and RBD are the MCRF, locus coeruleus/subcoeruleus (LC/SC) complex, PPN, LDTN, and the substantia nigra [145, 148]. Lesions in the LC/SC cause RSWA, and the site and severity of the lesion determines whether simple or complex behaviors result [3]. However, the rat sublateral dorsal nucleus (SLD), analogous to the cat SC, appears to have a vital role in the regulation of normal REM atonia, and when lesioned, mediating the pathophysiology of RSWA and RBD [149–153]. The SLD is located in the dorsal pontine tegmentum [152]. Lesions in the SLD result in RSWA in rats. The SLD contains primarily excitatory glutamatergic neurons with both caudal and rostral projections [153]. Rostral projections excite thalamocortical neurons, the basal forebrain, and lateral hypothalamus to generate REM sleep (EEG) [153]. Caudal projections to the ventromedial medulla (VMM), the premotor glycinergic/GABAergic neurons of the nucleus raphe magnus, and the ventral and lateral gigantocellular nuclei are responsible for activating hyperpolarization within spinal motorneurons and subsequent generation of REM sleep muscle atonia [153]. Rats also possess a “REM-on” and “REM-off” region [154]. The REM-on region is inhibited by GABAergic and galaninergic projections from the forebrain ventrolateral preoptic nucleus (VLPO), in addition to cholinergic projections from the PPN/LDTN which are active during wakefulness and non-REM sleep [153, 155]. The REM-off nuclei are activated by projections from the noradrenergic LC, serotonergic raphe nucleus (RN) and by hypocretinergic pathways from the lateral hypothalamus [3]. The REM-on nuclei in the SLD and the precoeruleus (PC) also interact with REM-off nuclei and are mutually inhibitory [154].
Recent research from a RBD transgenic mouse model has illustrated the importance of ionotropic glycine/GABAA, and metabotropic GABAB receptors for generation and maintenance of REM sleep muscle atonia [151]. Only simultaneous inhibition of both ionotropic and metabotropic receptors results in heightened REM sleep muscle tone similar to that of wakefulness [151]. Inhibition of only single neurotransmitter receptors results in incomplete muscle activation, and it is very likely that the large degree of variation of RSWA seen between different patients, and within the same patients on repeated studies over different nights, is a direct result of how many receptors are inhibited [151, 156]. While large lesions to the SLD result in RSWA, they also cause a large decrease in total REM sleep time [153, 154]. Therefore, because RBD patients often have a normal amount of REM sleep, it is unlikely that RBD and RSWA in iRBD result from lesions directly to the SLD [150, 153]. Two populations of SLD/SC neurons have been found in the cat brain; ascending glutamatergic projections responsible for generation of REM sleep EEG activity, and descending glutamatergic projections inducing REM sleep muscle atonia [153]. Possibly only the descending projections are disrupted in iRBD, resulting in RSWA and dream enactment, while the ascending projections are relatively spared [153]. Another possible hypothesis is that the selective degeneration of premotor REM-on GABA/glycinergic neurons of the nucleus raphe magnus and the ventral and alpha gigantocellular reticular nuclei occur in RBD as a result of alpha-synuclein aggregation, which is also in accordance with the Braak hypothesis [157]. Both noradrenergic and serotonergic influences also play a role in REM sleep muscle tone. Decreased serotonin and noradrenaline concentrations have been found in the nuclei of hypoglossal motor neurons, with a subsequent increase in REM muscle tone with direct injection of serotonin into these nuclei [153, 158]. On the other hand, noradrenaline does not appear to directly increase muscle tone, but instead amplifies glutamatergic driven muscle excitation [159]. Dream enactment behavior and phasic muscle twitches during REM sleep are hypothesized to result from direct or indirect glutamatergic inputs from the motor cortex projecting to spinal motor neurons [153]. The fact that movements during DEBs in patients with PD appear faster and less stereotyped than movements during wakefulness argues for direct activation of spinal motor neurons resulting from corticospinal and corticofugal projections bypassing the smoothing contributions of the basal ganglia during REM sleep [153, 160]. Ultimately, REM sleep muscle atonia likely results from a powerful combined glycinergic/GABAergic drive stimulated by glutamatergic projections from the SLD, with additional mediation by seratonergic and noradrengergic influences [153].
Since most of our understanding of REM sleep comes from animal models, there are very likely interspecies differences between humans and animals. However, lesional cases of RBD in humans also propose that human dorsomedial pontine structures analogous to the SLD and MCRF play similar roles to the animal models. Discrete dorsomedial pontine lesions in SLD-analogous structure lead to decreased motor inhibition, as well as decreased excitation of the MCRF, thereby potentially causing RBD symptoms [136]. It remains unclear whether lesions causing RBD in humans may involve the MCRF alone, or whether lesions must have additional involvement of the REM-off regions [154].
Because patients with narcolepsy frequently have RBD, the relationship between orexin-A and orexin-B (hypocretin-1 and hypocretin-2) and REM sleep muscle tone has also been examined. Decreased levels of CSF hypocretin-1 from damage to the lateral hypothalamus have been associated with a greater amount of muscle activation during REM and non-REM sleep in patients with narcolepsy with and without cataplexy [161]. Hypocretin has also been shown to play a role in REM sleep muscle activation by directly acting on LC neurons, in addition to REM sleep muscle tone suppression by action on the pontine inhibitory area [162, 163], suggesting that hypocretin likely plays a large role in the stabilization of REM sleep muscle control, and that disruptions in hypocretin expression, whether deficient or excessive, could result in RSWA [161, 162, 163]. Decreased hypocretin levels have been associated with RSWA in narcolepsy, while increased hypocretin levels have been associated with RSWA in PD [161, 164]. In addition, in a small case study of patients with idiopathic RBD, hypocretin levels were normal, so further research on the role of hypocretin in mediating RSWA and RBD is needed [165]. Dopamine may also play a stabilizing role in the control of REM sleep muscle tone, with both dopamine deficiency and excessive levels of dopamine resulting in increased REM muscle tone [166, 167]. Because RSWA is occasionally seen in PD patients without RBD, it is possible that dopaminergic dysfunction in these patients contributes to the RSWA without DEB [55]. Further exploration of this relationship is needed.
The Braak Staging Hypothesis and RBD
Because of the strong association of RBD with PD, the Braak staging hypothesis of PD may provide a possible explanation for the occurrence of RSWA and RBD symptoms in PD and perhaps also in DLB [3, 5]. Braak has postulated six stages of progressive and selective Lewy body deposition and Lewy neurite accumulation based on the clinical phenotype of PD, corroborated by pathologic examination [157]. Each successive stage has increased Lewy bodies and Lewy neurites compared to the previous stage. In addition, some structures are almost never affected in the six stages, while others are affected in most, indicating a selective vulnerability in the synucleinopathy neurodegeneration [168]. In stage 1, Lewy bodies and neurites begin to accumulate in the dorsal motor nucleus of the vagus in the medulla, while in stage 2, Lewy bodies and neurite progress rostrally through the magnocellularis reticular nucleus, subceruleus–ceruleus complex, olfactory bulb, and anterior olfactory nucleus. RBD symptoms are also considered to be part of stage 2, given common impairments in olfaction and cardiac denervation reported in idiopathic RBD patients [83]. Stages 1 and 2 are considered the “preclinical” phase of parkinsonism, preceding the evolution of motor or cognitive symptoms, in which RBD is the most distinguishing feature [168]. The parkinsonian stage is encompassed by stages 3 and 4 with the progression of Lewy deposits to the substantia nigra, the pedunculopontine nucleus, and the amygdala occurring in stage 3, while stage 4 results from progression of Lewy deposits into the temporal mesocortex. Widespread neurodegeneration occurs in stages 5 and 6, with Lewy deposits affecting the neocortex, causing cognitive impairment [157]. Further evidence for the relationship between RBD and Braak staging stems from reportedly impaired olfaction and dysautonomia in patients with idiopathic RBD, indicating that RBD may be a precursor of PD [81–83, 94, 103]. As PD progresses from stage 1 to stage 6, there is an increasing loss of hypocretin cells, possibly supporting the hypothesis that hypocretin deficiency either plays a role in RBD pathology, or occurs concurrently and causes other PD-associated features [169]. However, a recent small case series reported increased levels of RSWA associated with increased hypocretin, so this relationship remains incompletely understood [164]. In addition, Braak staging does not well explain why some Lewy body disease patients never display RBD symptomatology, nor why some RBD patients never develop PD.
