
Abstract
Using TALEN or CRISPR/Cas system to induce small indels into coding sequences has been implicated in broad applications for reverse genetic studies of many organisms including zebrafish. However, complete deletion of a large gene or noncoding gene(s) or removing a large genomic fragment spanning several genes or other chromosomal elements is preferred in various cases, as well as inducing chromosomal inversions. Here, we describe the detailed protocols for the generation of chromosomal deletion mutations mediated by Cas9 and a pair of gRNAs and the evaluation for the efficiencies in F0 founder fish and of germline transmission.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
As an ideal vertebrate model organism, zebrafish (Danio rerio) is valuable to study gene functions during embryonic development and organ regeneration, as well as in modeling human diseases. Transcription activator-like effector nucleases (TALENs) or clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems have been widely used to induce double-strand breaks (DSBs) in target genes and subsequent nonhomologous end-joining (NHEJ)-mediated repairing mutagenesis, leading to disruption of protein-coding genes, usually due to frameshift, in a variety of organisms including zebrafish [1–4]. NHEJ is error prone and tends to generate different types of small insertions and/or deletions (indels) around the DSB region, with the length ranging from a few to tens, or sometimes hundreds, of nucleotides .
However, the indel mutation strategy is not suitable or satisfactory for all purposes. For example, it is usually not sufficient to disrupt the function of noncoding genes, untranslated or regulatory regions in a genome; it is also tricky to use this approach to disrupt multiple adjacent genes or gene clusters simultaneously. Even for a single coding gene, indel mutations may not guarantee to disrupt the function of the target gene completely, especially for large genes, due to the existence of multiple transcripts or splice variants, or unexpected upstream/downstream alternative start codons. In all these cases, complete deletion of the whole gene(s) or sequence should be a reliable and better strategy to solve the above problems, which can be easily achieved by using two pairs of TALENs or two guide RNAs (gRNAs ) with Cas9 targeting two sites flanking the region to be deleted. This strategy may also be used to precisely remove specific functional elements, e.g., a protein domain, from the coding sequence without affecting other parts of the gene, which is difficult to achieve for a single pair of TALENs or one CRISPR/Cas (or called Cas9/gRNA) system when the target sequence is more than several hundreds of nucleotides long. Furthermore, in order to disrupt the target gene completely, the first several exons are usually selected for targeting to generate indel mutations and screen for frameshift alleles which could lead to early premature stop codons. However, this restriction for the targeting region may prevent the selection for high-efficient target sites. In contrast, the targeted deletion strategy provides more choices for the target sites, which could extend the options for the target site to introns and intergenic sequences , in addition to exons [5]. In addition to generating large genomic deletions, one can also create genomic inversions by using two pairs of TALENs or paired gRNAs with Cas9. In this chapter, we provide the detailed protocols for using two gRNAs together with the Cas9 mRNA to generate large chromosomal deletions in the zebrafish genome.
2 Materials
2.1 Reagents and Solutions for Molecular Biology Experiments
-
1.
zCas9 expression vector (pGH-T7-NLS-zCas9-NLS) [6].
-
2.
gRNA template plasmid (e.g., pX459-V2.0, Addgene #62988 [7]).
-
3.
mMessage mMachine T7 kit or mMessage mMachine T7 Ultra kit (Ambion, USA).
-
4.
MEGAshortscript T7 Kit (Ambion, USA).
-
5.
mirVana miRNA Isolation Kit (Ambion, USA).
-
6.
XbaI restriction enzyme (New England Biolabs, USA).
-
7.
PCR purification kit (Qiagen, USA).
-
8.
Gel extraction kit (Invitrogen, USA).
-
9.
TOPO TA-cloning vectors (Invitrogen, USA).
-
10.
50 mM NaOH.
-
11.
1 M Tris–HCl (pH 8.0).
-
12.
Nuclease-free water or RNase-free water.
-
13.
Hi-Taq DNA polymerase master mix.
-
14.