Treatment of RBD
Suppression of nightmares and prevention of patient and bed partner from injuries are the main goals of RBD treatment. Prediction of injury occurrence in RBD is very difficult. Patients with idiopathic RBD and those who remember their dreams have been recently found to be at significantly greater risk of injury [41]. However, frequency of dream enactment behaviors has not been shown to be predictive of possible injury, highlighting the importance of RBD treatment, even in patients with infrequent episodes of dream enactment [41]. All dangerous objects should be removed from the bedroom. Moving furniture away from the bed, padding the corners of bedside tables, and placing a mattress next to the bed may help prevent injury if the patient falls or leaves the bed during dream enactment [44]. If injury to the bed partner occurs, sleeping in separate beds or separate rooms minimizes risk of injury. Bed rails or barriers between the patient and bed partner may also be utilized, and bed alarms may be useful if the bedpartner has had to move to another room. A novel bed alarm designed to provide RBD patients with a calming instruction to return to sleep has been effective in reducing DEB in medication refractory patients [170]. The alarm system includes a pressure sensitive pad underneath the shoulders of the patient, as well as a clothing tether that is attached magnetically to the alarm system that detaches from the alarm system, activating the alarm and waking the patient from sleep, thereby averting the RBD episode [170]. The device is most effective in patients with a history of leaving the bed during dream enactment behavior. Confusional arousals resulting from obstructive sleep apnea, a common comorbidity in RBD patients, may mimic RBD episodes [171]. Treatment of apnea with continuous positive airway pressure may result in improvement in the frequency and severity of these episodes; however, treatment of OSA has not been shown to decrease RSWA, indicating there may be no direct effect of treatment of comorbid OSA on the pathophysiology of RBD [54].
Clonazepam and Other GABA Agonists
Melatonin and clonazepam are considered to be the two mainstays of pharmacologic treatment for RBD [32, 66, 68, 172, 173]. Melatonin and clonazepam appear to be equally effective in reducing frequency and severity of DEB, but only melatonin has been reported to significantly reduce injuries, with fewer side effects than clonazepam [66]. The GABAA receptor modulator clonazepam was the initial treatment reported as effective for decreasing RBD symptoms, with a median effective dose of 0.5 mg and a range of 0.25–2.0 mg given approximately 30 min before bedtime [8, 28, 59, 66–68, 75, 108]. Of note, while patients on clonazepam have decreased motor activity during REM, REM sleep muscle atonia is not restored, indicating that clonazepam may act on the motor cortex to inhibit glutamate induced phasic muscle bursts, but not on regions responsible for control of REM sleep muscle atonia [153, 174–176]. While clonazepam effectively reduces RBD symptoms, it can also increase the severity of obstructive sleep apnea and cognitive impairment. Additional side effects include sleepiness, dizziness, unsteadiness, and sexual dysfunction [32, 66, 176]. Therefore, patients with cognitive impairment, dementia, and sleep apnea must be monitored carefully when treated with clonazepam. While most patients with RBD associated with neurodegenerative disorders tolerate clonazepam well and benefit from its use, melatonin has been shown to be more effective in treating this population with fewer side effects [66]. Other GABA receptor modulators may also be useful in treating RBD; however, very little research on the effectiveness of these drugs on RBD symptoms exists [177]. Zopiclone has been reported to be effective in reducing RBD symptoms in a small number of patients [35, 177]. Zopiclone is a selective GABA α-1 and α-5 agonist with a short half-life and may impact the pathophysiology of RBD and reduce REM muscle tone by selectively acting on GABAA receptors [35]. However, further research is needed to validate this hypothesis.
Melatonin
Melatonin has recently been shown to be equally effective for RBD treatment, with fewer side effects than clonazepam [32, 66, 172, 173]. Adverse effects are dose related and primarily include daytime sleepiness; however, headache and hallucinations have been reported [32, 66, 178]. Melatonin may be a more efficacious treatment for patients with symptomatic RBD due to its more tolerable side effect profile [66]. The median effective dose of melatonin has been reported to be 6 mg at bedtime [66]. However, ranges of 3–25 mg have also been reported as tolerable and effective [66]. Melatonin has been shown to increase REM sleep atonia during treatment; however, the mechanism by which this occurs remains unknown [35, 172, 173].
Other Potential Treatments of RBD
Other treatments of RBD have been limited to case reports and small case series. The association between RBD and PD has prompted interest in the role of dopamine in the pathophysiology of RBD. While levodopa decreases the amount of REM sleep time, possibly providing less opportunity for dream enactment behaviors, high doses of levodopa have been shown to increase both REM muscle tone and hallucinations which may be mistaken for RBD [166, 179]. Pramipexole has shown mixed results, varying from little or no effect, to reduction of RBD episodes [180–182]. In a recent large series, 62 % of patients treated with pramipexole monotherapy reported improvement in RBD symptoms, compared to 88 % improvement reported by patients treated with clonazepam, suggesting that pramipexole is likely not a suitable first-line therapy for RBD [183]. In this study, pramipexole was more effective in treating patients with lower amounts of RSWA compared to patients with significant amounts of RSWA, indicating that dopaminergic agents may help stabilize control of REM muscle tone early in the early course of PD [183]. Zonisamide, which is used to treat motor symptoms of PD, has been reported to decrease DEB in one PD patient [184]. Zonisamide has been shown to increase GABA-ergic neurotransmission and decrease glutamate in the rat brain, indicating that zonisamide could influence RBD symptoms both by inhibition of glutamatergic phasic muscle bursts as well as through GABA induced hyperpolarization of motor neurons [185, 186]. Given the high frequency of RBD in PD, further research on the effects of zonisamide on RBD symptoms is warranted. If efficacious in treatment of RBD symptoms, zonisamide may allow for the use of one treatment for both motor dysfunction and RBD symptoms, eliminating the need for polytherapy in a cognitively vulnerable patient population that may be impacted more severely by polypharmacy. Donepezil has also rarely been reported to reduce motor events related to RBD [187, 188]. Agomelatine is a novel antidepressant with a melatoninergic mechanism that has been reported to effectively decrease RBD symptoms with minimal side effects [189]. In addition, RSWA decreased in patients taking agomelatine, suggesting a relatively potent melatoninergic effect [189]. Because agomelatine has also shown similar efficacy to other antidepressant medications with fewer side effects, agomelatine may be considered a very useful medication in RBD patients with depression [190]. In addition, the herbal supplement Yi-Gan San has been shown to suppress RBD symptoms in three patients, possibly by acting on serotonergic and GABAergic systems [191]. While clonazepam and melatonin are effective in reducing RBD symptoms, only rarely do these or other treatments completely eliminate potentially injurious dream enactment behaviors. Therefore, future large, prospective comparative treatment trials are needed to develop better and more tolerable treatments that completely eliminate injury potential (Table 49.2).
Conclusions
RBD is a potentially injurious parasomnia that can reflect an early manifestation of synucleinopathy neurodegenerative disorders in many individuals, especially Parkinson disease, Lewy body dementia, and multiple system atrophy. Injuries are frequent to both patients and bedpartners, occurring in at least 50 % of cases, and not infrequently, injuries are serious enough to require medical intervention [1, 66, 67]. Currently, addressing bedroom safety in addition to pharmacologic treatment is the most effective form of therapy. A 6 mg bedtime dose of melatonin, with its relatively benign side effect profile and comparable efficacy to clonazepam, may be considered the first-line treatment of RBD, especially in patients with neurodegenerative disorders. Unfortunately, no neuroprotective therapies to prevent the progression of RBD to other clinical manifestations of neurodegenerative disorders exist at this time.
Early identification of patients with RBD could enable entry of individuals vulnerable to developing future synucleinopathy neurodegenerative disorders into clinical trials of neuroprotective therapies, with the goal of preventing further neurodegeneration and arresting its course at a time point before clinically significant motor or cognitive sequelae unfold, or at least delaying the onset of these features [192]. Symptomatic treatment of RBD is focused on injury prevention by advising environmental measures to assure bedroom safety, and reduction of the frequency and severity of RBD episodes through administration of pharmacotherapy with clonazepam or melatonin. Future research of RSWA and RBD is necessary to determine sensitivity and specificity as biomarkers for conversion to overt synucleinopathy, to more effectively manage dream enactment so as to prevent injury, and to better understand the time course and causes of progressive neurodegeneration to inform design of future neuroprotective treatment trials.
References
Schenck CH, Bundlie SR, Ettinger MG, Mahowald MW (1986) Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep 9(2):293–308
Boeve BF (2010) REM sleep behavior disorder: updated review of the core features, the REM sleep behavior disorder-neurodegenerative disease association, evolving concepts, controversies, and future directions. Ann N Y Acad Sci 1184:15–54
Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE et al (2007) Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 130 (Pt 11):2770–2788
Chiu HF, Wing YK, Lam LC, Li SW, Lum CM, Leung T et al (2000) Sleep-related injury in the elderly—an epidemiological study in Hong Kong. Sleep 23(4):513–517
Claassen DO, Josephs KA, Ahlskog JE, Silber MH, Tippmann-Peikert M, Boeve BF (2010) REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology. 75(6):494–499
Frauscher B, Gschliesser V, Brandauer E, Marti I, Furtner MT, Ulmer H et al (2010) REM sleep behavior disorder in 703 sleep-disorder patients: the importance of eliciting a comprehensive sleep history. Sleep Med 11(2):167–171
Iranzo A, Lomena F, Stockner H, Valldeoriola F, Vilaseca I, Salamero M et al (2010) Decreased striatal dopamine transporter uptake and substantia nigra hyperechogenicity as risk markers of synucleinopathy in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a prospective study [corrected]. Lancet Neurol 9(11):1070–1077
Iranzo A, Molinuevo JL, Santamaria J, Serradell M, Marti MJ, Valldeoriola F et al (2006) Rapid-eye-movement sleep behaviour disorder as an early marker for a neurodegenerative disorder: a descriptive study. Lancet Neurol 5(7):572–577
Montplaisir J, Gagnon JF, Fantini ML, Postuma RB, Dauvilliers Y, Desautels A et al (2010) Polysomnographic diagnosis of idiopathic REM sleep behavior disorder. Mov Disord 25(13):2044–2051
Nightingale S, Orgill JC, Ebrahim IO, de Lacy SF, Agrawal S, Williams AJ (2005) The association between narcolepsy and REM behavior disorder (RBD). Sleep Med 6(3):253–258
Ohayon MM, Schenck CH (2010) Violent behavior during sleep: prevalence, comorbidity and consequences. Sleep Med 11(9):941–946
Sixel-Doring F, Trautmann E, Mollenhauer B, Trenkwalder C (2011) Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology 77(11):1048–1054
Cornelius JR, Pittock SJ, McKeon A, Lennon VA, Aston PA, Josephs KA et al (2011) Sleep manifestations of voltage-gated potassium channel complex autoimmunity. Arch Neurol 68(6):733–738
Dauvilliers Y, Rompre S, Gagnon JF, Vendette M, Petit D, Montplaisir J (2007) REM sleep characteristics in narcolepsy and REM sleep behavior disorder. Sleep 30(7):844–849
Iranzo A, Graus F, Clover L, Morera J, Bruna J, Vilar C et al (2006) Rapid eye movement sleep behavior disorder and potassium channel antibody-associated limbic encephalitis. Ann Neurol. 59(1):178–181
Iranzo A, Aparicio J (2009) A lesson from anatomy: focal brain lesions causing REM sleep behavior disorder. Sleep Med 10(1):9–12
Jianhua C, Xiuqin L, Quancai C, Heyang S, Yan H (2013) Rapid eye movement sleep behavior disorder in a patient with brainstem lymphoma. Intern Med 52(5):617–621
Lai YY, Hsieh KC, Nguyen D, Peever J, Siegel JM (2008) Neurotoxic lesions at the ventral mesopontine junction change sleep time and muscle activity during sleep: an animal model of motor disorders in sleep. Neuroscience 154(2):431–443
Limousin N, Dehais C, Gout O, Heran F, Oudiette D, Arnulf I (2009) A brainstem inflammatory lesion causing REM sleep behavior disorder and sleepwalking (parasomnia overlap disorder). Sleep Med 10(9):1059–1062
Lin FC, Liu CK, Hsu CY (2009) Rapid-eye-movement sleep behavior disorder secondary to acute aseptic limbic encephalitis. J Neurol 256(7):1174–1176
Luppi PH, Clement O, Sapin E, Gervasoni D, Peyron C, Leger L et al (2011) The neuronal network responsible for paradoxical sleep and its dysfunctions causing narcolepsy and rapid eye movement (REM) behavior disorder. Sleep Med Rev 15(3):153–163
Mathis J, Hess CW, Bassetti C (2007) Isolated mediotegmental lesion causing narcolepsy and rapid eye movement sleep behaviour disorder: a case evidencing a common pathway in narcolepsy and rapid eye movement sleep behaviour disorder. J Neurol Neurosurg Psychiatry 78(4):427–429
Plazzi G, Montagna P (2002) Remitting REM sleep behavior disorder as the initial sign of multiple sclerosis. Sleep Med 3(5):437–439
Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ (2008) Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 70(20):1883–1890
Tippmann-Peikert M, Boeve BF, Keegan BM (2006) REM sleep behavior disorder initiated by acute brainstem multiple sclerosis. Neurology 66(8):1277–1279
Xi Z, Luning W (2009) REM sleep behavior disorder in a patient with pontine stroke. Sleep Med 10(1):143–146
Vorona RD, Ware JC (2002) Exacerbation of REM sleep behavior disorder by chocolate ingestion: a case report. Sleep Med 3(4):365–367
Bonakis A, Howard RS, Ebrahim IO, Merritt S, Williams A (2009) REM sleep behaviour disorder (RBD) and its associations in young patients. Sleep Med 10(6):641–645
Ju YE, Larson-Prior L, Duntley S (2011) Changing demographics in REM sleep behavior disorder: possible effect of autoimmunity and antidepressants. Sleep Med 12(3):278–283
Teman PT, Tippmann-Peikert M, Silber MH, Slocumb NL, Auger RR (2009) Idiopathic rapid-eye-movement sleep disorder: associations with antidepressants, psychiatric diagnoses, and other factors, in relation to age of onset. Sleep Med 10(1):60–65
Suzuki K, Miyamoto T, Miyamoto M, Suzuki S, Watanabe Y, Takashima R, Hirata K (2013) Dream-enacting behaviour is associated with impaired sleep and severe headache-related disability in migraine patients. Cephalagia 33(10):868–878
Boeve BF, Silber MH, Ferman TJ (2003) Melatonin for treatment of REM sleep behavior disorder in neurologic disorders: results in 14 patients. Sleep Med 4(4):281–284
Postuma RB, Gagnon JF, Rompre S, Montplaisir JY (2010) Severity of REM atonia loss in idiopathic REM sleep behavior disorder predicts Parkinson disease. Neurology 74(3):239–244
Wing YK, Lam SP, Li SX, Yu MW, Fong SY, Tsoh JM et al (2008) REM sleep behaviour disorder in Hong Kong Chinese: clinical outcome and gender comparison. J Neurol Neurosurg Psychiatry 79(12):1415–1416
Anderson KN, Shneerson JM (2009) Drug treatment of REM sleep behavior disorder: the use of drug therapies other than clonazepam. J Clin Sleep Med 5(3):235–239
Schenck CH, Boeve BF, Mahowald MW (2013) Delayed emergence of a parkinsonian disorder or dementia in 81 % of older males initially diagnosed with idiopathic REM sleep behavior disorder (RBD): 16 year update on a previously reported series. Sleep Med 14(8):744–748.