Forward primer to amplify the DNA template for gRNA synthesis: 5′-TAATACGACTCACTATA

GTTTTAGAGCTAGAAATAGC-3′. The nucleotides in bold letters (and framed) represent the 20-nt protospacer sequence and are variable according to different target sites.
-
15.
Reverse primer to amplify the DNA template for gRNA synthesis: 5′-AAAAAAGCACCGACTCGGTGCCAC-3′. Universal for all gRNAs.
-
16.
Real-time PCR primers and probes: For evaluation of the efficiency of chromosomal deletions . Variable according to different target regions.

2.2 Equipment, Reagents, and Consumable Materials for Zebrafish Husbandry and Microinjection
-
1.
Wild-type zebrafish or other desired zebrafish strains.
-
2.
E3 embryo buffer: 5 mmol/L NaCl, 0.17 mmol/L KCl, 0.33 mmol/L CaCl2, 0.33 mmol/L MgSO4.
-
3.
0.5 % Phenol red.
-
4.
Glass capillaries: For making injection needles (e.g., O.D. 1.0 mm, I.D. 0.58 mm; Harvard Apparatus, USA).
-
5.
Dumont #5 Tweezer (Inox, 11 cm) (Word Precision Instruments, Inc.) or other equivalents.
-
6.
1-μL disposable capillaries (R: 0.25 %, CV: 0.6 %) (CAMAG, Switzerland).
-
7.
Microloader tips (Eppendorf, USA).
-
8.
Microinjection molds or other equivalents to hold zebrafish embryos for microinjection .
-
9.
Mating tanks.
-
10.
Stereo microscope.
-
11.
28.5 °C incubator.
-
12.
PN-30 Puller (Narishige, Japan) or other equivalents.
-
13.
Nitrogen gas and tank.
-
14.
MPPI-2 Pressure Injector (Applied Scientific Instrumentation, USA), or PLI-90Pico-Injector (Harvard Apparatus, USA) or other equivalents.
3 Methods
3.1 Design and Verification of Cas9/gRNA Target Sites
-
1.
Each Cas9/gRNA target site (Cas9/gRNA recognition and binding site) consists of a 20-nt protospacer followed by a 3-nt PAM (protospacer adjacent motif) sequence (i.e., 5′-NGG-3′). Therefore, the general formula for a single target site is 5′-

-NGG-3′, where N represents any nucleotide and the first 20 nucleotides (framed) represent the sequence of the protospacer. In reality, T7 RNA polymerase is usually employed for the synthesis of gRNAs (beginning with the 20-nt protospacer sequence) by in vitro transcription. Since the T7 polymerase requires at least the first transcribed nucleotide to be G for efficient transcription, the target site or the protospacer sequence should begin with a G, and the practical formula for a target site should therefore be modified as 5′-G-(N)19-NGG-3′, or further simplified as 5′-G-(N)20-GG-3′(see Note 1 ).
-
2.
To delete a large fragment of chromosomal sequence , one needs to design two gRNAs targeting the 5′- and 3′-ends of the region to be deleted, respectively. Individual Cas9/gRNA target site can be selected manually using the above formula or identified by using web tools, such as CRISPR Design Tool (http://crispr.mit.edu/) [8] or ZiFiT Targeter (http://zifit.partners.org/ZiFiT/CSquare9Nuclease.aspx) [9]. The Cas9/gRNA target sites can be designed to locate in either exonic, intronic, or intergenic regions. Unless the purpose is to delete a defined genomic element (i.e., not much choice for the target sites), the target sites within exons are preferred since they have much less sequence polymorphism.
-
3.
(Optional) For a given sequence, usually there are quite a few choices for the Cas9/gRNA target sites. To simplify the evaluation of targeting efficiency of single gRNA site (and screening for indel mutations in other applications), the target sequence containing a unique restriction enzyme site is usually preferred, which is necessary for restriction enzyme (RE)-resistance assay (see Note 2 ).
-
4.
Ensure that each selected target site is unique and highly specific in the zebrafish genome (see Note 3 ). Some web tools or software are available to help search for potential off-target sites in the zebrafish genome [8, 10, 11].