Boeve BF, Silber MH, Ferman TJ, Lin SC, Benarroch EE, Schmeichel AM, et al (2013) Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med 14(8):754–762
Medicine AAoS (2014) International classification of sleep disorders, 2nd edition (2005): diagnostic and coding manual. American Academy of Sleep Medicine, West Chester, IL, USA
Medicine AAoS (2005) International classification of sleep disorders, 3rd edition (2014): American Academy of Sleep Medicine, Darien, IL, USA
Boeve BF, Molano JR, Ferman TJ, Smith GE, Lin SC, Bieniek K et al (2011) Validation of the Mayo Sleep Questionnaire to screen for REM sleep behavior disorder in an aging and dementia cohort. Sleep Med 12(5):445–453
Valli K, Frauscher B, Gschliesser V, Wolf E, Falkenstetter T, Schonwald SV et al (2012) Can observers link dream content to behaviours in rapid eye movement sleep behaviour disorder? A cross-sectional experimental pilot study. J Sleep Res 21(1):21–29
McCarter SJ, St. Louis EK, Boswell CL, Dueffert LG, Slocumb N, Boeve BF, et al (2014) Factors associated with injury in REM sleep behavior disorder. Sleep Med 15(11):1332–1338
Uguccioni G, Golmard JL, de Fontreaux AN, Leu-Semenescu S, Brion A, Arnulf I (2013) Fight or flight? Dream content during sleepwalking/sleep terrors vs. rapid eye movement sleep behavior disorder. Sleep Med 14(5):391–398
Li SX, Wing YK, Lam SP, Zhang J, Yu MW, Ho CK et al (2010) Validation of a new REM sleep behavior disorder questionnaire (RBDQ-HK). Sleep Med 11(1):43–48
McCarter SJ (2012) St Louis EK, Boeve BF. REM sleep behavior disorder and REM sleep without atonia as an early manifestation of degenerative neurological disease. Curr Neurol Neurosci Rep 12(2):182–192
Stiasny-Kolster K, Mayer G, Schafer S, Moller JC, Heinzel-Gutenbrunner M, Oertel WH (2007) The REM sleep behavior disorder screening questionnaire—a new diagnostic instrument. Mov Disord 22(16):2386–2393
Postuma RB, Arnulf I, Hogl B, Iranzo A, Miyamoto T, Dauvilliers Y et al (2012) A single-question screen for rapid eye movement sleep behavior disorder: a multicenter validation study. Mov Disord 27(7):913–916
Sasai T, Matsuura M, Wing YK, Inoue Y (2012) Validation of the Japanese version of the REM sleep behavior disorder questionnaire (RBDQ-JP). Sleep Med 13(7):913–918
Boeve BF, Molano JR, Ferman TJ, Lin SC, Bieniek K, Tippmann-Peikert M et al (2013) Validation of the Mayo Sleep Questionnaire to screen for REM sleep behavior disorder in a community-based sample. J Clin Sleep Med 9(5):475–480
Frauscher B, Ehrmann L, Zamarian L, Auer F, Mitterling T, Gabelia D et al (2012) Validation of the Innsbruck REM sleep behavior disorder inventory. Mov Disord 27(13):1673–1678
Husain AM, Miller PP, Carwile ST (2001) Rem sleep behavior disorder: potential relationship to post-traumatic stress disorder. J Clin Neurophysiol 18(2):148–157
Lam SP, Li SX, Chan JW, Mok V, Tsoh J, Chan A, et al (2012) Does REM sleep behavior disorder exist in psychiatric populations? A clinical and polysomnographic case-control study. Sleep Med 14(8):788–794
Frauscher B, Iranzo A, Hogl B, Casanova-Molla J, Salamero M, Gschliesser V et al (2008) Quantification of electromyographic activity during REM sleep in multiple muscles in REM sleep behavior disorder. Sleep 31(5):724–731
Iranzo A, Frauscher B, Santos H, Gschliesser V, Ratti L, Falkenstetter T et al (2011) Usefulness of the SINBAR electromyographic montage to detect the motor and vocal manifestations occurring in REM sleep behavior disorder. Sleep Med 12(3):284–288
McCarter S, St. Louis E, Duwell E, Timm P, Sandness D, Boeve B, et al (2014) Diagnostic thresholds for quantitative rem sleep phasic burst duration, phasic and tonic muscle activity, and rem atonia index in rem sleep behavior disorder with and without comorbid obstructive sleep apnea. Sleep 37(10):1649–1662
Gagnon JF, Bedard MA, Fantini ML, Petit D, Panisset M, Rompre S et al (2002) REM sleep behavior disorder and REM sleep without atonia in Parkinson’s disease. Neurology 59(4):585–589
Ferri R, Bruni O, Fulda S, Zucconi M, Plazzi G (2011) A quantitative analysis of the submentalis muscle electromyographic amplitude during rapid eye movement sleep across the lifespan. J Sleep Res 21(3):257–263
Dauvilliers Y, Jennum P, Plazzi G (2012) REM sleep behavior disorder and REM sleep without atonia in narcolepsy. Sleep Med 14(8):775–781
Ferri R, Rundo F, Manconi M, Plazzi G, Bruni O, Oldani A et al (2010) Improved computation of the atonia index in normal controls and patients with REM sleep behavior disorder. Sleep Med 11(9):947–949
Lapierre O, Montplaisir J (1992) Polysomnographic features of REM sleep behavior disorder: development of a scoring method. Neurology 42(7):1371–1374
Mayer G, Kesper K, Ploch T, Canisius S, Penzel T, Oertel W et al (2008) Quantification of tonic and phasic muscle activity in REM sleep behavior disorder. J Clin Neurophysiol 25(1):48–55
Frauscher B, Iranzo A, Gaig C, Gschliesser V, Guaita M, Raffelseder V et al (2012) Normative EMG values during REM sleep for the diagnosis of REM sleep behavior disorder. Sleep 35(6):835–847
Ferri R, Fulda S, Cosentino FI, Pizza F, Plazzi G (2012) A preliminary quantitative analysis of REM sleep chin EMG in Parkinson’s disease with or without REM sleep behavior disorder. Sleep Med 13(6):707–713
Ferri R, Manconi M, Plazzi G, Bruni O, Vandi S, Montagna P et al (2008) A quantitative statistical analysis of the submentalis muscle EMG amplitude during sleep in normal controls and patients with REM sleep behavior disorder. J Sleep Res 17(1):89–100
Frauscher B, Ehrmann L, Hogl B (2012) Defining muscle activities for assessment of REM sleep behavior disorder: from a qualitative to a quantitative diagnostic level. Sleep Med 14(8):729–733
Dauvilliers Y, Postuma RB, Ferini-Strambi L, Arnulf I, Hogl B, Manni R et al (2013) Family history of idiopathic REM behavior disorder: a multicenter case-control study. Neurology 80(24):2233–2235
McCarter SJ, Boswell CL, St Louis EK, Dueffert LG, Slocumb N, Boeve BF et al (2013) Treatment outcomes in REM sleep behavior disorder. Sleep Med 14(3):237–242