-
5.
For each target site, design a pair of primers for the amplification of the genomic region spanning the target site. Choose the primers which are at least 100 bp away from the target site and which give rise to the PCR product of less than 500 bp. In addition, since we have to use a particular pair of primers, one from the upstream target site and the other from the downstream target site, to detect the large deletions, try to choose the two pairs of primers for the two target sites to have similar annealing temperature.
-
6.
PCR amplify the genomic DNA from parental fish and make sure only one single band is visible after agarose gel electrophoresis . Since mismatches in the target sites will affect the mutagenesis activity, to exclude polymorphisms in the target sites, it is important to confirm their sequences from your in-house fish stock by PCR and direct sequencing, and use the confirmed zebrafish to collect embryos for the gene-targeting experiments.
-
7.
(Optional) Establish RE-resistance assay for the evaluation of targeting efficiency: If one plans to use RE-resistance assay to determine the targeting efficiency, PCR amplify the genomic sequence of the parental fish with the selected primer pairs, perform RE digestion, and evaluate by agarose gel electrophoresis. The RE digestion should be complete and the digestion products should be easily identifiable and distinguishable with the undigested PCR product.

3.2 Preparation of zCas9 mRNA by In Vitro Transcription
-
1.
We recommend to use zebrafish codon-optimized Cas9 (zCas9) rather than other versions of Cas9 (e.g., hCas9, the human codon-optimized Cas9) since zCas9 gives higher targeting efficiency [6]. Our zCas9 plasmid (pGH-T7-NLS-zCas9-NLS) contains a T7 promoter upstream to the NLS-zCas9-NLS coding region and can be linearized for making mRNA through in vitro transcription . For linearization, 10 μg zCas9 plasmids are digested by XbaI in a 10 μL system overnight at 37 °C. To monitor the extent of linearization, load 0.5 μL of the reaction mixture to 0.8 % agarose gel and examine by electrophoresis (see Note 4 ).
-
2.
When the digestion is complete, use a DNA purification kit to purify the linearized plasmids and elute with 20 μL nuclease-free water (see Note 5 ). Determine the concentration of the linearized plasmid by a spectrophotometer. (Optional: The linearized plasmid can be stored at −20 °C and used later.)
-
3.
Prepare capped zCas9 mRNA by using the mMessage mMachine T7 Kit (or mMessage mMachine T7 Ultra Kit) (see Notes 6 and 7 ). Reaction mixture: 1 μg linearized DNA from the above step, 10 μL 2× NTP/CAP (or NTP/ARCA), 2 μL 10× reaction buffer, and 2 μL T7 enzyme mix, and supplement the volume to 20 μL with nuclease-free water; mix well by pipetting. Incubate the mixture at 37 °C for 2–3 h. To monitor the mRNA synthesis, load 0.5 μL reaction mixture to 1 % agarose gel and examine by electrophoresis (see Note 8 ).
-
4.
If the transcription is successful, add 1 μL TURBO DNase I supplied by the kit and incubate at 37 °C for 15 min to remove the DNA template.
-
5.
Purify the zCas9 mRNA. Option I: According to the manual of the kit, stop reaction by adding 30 μL LiCl and 30 μL nuclease-free water provided by the kit. Mix well and store at −20 °C for at least 30 min. Then centrifuge at 4 °C for 15 min at top speed. The RNA pellet should be visible at the bottom of the Eppendorf tube. Remove the supernatant and wash with 1 mL cold 70 % ethanol. Centrifuge at 4 °C for 10 min at top speed. Remove the 70 % ethanol, air-dry the pellet, and add 30–50 μL nuclease-free water to dissolve the pellet. Option II: use RNeasy kit to purify the zCas9 mRNA following the manufacturer’s instructions. After purification, determine the concentration of mRNA by a spectrophotometer (typically 500–800 ng/μL; see Note 9 ), and aliquot them into small volumes (e.g., 5 μL). Store the aliquots at −80 °C for later use and long-term storage (see Note 10 ).
3.3 Preparation of gRNAs by In Vitro Transcription
-
1.
We use purified PCR product as the in vitro transcription template for making gRNAs. Any plasmid containing a full gRNA scaffold sequence can be used as the PCR template (e.g., Addgene plasmid pX459-V2.0 #62988 [7]; see Note 11 ). Synthesize (by order from company service) a 57-nt oligo for each gRNA with the sequence 5′-TAATACGACTCACTATA

GTTTTAGAGCTAGAAATAGC-3′, where the nucleotides framed in bold represent the 20-nt protospacer sequence of each particular gRNA, preceded by a 17-nt T7 promoter sequence at the 5′-upstream; and synthesize a universal reverse primer oligo with the sequence 5′-AAAAAAGCACCGACTCGGTGCCAC-3′. PCR reaction mixture: 2 ng plasmid template (see Note 12 ), 4 μL 10 μmol/L oligos each, and 20 μL 2× hi-fidelity DNA polymerase master mix, and supplement the volume to 40 μL with nuclease-free water. PCR program: 95 °C 5 min, then 45 cycles of (95 °C 20 s, 55 °C 20 s, 72 °C 30 s), and then 72 °C 6 min. The PCR products can be examined by electrophoresis, the length of which should be 119 bp (17 bp T7 promotor + 102 bp gRNA template).
-
2.
Purify the PCR product by ethanol (EtOH) precipitation (see Note 13 ). Adjust the volume by water to 150 μL, add 2.5× volume (375 μL) EtOH and 0.1× volume (15 μL) 3 mol/L NaOAc, and mix well. Store at −20 °C for at least 1 h. Then centrifuge at 4 °C for 10 min at top speed. The pellet of PCR product should be visible at the bottom of the tube. Remove the supernatant and wash with 1 mL cold 70 % ethanol. Centrifuge at 4 °C for 10 min at top speed. Remove the 70 % ethanol, air-dry the pellet, and add 30 μL nuclease-free water to dissolve the pellet. Determine the concentration of gRNA DNA template by a spectrophotometer (typical 200–300 ng/μL) (Optional: The PCR product can be stored at −20 °C and used later.).
-
3.
Prepare gRNAs by using the MEGAshortscript T7 Kit or other T7 in vitro transcription system (Fig. 1; see Note 6 ). Reaction mixture: 1 μg DNA template from the above step, 2 μL each ATP/CTP/GTP/UTP, 2 μL 10× reaction buffer, and 2 μL T7 enzyme mix, and supplement the volume to 20 μL with nuclease-free water; mix well by pipetting. Incubate the mixture at 37 °C for 2–3 h. Add 1 μL TURBO DNase I supplied by the kit and incubate at 37 °C for 15 min to remove the DNA template.
Fig. 1 
The schematic diagram of the steps to prepare gRNAs
-
4.
Purify the short gRNAs with mirVana miRNA Isolation Kit, following the manufacturer’s instruction. Elute the gRNAs with 50 μL elution buffer. Determine the concentration of gRNA by a spectrophotometer (typically varies from 300 to >1000 ng/μL; see Note 9 ).


3.4 Generation of Founder Fish by Microinjection of zCas9 mRNA and gRNA Pairs into Zebrafish Embryos
-
1.
The parental zebrafish intended to give rise to founder embryos for gene targeting should be genotyped to confirm the sequence of the target sites (see Note 14 ). One day before the injection day, set up mating tanks and put one pair of zebrafish in each tank and separate by genders with a divider. In the day of injection, remove the divider from one tank each time and collect embryos after spawning.
-
2.
Prepare 3–5 μL injection mixture: ~300 ng/μL zCas9 mRNA and 20–50 ng/μL of each gRNA, add some phenol red to a final concentration of no more than 0.05 %, and supplement the final volume to 20 μL with nuclease-free water. Use disposable capillaries to calibrate the injection volumes by injecting several drops into it. The total volume can be measured by the length of the liquid in the capillary by using a ruler. Then the volume of each drop can be calculated by the inner diameter of the capillary, the length of liquid column made by injection, and the count of drops.