Olson EJ, Boeve BF, Silber MH (2000) Rapid eye movement sleep behaviour disorder: demographic, clinical and laboratory findings in 93 cases. Brain. 123(Pt 2):331–339
Schenck CH, Mahowald MW (1990) A polysomnographic, neurologic, psychiatric and clinical outcome report on 70 consecutive cases with REM sleep behavior disorder (RBD): sustained clonzepam efficacy in 89.5 % of 57 treated patients. Clev Clin J Med 57(Suppl):10–24
Schenck CH, Mahowald MW (2002) REM sleep behavior disorder: clinical, developmental, and neuroscience perspectives 16 years after its formal identification in SLEEP. Sleep 25(2):120–138
Boot BP, Boeve BF, Roberts RO, Ferman TJ, Geda YE, Pankratz VS et al (2012) Probable rapid eye movement sleep behavior disorder increases risk for mild cognitive impairment and Parkinson disease: a population-based study. Ann Neurol. 71(1):49–56
Kang SH, Yoon IY, Lee SD, Han JW, Kim TH, Kim KW (2013) REM sleep behavior disorder in the korean elderly population: prevalence and clinical characteristics. Sleep 36(8):1147–1152
Lloyd R, Tippmann-Peikert M, Slocumb N, Kotagal S (2012) Characteristics of REM sleep behavior disorder in childhood. J Clin Sleep Med 8(2):127–131
Sheldon SH, Jacobsen J (1998) REM-sleep motor disorder in children. J Child Neurol 13(6):257–260
Stores G (2008) Rapid eye movement sleep behaviour disorder in children and adolescents. Dev Med Child Neurol 50(10):728–732
Bodkin CL, Schenck CH (2009) Rapid eye movement sleep behavior disorder in women: relevance to general and specialty medical practice. J Womens Health (Larchmt). 18(12):1955–1963
Schenck CH, GarciaRill E, Segall M, Noreen H, Mahowald MW (1996) HLA class II genes associated with REM sleep behavior disorder. Ann Neurol. 39(2):261–263
Postuma RB, Montplaisir JY, Pelletier A, Dauvilliers Y, Oertel W, Iranzo A et al (2012) Environmental risk factors for REM sleep behavior disorder: a multicenter case-control study. Neurology 79(5):428–434
Fantini ML, Farini E, Ortelli P, Zucconi M, Manconi M, Cappa S et al (2011) Longitudinal study of cognitive function in idiopathic REM sleep behavior disorder. Sleep 34(5):619–625
Iranzo A, Tolosa E, Gelpi E, Molinuevo JL, Valldeoriola F, Serradell M et al (2013) Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: an observational cohort study. Lancet Neurol 12(5):443–453
Postuma RB, Gagnon JF, Vendette M, Fantini ML, Massicotte-Marquez J, Montplaisir J (2009) Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology 72(15):1296–1300
Fantini ML, Postuma RB, Montplaisir J, Ferini-Strambi L (2006) Olfactory deficit in idiopathic rapid eye movements sleep behavior disorder. Brain Res Bull 70(4–6):386–390
Postuma RB, Gagnon JF, Vendette M, Montplaisir JY (2009) Markers of neurodegeneration in idiopathic rapid eye movement sleep behaviour disorder and Parkinson’s disease. Brain 132(Pt 12):3298–3307
Postuma RB, Gagnon JF, Vendette M, Desjardins C, Montplaisir JY (2011) Olfaction and color vision identify impending neurodegeneration in rapid eye movement sleep behavior disorder. Ann Neurol. 69(5):811–818
Marques A, Dujardin K, Boucart M, Pins D, Delliaux M, Defebvre L et al (2010) REM sleep behaviour disorder and visuoperceptive dysfunction: a disorder of the ventral visual stream? J Neurol 257(3):383–391
Terzaghi M, Sinforiani E, Zucchella C, Zambrelli E, Pasotti C, Rustioni V et al (2008) Cognitive performance in REM sleep behaviour disorder: a possible early marker of neurodegenerative disease? Sleep Med 9(4):343–351
Massicotte-Marquez J, Decary A, Gagnon JF, Vendette M, Mathieu A, Postuma RB et al (2008) Executive dysfunction and memory impairment in idiopathic REM sleep behavior disorder. Neurology 70(15):1250–1257
Manni R, Sinforiani E, Pacchetti C, Zucchella C, Cremascoli R, Terzaghi M (2013) Cognitive dysfunction and REM sleep behavior disorder: key findings in the literature and preliminary longitudinal findings. Int J Psychophysiol 89(2):213–217
Fantini ML, Gagnon JF, Petit D, Rompre S, Decary A, Carrier J et al (2003) Slowing of electroencephalogram in rapid eye movement sleep behavior disorder. Ann Neurol 53(6):774–780
Iranzo A, Isetta V, Molinuevo JL, Serradell M, Navajas D, Farre R et al (2010) Electroencephalographic slowing heralds mild cognitive impairment in idiopathic REM sleep behavior disorder. Sleep Med 11(6):534–539
Brazète J, Montplaisir J, Petit D, Postuma RB, Bertrand J, Marchand D, et al (2013) Electroencephalogram slowing in rapid eye movement sleep behavior disorder is associated with mild cognitive impairment. Sleep Med 14(11):1059–1063
McCarter SJ, St Louis EK, Boeve BF (2013) Is rapid eye movement sleep behavior disorder in Parkinson disease a specific disease subtype? Sleep Med 14(10):931–933
Postuma RB, Gagnon JF, Pelletier A, Montplaisir J (2013) Prodromal autonomic symptoms and signs in Parkinson’s disease and dementia with Lewy bodies. Mov Disord 28(5):597–604
Frauscher B, Nomura T, Duerr S, Ehrmann L, Gschliesser V, Wenning GK, et al (2011) Investigation of autonomic function in idiopathic REM sleep behavior disorder. J Neurol 259(6):1056–1061
Postuma RB, Montplaisir J, Lanfranchi P, Blais H, Rompre S, Colombo R et al (2011) Cardiac autonomic denervation in Parkinson’s disease is linked to REM sleep behavior disorder. Mov Disord 26(8):1529–1533
Postuma RB, Bertrand JA, Montplaisir J, Desjardins C, Vendette M, Rios Romenets S et al (2012) Rapid eye movement sleep behavior disorder and risk of dementia in Parkinson’s disease: a prospective study. Mov Disord 27(6):720–726
Romenets SR, Gagnon JF, Latreille V, Panniset M, Chouinard S, Montplaisir J et al (2012) Rapid eye movement sleep behavior disorder and subtypes of Parkinson’s disease. Mov Disord 27(8):996–1003
Uribe-San Martin R, Venegas Francke P, Lopez Illanes F, Jones Gazmuri A, Salazar Rivera J, Godoy Fernndez J, et al (2013) Plasma urate in REM sleep behavior disorder. Mov Disord 28(8):1150–1151
Dang-Vu TT, Gagnon JF, Vendette M, Soucy JP, Postuma RB, Montplaisir J (2012) Hippocampal perfusion predicts impending neurodegeneration in REM sleep behavior disorder. Neurology 79(24):2302–2306