-
3.
Inject 2 nL mixture into the cytoplasm of one-cell stage zebrafish embryos, i.e., ~600 pg zCas9 mRNA and 40–100 pg of each gRNA per embryo (see Notes 15 and 16 ). When using different batches of zCas9 mRNA, we recommend reevaluating the optimal injection dosages (see Note 17 ).
-
4.
After injection, incubate the embryos in E3 embryo buffer at 28.5 °C. Save some uninjected sibling embryos as a control and process them the same as the injected ones for the determination of the efficiency of individual gRNA. The dead and deformed embryos are counted and removed at 5–6 h post-fertilization (hpf) and 1-day post-fertilization (dpf).
3.5 (Optional) Evaluation of Targeting Efficiency of Individual gRNA in Founder Embryos
Evaluation of individual gRNA for its targeting efficiency can help in choosing proper gRNAs for the chromosomal deletion experiments or troubleshooting if the deletion is not successful. We generally use RE-resistance assay to evaluate the efficiency of individual gRNA (Option I) [12]. Other methods can also be used, such as Surveyor (CEL-I) or T7E1 assay (Option II), melting curve assay, and direct sequencing.
-
1.
The genomic DNA of control (uninjected siblings) and injected embryos is extracted using NaOH lysis method [13]. Five to ten normally developing 2–4 dpf embryos are pooled into one PCR tube. Remove extra buffer and add 50 μL 50 mmol/L NaOH. The embryos are lysed by heating for 10–30 min at 95 °C using a PCR machine; then cool down to 4 °C. Vortex the tubes briefly once or twice to break up the embryos. Then add 5 μL Tris–HCl (pH 8.0) to neutralize NaOH and centrifuge for 5 min at 14,000 × g. The supernatant contains crude genomic DNA and is ready to be used as PCR templates.
-
2.
Take 1 μL crude genomic DNA extract as template and assemble a 10 μL PCR reaction (see Note 18 ). Recover the PCR products.
-
3.
(Option I) RE-resistance assay: Digest 2 μL PCR products with the proper RE and buffer (see Note 19 ). Load reaction mixture to 2–3 % agarose gel and examine the digestion by electrophoresis , using undigested PCR products as a reference. If the DNA from control embryos is digested completely, the efficiency of the gRNA can be estimated with the percentage of the resistant (undigested) band by measuring their intensity. The resistant band can be extracted from the gel and verified by sequencing after cloning into TA-cloning vectors.
-
4.
(Option II) Surveyor (CEL-I) or T7E1 assay: Mix ~250 ng PCR product (from control or Cas9/gRNA injected embryos, respectively) with 1 μL NEB Buffer 2; supplement the volume to 9.5 μL with nuclease-free water. Slowly anneal the DNA in a thermocycler with the following program: 95 °C, 5 min; 95–25 °C at −0.1 °C/s; hold at 4 °C. Then add 0.5 μL Surveyor or T7E1 enzyme on ice. Mix well; incubate in 37 °C for 45 min. Add 1 μL 0.5 mol/L EDTA to stop reaction. Then load reaction mixture to 2–3 % agarose gel and examine the digestion by electrophoresis. If the DNA from control embryos is not digested and the DNA from injected ones is, that means the Cas9/gRNA is functional and small indels have been generated (see Note 20 ). The efficiency of mutated allele can be estimated by the corresponding formula [7].
3.6 Detection and Evaluation of Chromosomal Deletion Efficiency in Founder Embryos
-
1.
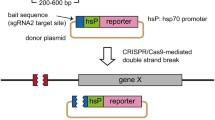
To examine the success of the chromosomal deletion , the genomic regions containing the target sites are amplified by PCR with the primers outside the region to be deleted (Fig. 2a). If the distance of the two primers at the chromosome is short enough, the DNA from control embryos can be amplified with a single amplicon, and DNA from injected embryos may have two amplicons : one with the same length as the control and the other should be shorter, which represents the alleles bearing deletions (Fig. 2b). If the distance of the two primers is too long, there may be no amplicon from control DNA, and only a short amplicon from injected embryos can be detected (Fig. 2c). Recover the shorter band and verify the deletions by sequencing after cloning into TA-cloning vectors.
Fig. 2 
Detection of chromosomal deletions induced by two Cas9/gRNAs. (a) The schematic diagram of the target region and PCR primers used to amplify the genomic fragment. (b) In wild-type embryos, if the genomic fragment between the two target sites is short enough to be amplified by PCR, one single PCR product (band A) will be obtained from the control (uninjected) embryos. In the injected embryos, an additional and shorter PCR product (band B), which corresponds to the deletion allele, should be clearly visible if the two Cas9/gRNAs are functional. (c) If the genomic fragment between the two target sites in wild-type embryos is too long to be amplified, no PCR product will be obtained from the control embryos, while only one single band could be detected from the injected embryos if the two Cas9/gRNAs are functional. Ctrl control
-
2.
The general PCR is not accurate in revealing the exact efficiency of chromosomal deletions [5]. To accurately evaluate the deletion efficiency, design quantitative real-time PCR primers and probes for wild-type only alleles (one primer located outside the deletion region and the other located inside) and deletion alleles (primers located at each sides out side the deletion region) (Fig. 3). Use the same amount of DNA template and evaluate the relative ratio of different alleles by ΔCt approach [14] (see Note 21 ).
Fig. 3 
The schematic diagram of the real-time PCR primers to quantify the ratio of wild-type and deletion alleles


3.7 Evaluation of Germline Transmission Efficiency of Chromosomal Deletions and Screen for Mutants
-
1.
If the chromosomal deletion efficiency is acceptable (e.g., >1 %; see Note 22 ), raise enough amount of the same batch of evaluated injected embryos to adulthood as founder (F0) fish for screening of heritable mutations.
-
2.
Out-cross the mosaic F0 fish with wild-type zebrafish. After breeding, each F0 fish is placed and raised separately in a single tank until the F1 embryos are evaluated. Collect F1 embryos from each individual F0, prepare the genomic DNA , and detect the chromosomal deletion with the same strategy as described above. We normally screen 50–100 F1 embryos (five to ten PCR tubes of ten pooled embryos) for each F0 fish (see Note 23 ).
-
3.
If heritable deletion alleles were identified and confirmed by sequencing, breed the positive F0 fish to get more F1 offspring. F1 zebrafish heterozygous for the chromosomal deletion mutations are identified by genotyping of the genomic DNA from fin clips from each individual, with the same strategy as described above.
-
4.
Homozygous zebrafish mutants can be obtained by in-cross of pairs of heterozygous carriers. They can be verified again with the same process.
4 Notes
-
1.
The promoter sequence for the T7 RNA polymerase is 5′-TAATACGACTCACTATAGGG-3′, where the last triple G (GGG) will also serve as the transcription start site and be incorporated into the RNA transcript; however, one or two G’s (i.e., GNN or GGN) is also acceptable for the normal function of T7 RNA polymerase. Alternatively, if the protospacer sequence in the target site does not begin with G, one can simply replace its first nucleotide with G for the synthesis of gRNA with T7 polymerase. The mismatch between the gRNA and the target sequence for the first nucleotide does not seem to significantly compromise the targeting activity of the Cas9/gRNA system (Unpublished observation).
-
2.
The Cas9 protein theoretically cleaves the target sequence between the third and fourth nucleotides upstream of the PAM. The RE recognition and cleavage sites used for the RE-resistance assay should not be too far from this Cas9 cleavage position, since indel mutations may only affect a small region around this position.
-
3.
Since the chromosomal deletion strategy introduces more than one type of gRNA simultaneously in the same embryo, one should be more cautious about the potential off-targeting effects of the Cas9/gRNA system.
-
4.