Nardone R, Bergmann J, Kunz A, Christova M, Brigo F, Tezzon F et al (2012) Cortical afferent inhibition is reduced in patients with idiopathic REM sleep behavior disorder and cognitive impairment: a TMS study. Sleep Med 13(7):919–925
Hanyu H, Inoue Y, Sakurai H, Kanetaka H, Nakamura M, Miyamoto T et al (2012) Voxel-based magnetic resonance imaging study of structural brain changes in patients with idiopathic REM sleep behavior disorder. Parkinsonism Relat Disord. 18(2):136–139
Iranzo A, Ratti PL, Casanova-Molla J, Serradell M, Vilaseca I, Santamaria J (2009) Excessive muscle activity increases over time in idiopathic rem sleep behavior disorder. Sleep 32(9):1149–1153
Tachibana N, Oka Y (2004) Longitudinal change in REM sleep components in a patient with multiple system atrophy associated with REM sleep behavior disorder: paradoxical improvement of nocturnal behaviors in a progressive neurodegenerative disease. Sleep Med 5(2):155–158
Schenck CH, Bundlie SR, Mahowald MW (1996) Delayed emergence of a parkinsonian disorder in 38 % of 29 older men initially diagnosed with idiopathic rapid eye movement sleep behaviour disorder. Neurology 46(2):388–393
Bugalho P, da Silva JA, Neto B (2011) Clinical features associated with REM sleep behavior disorder symptoms in the early stages of Parkinson’s disease. J Neurol 258(1):50–55
Poryazova R, Oberholzer M, Baumann CR, Bassetti CL (2013) REM sleep behavior disorder in parkinson’s disease: a questionnaire-based survey. J Clin Sleep Med 9(1):55–9A
Cummings JL (1992) Depression and Parkinson’s disease: a review. Am J Psychiatry 149(4):443–454
McCarter S, St. Louis E, Sandness D, Arndt K, Erickson M, Tabatabai G, et al (2014) Antidepressants increase rem sleep muscle tone in patients with and without REM sleep behavior disorder. Sleep 38(6):907–917
Wing YK, Li SX, Mok V, Lam SP, Tsoh J, Chan A et al (2012) Prospective outcome of rapid eye movement sleep behaviour disorder: psychiatric disorders as a potential early marker of Parkinson’s disease. J Neurol Neurosurg Psychiatry 83(4):470–472
Nomura T, Inoue Y, Kagimura T, Nakashima K (2013) Clinical significance of REM sleep behavior disorder in Parkinson’s disease. Sleep Med 14(2):131–135
Bjornara KA, Dietrichs E, Toft M (2013) REM sleep behavior disorder in Parkinson’s disease—is there a gender difference? Parkinsonism Relat Disord. 19(1):120–122
Lavault S, Leu-Semenescu S, Tezenas du Montcel S, Cochen de Cock V, Vidailhet M, Arnulf I (2010) Does clinical rapid eye movement behavior disorder predict worse outcomes in Parkinson’s disease? J Neurol 257(7):1154–9
Gjerstad MD, Boeve B, Wentzel-Larsen T, Aarsland D, Larsen JP (2008) Occurrence and clinical correlates of REM sleep behaviour disorder in patients with Parkinson’s disease over time. J Neurol Neurosurg Psychiatry 79(4):387–391
Nomura T, Inoue Y, Hogl B, Uemura Y, Yasui K, Sasai T et al (2011) Comparison of the clinical features of rapid eye movement sleep behavior disorder in patients with Parkinson’s disease and multiple system atrophy. Psychiatry Clin Neurosci 65(3):264–271
Cochen De Cock V (2012) Recent data on rapid eye movement sleep behavior disorder in patients with Parkinson’s disease: analysis of behaviors, movements and periodic leg movements. Sleep Med 14(8):749–753
Ferman TJ, Boeve BF, Smith GE, Silber MH, Kokmen E, Petersen RC et al (1999) REM sleep behavior disorder and dementia: cognitive differences when compared with AD. Neurology 52(5):951–957
Ferman TJ, Smith GE, Boeve BF, Graff-Radford NR, Lucas JA, Knopman DS et al (2006) Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol 20(4):623–636
Ferman TJ, Boeve BF, Smith GE, Lin SC, Silber MH, Pedraza O et al (2011) Inclusion of RBD improves the diagnostic classification of dementia with Lewy bodies. Neurology 77(9):875–882
Lippa CF, Duda JE, Grossman M, Hurtig HI, Aarsland D, Boeve BF et al (2007) DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology 68(11):812–819
Pao WC, Boeve BF, Ferman TJ, Lin SC, Smith GE, Knopman DS et al (2013) Polysomnographic findings in dementia with Lewy bodies. Neurologist 19(1):1–6
Molano J, Boeve B, Ferman T, Smith G, Parisi J, Dickson D et al (2010) Mild cognitive impairment associated with limbic and neocortical Lewy body disease: a clinicopathological study. Brain 133(Pt 2):540–556
Dugger BN, Boeve BF, Murray ME, Parisi JE, Fujishiro H, Dickson DW et al (2012) Rapid eye movement sleep behavior disorder and subtypes in autopsy-confirmed dementia with Lewy bodies. Mov Disord 27(1):72–78
Plazzi G, Cortelli P, Montagna P, De Monte A, Corsini R, Contin M et al (1998) REM sleep behaviour disorder differentiates pure autonomic failure from multiple system atrophy with autonomic failure. J Neurol Neurosurg Psychiatry 64(5):683–685
De Cock VC, Debs R, Oudiette D, Leu S, Radji F, Tiberge M et al (2011) The improvement of movement and speech during rapid eye movement sleep behaviour disorder in multiple system atrophy. Brain 134(Pt 3):856–862
Iranzo A, Santamaria J, Rye DB, Valldeoriola F, Marti MJ, Munoz E et al (2005) Characteristics of idiopathic REM sleep behavior disorder and that associated with MSA and PD. Neurology 65(2):247–252
Muntean ML, Sixel-Doring F, Trenkwalder C (2013) No Difference in Sleep and RBD between Different Types of Patients with Multiple System Atrophy: A Pilot Video-Polysomnographical Study. Sleep Disord 2013:258390
Gagnon JF, Petit D, Fantini ML, Rompre S, Gauthier S, Panisset M et al (2006) REM sleep behavior disorder and REM sleep without atonia in probable Alzheimer disease. Sleep 29(10):1321–1325
Cooper AD, Josephs KA (2009) Photophobia, visual hallucinations, and REM sleep behavior disorder in progressive supranuclear palsy and corticobasal degeneration: a prospective study. Parkinsonism Relat Disord 15(1):59–61
Sixel-Doring F, Schweitzer M, Mollenhauer B, Trenkwalder C (2009) Polysomnographic findings, video-based sleep analysis and sleep perception in progressive supranuclear palsy. Sleep Med 10(4):407–415
Gatto EM, Uribe Roca MC, Martinez O, Valiensi S, Hogl B (2007) Rapid eye movement (REM) sleep without atonia in two patients with corticobasal degeneration (CBD). Parkinsonism Relat Disord 13(2):130–132