Make sure all the circular plasmids are digested completely. Use the original (undigested) plasmids as a control in electrophoresis , and there should be no visible bands at the same position as the original plasmid in the lanes loaded with the linearized plasmids. Insufficient linearization will lead to low yield of mRNA from in vitro transcription.
-
5.
Follow the supplier’s protocol of the purification kit. Avoid loading too much linearized plasmids into the column, or it may be overloaded and lead to low yield of purified product.
-
6.
All the reagents and consumables used for in vitro transcription should be free of RNase.
-
7.
Both the T7 Kit and T7 Ultra Kit from Ambion are acceptable. The T7 Ultra Kit uses Anti-Reverse Cap Analog (ARCA) instead of the general Cap in the T7 Kit. We found the zCas9 mRNA prepared by using the ARCA Kit showed higher targeting efficiency [6].
-
8.
The size of mRNA might not be accurately revealed by the agarose gel electrophoresis and may show up as multiple bands, possibly due to the formation of secondary structures of the mRNA.
-
9.
If the concentration of zCas9 mRNA or gRNA is not high enough, open the cap of the tubes and carefully put the tubes in a 37 °C incubator (avoid RNase contamination) for a few hours to concentrate the liquid. If the original concentration is too low, check the troubleshooting section in the manual of the kit and make new reactions.
-
10.
We suggest using a new aliquot of mRNA each time for microinjection, since it may degrade if thawing and freezing frequently.
-
11.
Most of the popular Addgene Cas9 plasmids from Feng Zhang Lab (pX330, pX458, pX459-V2.0, etc. [7, 15]) can be used as the PCR template for making gRNA in vitro transcription template, since they contain the gRNA (or called chimeric RNA) scaffold sequence. Avoid using the first-generation Cas9 plasmids (such as pX260), since they do not contain the gRNA scaffold, but use crRNA and tracrRNA instead.
-
12.
Avoid adding too much plasmid template; otherwise it will remain as the main component in the final PCR product.
-
13.
Most PCR purification kits are not designed to recover small fragments. Read the manual carefully and make sure it is suitable for recovering the 119-bp gRNA template if you want to use a kit.
-
14.
Mismatches in the Cas9/gRNA target sites may dramatically decrease targeting efficiency. Due to the strong sequence polymorphisms of the zebrafish genome, we strongly recommend to confirm the actual sequence of each selected target site in each individual fish before using their offspring to perform the mutagenesis experiments.
-
15.
Adjust the parameters of pressure and duration time of the microinjection machine to calibrate the volume of each drop to be 2 nL. 1 nL is also acceptable; however, larger injection volumes make relatively smaller variations between each shot. The injection needle should be recalibrated after any change in the balance pressure, eject pressure, duration time, or re-breaking of the tips of the needles. Avoid plugging of the needle tips, and occasionally try to inject several drops into the medium to see whether the needle still work normally.
-
16.
The dosage of the zCas9 mRNA and gRNAs injected into the embryo varies by different target sites and needs to be optimized. We always choose the dosage which gives no less than 50 % embryo survival rate at 2 dpf. We suggest better to inject the RNAs directly into the cytoplasm instead of the yolk.
-
17.
For different batches of zCas9 mRNA, even though the concentrations of mRNA might be the same, the activity may still be different due to variations of the capping efficiency during in vitro transcription.
-
18.
The crude lysate obtained from this fast genomic DNA extraction method is sufficient for the amplification of small fragment (<500 bp).
-
19.
The RE needs to be tested in advance to make sure it can completely digest the PCR product from control embryos (can be done at the step of target sequences confirmation). It is even better if the RE can function with the unpurified PCR system, which can save time by omitting purification steps.
-
20.
The ratio of PCR products and enzymes is important for a successful assay. If the PCR products from control (uninjected) embryos are digested and showed a smear pattern, the concentration of the enzymes should be reduced (particularly for T7E1).
-
21.
Designing good real-time PCR primers and probes for a specific genomic region is an important but elaborate process. Many different types of PCR probes are available; refer to the supplier’s instructions or tools for choosing and preparing good probes.
-
22.
The efficiency of large genomic deletions depends on both the activity of individual Cas9/gRNA and the size of deletion and also other undefined factors. We have successfully generated deletions with the length up to 122 kb with paired customized endonucleases and found ~1/10 of the founder fish can transmit the mutations to offspring with an average mosaicism of 14 %.
-
23.
Since the purpose of this detection approach is to get a positive PCR signal, more embryos (up to 50 as we have tested) can be pooled in one PCR tube to reduce the amount of samples. One should ensure that all the embryos are broken up completely when preparing the genomic DNA.
References
Huang P, Zhu Z, Lin S et al (2012) Reverse genetic approaches in zebrafish. J Genet Genomics 39(9):421–433. doi:10.1016/j.jgg.2012.07.004
Xiao A, Wu Y, Yang Z et al (2013) EENdb: a database and knowledge base of ZFNs and TALENs for endonuclease engineering. Nucleic Acids Res 41(Database issue):D415–D422. doi:10.1093/nar/gks1144
Hisano Y, Ota S, Kawahara A (2014) Genome editing using artificial site-specific nucleases in zebrafish. Dev Growth Differ 56(1):26–33. doi:10.1111/dgd.12094
Bedell VM, Ekker SC (2015) Using engineered endonucleases to create knockout and knockin zebrafish models. Methods Mol Biol 1239:291–305. doi:10.1007/978-1-4939-1862-1_17
Xiao A, Wang Z, Hu Y et al (2013) Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res 41(14):e141. doi:10.1093/nar/gkt464
Liu D, Wang Z, Xiao A et al (2014) Efficient gene targeting in zebrafish mediated by a zebrafish-codon-optimized cas9 and evaluation of off-targeting effect. J Genet Genomics 41(1):43–46. doi:10.1016/j.jgg.2013.11.004
Ran FA, Hsu PD, Wright J et al (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11):2281–2308. doi:10.1038/nprot.2013.143
Hsu PD, Scott DA, Weinstein JA et al (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31(9):827–832. doi:10.1038/nbt.2647
Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D (2010) ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res 38(Web Server issue):W462–W468. doi:10.1093/nar/gmk319
Xiao A, Cheng Z, Kong L et al (2014) CasOT: a genome-wide Cas9/gRNA off-target searching tool. Bioinformatics. doi:10.1093/bioinformatics/btt764
O’Brien A, Bailey TL (2014) GT-Scan: identifying unique genomic targets. Bioinformatics 30(18):2673–2675. doi:10.1093/bioinformatics/btu354
Huang P, Xiao A, Tong X et al (2014) TALEN construction via “Unit Assembly” method and targeted genome modifications in zebrafish. Methods 69(1):67–75. doi:10.1016/j.ymeth.2014.02.010
Meeker ND, Hutchinson SA, Ho L et al (2007) Method for isolation of PCR-ready genomic DNA from zebrafish tissues. Biotechniques 43(5):610, 612, 614
Zheng Q, Cai X, Tan MH et al (2014) Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 57(3):115–124. doi:10.2144/000114196
Cong L, Ran FA, Cox D et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823. doi:10.1126/science.1231143
Acknowledgment
We thank Zhanxiang Wang, Da Liu, and other members in our lab for their efforts on optimizing the CRISPR/Cas applications in zebrafish. This work was partially supported by the National Natural Science Foundation of China (31110103904, 81371264), the 973 Program of the Ministry of Science and Technology of China (2012CB945101, 2015CB942803), and the Seeding Grant for Medicine and Life Sciences of Peking University (2014-MB-06).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Xiao, A., Zhang, B. (2016). Generation of Targeted Genomic Deletions Through CRISPR/Cas System in Zebrafish. In: Kawakami, K., Patton, E., Orger, M. (eds) Zebrafish. Methods in Molecular Biology, vol 1451. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3771-4_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3771-4_5
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3769-1
Online ISBN: 978-1-4939-3771-4
eBook Packages: Springer Protocols