Arnulf I, Merino-Andreu M, Bloch F, Konofal E, Vidailhet M, Cochen V et al (2005) REM sleep behavior disorder and REM sleep without atonia in patients with progressive supranuclear palsy. Sleep 28(3):349–354
Nomura T, Inoue Y, Takigawa H, Nakashima K (2012) Comparison of REM sleep behaviour disorder variables between patients with progressive supranuclear palsy and those with Parkinson’s disease. Parkinsonism Relat Disord 18(4):394–396
Cipolli C, Franceschini C, Mattarozzi K, Mazzetti M, Plazzi G (2011) Overnight distribution and motor characteristics of REM sleep behaviour disorder episodes in patients with narcolepsy-cataplexy. Sleep Med 12(7):635–640
Delrosso LM, Chesson AL Jr, Hoque R (2013) Characterization of REM Sleep without Atonia in Patients with Narcolepsy and Idiopathic Hypersomnia using AASM Scoring Manual Criteria. J Clin Sleep Med 9(7):675–680
Josephs KA, Silber MH, Fealey RD, Nippoldt TB, Auger RG, Vernino S (2004) Neurophysiologic studies in Morvan syndrome. J Clin Neurophysiol 21(6):440–445
Flanagan EP, Gavrilova RH, Boeve BF, Kumar N, Jelsing EJ, Silber MH (2013) Adult-onset autosomal dominant leukodystrophy presenting with REM sleep behavior disorder. Neurology 80(1):118–120
St Louis EK, McCarter SJ, Boeve BF, Silber MH, Kantarci K, Benarroch EE, et al (2014) Lesional REM sleep behavior disorsomedial pons. Neurology 83(20):1871–1873
Verma A, Anand V, Verma NP (2007) Sleep disorders in chronic traumatic brain injury. J Clin Sleep Med 3(4):357–362
Ju YE, Larson-Prior L, Duntley S (2012) RBD and antidepressants. Sleep Med. 13(2):211–212
Postuma RB, Gagnon JF, Tuineaig M, Bertrand JA, Latreille V, Desjardins C et al (2013) Antidepressants and REM sleep behavior disorder: isolated side effect or neurodegenerative signal? Sleep 36(11):1579–1585
Schuurman AG, van den Akker M, Ensinck KT, Metsemakers JF, Knottnerus JA, Leentjens AF et al (2002) Increased risk of Parkinson’s disease after depression: a retrospective cohort study. Neurology 58(10):1501–1504
Leentjens AF, Van den Akker M, Metsemakers JF, Lousberg R, Verhey FR (2003) Higher incidence of depression preceding the onset of Parkinson’s disease: a register study. Mov Disord 18(4):414–418
Remy P, Doder M, Lees A, Turjanski N, Brooks D (2005) Depression in Parkinson’s disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain 128(Pt 6):1314–1322
Winkelman JW, James L (2004) Serotonergic antidepressants are associated with REM sleep without atonia. Sleep 27(2):317–321
Applebee GA, Attarian HP, Schenck CH (2009) An angry bed partner. J Clin Sleep Med 5(5):477–479
Jouvet M (1965) F D. Locus coeruleus et sommeil paradoxal. C R Soc Biol 159:895–899
Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB et al (2011) Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS ONE 6(10):e24998
Sakai K, Sastre JP, Salvert D, Touret M, Tohyama M, Jouvet M (1979) Tegmentoreticular projections with special reference to the muscular atonia during paradoxical sleep in the cat: an HRP study. Brain Res 176(2):233–254
Jouvet M (1967) Structures and mechanisms for paradoxical sleep. Neurophysiology of the States of Sleep. The American Physiological Society, USA, pp 146–157
Brooks PL, Peever JH (2008) Unraveling the mechanisms of REM sleep atonia. Sleep 31(11):1492–1497
Brooks PL, Peever JH (2011) Impaired GABA and glycine transmission triggers cardinal features of rapid eye movement sleep behavior disorder in mice. J Neurosci 31(19):7111–7121
Brooks PL, Peever JH (2012) Identification of the transmitter and receptor mechanisms responsible for REM sleep paralysis. J Neurosci 32(29):9785–9795
Krenzer M, Lu J, Mayer G, Oertel W (2013) From bench to bed: putative animal models of REM sleep behavior disorder (RBD). J Neural Transm 120(4):683–688
Luppi PH, Clement O, Valencia Garcia S, Brischoux F, Fort P (2013) New aspects in the pathophysiology of rapid eye movement sleep behavior disorder: the potential role of glutamate, gamma-aminobutyric acid, and glycine. Sleep Med 14(8):714–718
Lu J, Sherman D, Devor M, Saper CB (2006) A putative flip-flop switch for control of REM sleep. Nature 441(7093):589–594
Fort P, Bassetti CL, Luppi PH (2009) Alternating vigilance states: new insights regarding neuronal networks and mechanisms. Eur J Neurosci 29(9):1741–1753
Cygan F, Oudiette D, Leclair-Visonneau L, Leu-Semenescu S, Arnulf I (2010) Night-to-night variability of muscle tone, movements, and vocalizations in patients with REM sleep behavior disorder. J Clin Sleep Med 6(6):551–555
Braak H, Ghebremedhin E, Rub U, Bratzke H, Del Tredici K (2004) Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res 318(1):121–134
Neuzeret PC, Sakai K, Gormand F, Petitjean T, Buda C, Sastre JP et al (2009) Application of histamine or serotonin to the hypoglossal nucleus increases genioglossus muscle activity across the wake-sleep cycle. J Sleep Res 18(1):113–121
Schwarz PB, Yee N, Mir S, Peever JH (2008) Noradrenaline triggers muscle tone by amplifying glutamate-driven excitation of somatic motoneurones in anaesthetized rats. J Physiol 586(23):5787–5802
Arnulf I (2012) REM sleep behavior disorder: motor manifestations and pathophysiology. Mov Disord 27(6):677–689
Knudsen S, Gammeltoft S, Jennum PJ (2010) Rapid eye movement sleep behaviour disorder in patients with narcolepsy is associated with hypocretin-1 deficiency. Brain 133(Pt 2):568–579
Kiyashchenko LI, Mileykovskiy BY, Lai YY, Siegel JM (2001) Increased and decreased muscle tone with orexin (hypocretin) microinjections in the locus coeruleus and pontine inhibitory area. J Neurophysiol 85(5):2008–2016
Willie JT, Takahira H, Shibahara M, Hara J, Nomiyama M, Yanagisawa M et al (2011) Ectopic overexpression of orexin alters sleep/wakefulness states and muscle tone regulation during REM sleep in mice. J Mol Neurosci 43(2):155–161
Bridoux A, Moutereau S, Covali-Noroc A, Margarit L, Palfi S, Nguyen JP et al (2013) Ventricular orexin-A (hypocretin-1) levels correlate with rapid-eye-movement sleep without atonia in Parkinson’s disease. Nat Sci Sleep. 5:87–91
Anderson KN, Vincent A, Smith IE, Shneerson JM (2010) Cerebrospinal fluid hypocretin levels are normal in idiopathic REM sleep behaviour disorder. Eur J Neurol 17(8):1105–1107
Garcia-Borreguero D, Caminero AB, De La Llave Y, Larrosa O, Barrio S, Granizo JJ et al (2002) Decreased phasic EMG activity during rapid eye movement sleep in treatment-naive Parkinson’s disease: effects of treatment with levodopa and progression of illness. Mov Disord 17(5):934–941
Verhave PS, Jongsma MJ, Van den Berg RM, Vis JC, Vanwersch RA, Smit AB et al (2011) REM sleep behavior disorder in the marmoset MPTP model of early Parkinson disease. Sleep 34(8):1119–1125
Boeve BF (2013) Idiopathic REM sleep behaviour disorder in the development of Parkinson’s disease. Lancet Neurol 12(5):469–482
Thannickal TC, Lai YY, Siegel JM (2007) Hypocretin (orexin) cell loss in Parkinson’s disease. Brain 130(Pt 6):1586–1595
Howell MJ, Arneson PA, Schenck CH (2011) A Novel Therapy for REM Sleep Behavior Disorder (RBD). J Clin Sleep Med 7(6):639–44A
Iranzo A, Santamaria J (2005) Severe obstructive sleep apnea/hypopnea mimicking REM sleep behavior disorder. Sleep 28(2):203–206
Kunz D, Bes F (1999) Melatonin as a therapy in REM sleep behavior disorder patients: an open-labeled pilot study on the possible influence of melatonin on REM-sleep regulation. Mov Disord 14(3):507–511
Kunz D, Mahlberg R (2010) A two-part, double-blind, placebo-controlled trial of exogenous melatonin in REM sleep behaviour disorder. J Sleep Res 19(4):591–596
Ferri R, Marelli S, Ferini-Strambi L, Oldani A, Colli F, Schenck CH et al (2013) An observational clinical and video-polysomnographic study of the effects of clonazepam in REM sleep behavior disorder. Sleep Med 14(1):24–29
Mahowald MW, CH Schenck (2005) REM sleep parasomnias In: Kryger MH, Roth T, Dement WC (eds) Principles and practice of sleep medicine 4 edn (897–916). Elsevier Saunders, Philadelphia
Schenck CH, Mahowald MW (1996) Long-term, nightly benzodiazepine treatment of injurious parasomnias and other disorders of disrupted nocturnal sleep in 170 adults. Am J Med 100(3):333–337
Aurora RN, Zak RS, Maganti RK, Auerbach SH, Casey KR, Chowdhuri S et al (2010) Best practice guide for the treatment of REM sleep behavior disorder (RBD). J Clin Sleep Med 6(1):85–95
Takeuchi N, Uchimura N, Hashizume Y, Mukai M, Etoh Y, Yamamoto K et al (2001) Melatonin therapy for REM sleep behavior disorder. Psychiatry Clin Neurosci 55(3):267–269
Gagnon JF, Postuma RB, Montplaisir J (2006) Update on the pharmacology of REM sleep behavior disorder. Neurology 67(5):742–747
Fantini ML, Gagnon JF, Filipini D, Montplaisir J (2003) The effects of pramipexole in REM sleep behavior disorder. Neurology 61(10):1418–1420
Kumru H, Iranzo A, Carrasco E, Valldeoriola F, Marti MJ, Santamaria J et al (2008) Lack of effects of pramipexole on REM sleep behavior disorder in Parkinson disease. Sleep 31(10):1418–1421
Schmidt MH, Koshal VB, Schmidt HS (2006) Use of pramipexole in REM sleep behavior disorder: results from a case series. Sleep Med 7(5):418–423
Sasai T, Matsuura M, Inoue Y (2013) Factors associated with the effect of pramipexole on symptoms of idiopathic REM sleep behavior disorder. Parkinsonism Relat Disord 19(2):153–157
Kataoka H, Ueno S (2012) Nightmare-enacting behavior responding to zonisamide in early Parkinson’s disease. Case Rep Neurol 4(1):31–33
Mimaki T, Suzuki Y, Tagawa T, Karasawa T, Yabuuchi H (1990) Interaction of zonisamide with benzodiazepine and GABA receptors in rat brain. Med J Osaka Univ 39(1–4):13–17
Ueda Y, Doi T, Tokumaru J, Willmore LJ (2003) Effect of zonisamide on molecular regulation of glutamate and GABA transporter proteins during epileptogenesis in rats with hippocampal seizures. Brain Res Mol Brain Res 116(1–2):1–6
Massironi G, Galluzzi S, Frisoni GB (2003) Drug treatment of REM sleep behavior disorders in dementia with Lewy bodies. Int Psychogeriatr 15(4):377–383
Ringman JM, Simmons JH (2000) Treatment of REM sleep behavior disorder with donepezil: a report of three cases. Neurology 55(6):870–871
Bonakis A, Economou NT, Papageorgiou SG, Vagiakis E, Nanas S, Paparrigopoulos T (2012) Agomelatine may improve REM sleep behavior disorder symptoms. J Clin Psychopharmacol 32(5):732–734
Corruble E, de Bodinat C, Belaidi C, Goodwin GM (2013) Efficacy of agomelatine and escitalopram on depression, subjective sleep and emotional experiences in patients with major depressive disorder: a 24-wk randomized, controlled, double-blind trial. Int J Neuropsychopharmacol 16(10):2219–2234
Shinno H, Kamei M, Nakamura Y, Inami Y, Horiguchi J (2008) Successful treatment with Yi-Gan San for rapid eye movement sleep behavior disorder. Prog Neuropsychopharmacol Biol Psychiatry 32(7):1749–1751
Schenck CH, Montplaisir JY, Frauscher B, Hogl B, Gagnon JF, Postuma R et al (2013) Rapid eye movement sleep behavior disorder: devising controlled active treatment studies for symptomatic and neuroprotective therapy-a consensus statement from the International Rapid Eye Movement Sleep Behavior Disorder Study Group. Sleep Med 14(8):795–806
Acknowledgments
This publication was made possible by the following grants: 1 UL1 RR024150 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and the NIH Roadmap for Medical Research; P50 AG016574, UO1 AG006786, RO1 AG015866, the Mangurian Foundation, and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer’s Disease Research Program of the Mayo Foundation Its contents are solely the responsibility of the author and do not necessarily represent the official view of NCRR or NIH. Information on NCRR is available at http://www.ncrr.nih.gov/. Information on Reengineering the Clinical Research Enterprise can be obtained from http://nihroadmap.nih.gov.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media New York
About this chapter
Cite this chapter
McCarter, S.J., St. Louis, E.K., Boeve, B.F. (2017). Rapid Eye Movement Sleep Behavior Disorder. In: Chokroverty, S. (eds) Sleep Disorders Medicine. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-6578-6_49
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6578-6_49
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-6576-2
Online ISBN: 978-1-4939-6578-6
eBook Packages: MedicineMedicine (R0)