
Abstract
We present a protocol to combine behavioral recording and imaging using 2-photon laser-scanning microscopy in head-fixed larval zebrafish that express a genetically encoded calcium indicator. The steps involve restraining the larva in agarose, setting up optics that allow projection of a visual stimulus and infrared illumination to monitor behavior, and analysis of the neuronal and behavioral data.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Larval zebrafish are small and transparent, and therefore, amongst vertebrate model organisms, they offer unique possibilities for studying neuronal activity using imaging techniques [1, 2]. As their brain is relatively small, roughly 800 × 500 × 300 μm, it is possible to monitor, at high spatial resolution, a large fraction of their brain in a single experiment. This allows experimenters to study circuits that extend all the way from the sensory input to the motor output and to ask questions about complete neural systems. This approach has been used to study sensorimotor processing, spontaneous activity patterns, and learning [3–6]. In this chapter, we present a primer on how to perform these experiments in a restrained, but behaving, 5–7 days post fertilization (dpf) larval zebrafish using scanning 2-photon microscopy. We also describe a simple method, based on linear correlation, which allows us to understand this neuronal activity in terms of the stimulus presented and the behavior measured.
2 Materials
2.1 Embedding Larvae
-
1.
5–7 dpf. Zebrafish larvae with a fluorescent calcium indicator in some or all neurons (e.g., elavl3:GCaMP5G [4]). In this protocol, we will assume the use of a similar GFP-based indicator.
-
2.
Fire-polished glass transfer pipette.
-
3.
35 mm. Petri dish.
-
4.
Sylgard 184 silicone elastomer (Dow Corning).
-
5.
Low melting point agarose (UltraPure LMP Agarose—Invitrogen).
-
6.
Forceps (e.g., Fine Science Tools Dumont #5).
-
7.
Fine scalpel (e.g., Sharpoint Stab knife, restricted depth straight; 15°, 5.0 mm blade).
-
8.
Heating block or water bath.
-
9.
Stereo dissecting microscope.
-
10.
E3 embryo medium: 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl, 0.33 mM MgSO4. To make a buffered E3 solution add Tris solution, adjusted to pH 7.0 with HCl, to a final concentration of 1 mM.
2.2 Imaging Rig
-
1.
Two-photon microscope. We use a custom-built microscope [7], but several flexible, commercially available systems would also be suitable.
-
2.
Compact digital projector for visual stimulation.
-
3.
Stage to hold the sample, fabricated from transparent acrylic.
-
4.
Diffusive screen for stimulus projection (e.g., Rosco filter 3026 [8]).
-
5.
Long pass filter for visual stimulation, e.g., Wratten #29 gel filter (Kodak).
-
6.
850 nm LEDs with mount and power supply.
-
7.
High-speed infrared-sensitive camera (e.g., Pike F-032B, AVT).
-
8.
C-mount camera objective lens that allows the appropriate field of view and working distance for your microscope (see Note 1 ).
-
9.
900 nm short-pass laser blocking filter appropriate to mount in front of the behavior camera objective.
-
10.
45° Hot mirror, 790 nm cut-off.
-
11.
Silver mirror to direct the visual stimulus onto the screen.
-
12.
Computer and software for stimulus production and camera acquisition (see Note 2 ).
2.3 Data Analysis
-
1.
High performance computer.
-
2.
Flexible software for analysis of numerical data and image processing (e.g., MATLAB from Mathworks).
3 Methods
3.1 Embedding Larvae
Zebrafish larvae in the age range of 5–7 dpf still receive nutrition from their yolk and can be embedded in low melting point agarose and survive in good health for more than a day, as long as they are immersed in well-aerated E3 embryo medium [9, 10]. With practice, it is possible to embed larvae quickly, without damage and with minimal tilt.
-
1.
At least 2 days before starting experiments, mix the Sylgard 184 components according to the manufacturer’s instructions and pour to a depth of about 3 mm into 35 mm petri dishes. Cover the dishes and allow 48 h for the Sylgard to polymerize completely.
-
2.
Place a larva (5–7 dpf) in a drop of E3 water in the center of a Sylgard-coated 35 mm Petri dish. Carefully suck off most of the E3 with a fire-polished glass pipette (see Note 3 ). To allow imaging in the brain, nacre mutant larvae, which lack skin melanophores should be used (see Note 4 ) [11].
-
3.
Cover the larva with 2 % low melting point agarose in E3 using a transfer pipette (see Note 5 ).
-
4.
Melt the agarose an hour or so before, in a microwave, and then keep it in a water bath at 35° Celsius. Check that the agarose is not too hot by touching the side of the transfer pipette. Ensure that the agarose and the E3 droplet mix well. The agarose will gel in a few seconds so the next steps should be executed rapidly.
-
5.
Use forceps to mount the larva dorsal side up (see Notes 6 – 8 ) close to the center of the dish by manipulating the agarose around the tail with the closed forcep tips: the larva will be dragged along (Fig. 1c). Pitch can be controlled by gently pressing down on the tail with the side of the forceps.
Fig. 1 
Larval zebrafish embedded in agarose. (a) A 6 dpf larval zebrafish embedded in agarose. The agarose has been removed to free the eyes and the tail. (b) A nacre −/− fish lacking melanophores, and with pan-neuronal expression of a green-fluorescent, genetically encoded calcium indicator, Tg(elavl3:GCaMP5G). Example of the field of view that can be imaged with a 20× objective is shown in red. (c) Typical maneuver (red arrow) that is performed with the forceps without directly touching the larva to fix unwanted roll when embedding
-
6.
Let the agarose set for 10–20 min and then carefully add E3 water to completely cover the agarose (see Note 9 ).
-
7.
Depending on the experiment to perform, and the behavior that will be monitored under the 2-photon microscope, it will be necessary to free the tail and/or the eyes. Do this only after adding E3 water to the dish. Using a scalpel, remove the agarose as shown in Fig. 1a. Using a scalpel, insert the blade vertically close to the fish and then cut away from the body (see Note 10 ). The length of the tail that is freed can be smaller for imaging versus behavioral experiments in order to minimize motion artifacts [6, 12].
-
8.
Under the dissecting microscope, check that the embedded larva is healthy before transferring to the imaging rig (see Note 11 ).

3.2 Two-Photon Microscope
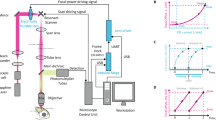
Two-photon microscopes may have widely varying capabilities and features. In this section, we describe a basic setup that may be adapted depending on the space available for the stage and the particular microscope that will be used. Such a setup is schematized in Fig. 2.

Two-photon microscope setup for presenting visual stimuli and tracking behavior. The fish sits on a transparent stage in a two-photon microscope, illuminated from the side by infrared LEDs. Visual stimuli are projected on a screen below the fish. A small hole in the screen allows the tail to be imaged with an infrared-sensitive camera. A hot (infrared reflecting, visible light transmitting) mirror selectively directs infrared light to the camera
-
1.
Present the visual stimulus on a diffusing screen below the embedded fish using a projector and mirror. The stage is cut to a custom shape from transparent acrylic.
-
2.
In order to prevent the visual stimulus interfering with collection of green fluorescence, modify the projector so that illumination is provided only by a red LED or simply place a long-pass filter directly in front of the output beam lens of the projector [13] (see Note 12 ).
-
3.
In order to track behavior, illuminate the larva using infrared LEDs (red arrows in Fig. 2), in the range of 790–850 nm. This will ensure that the illumination light is not visible to the larva but will pass through the filters used to block the laser before the high-speed camera. Place the LED as close as possible to the fish in order to use the lowest power that allows good behavioral tracking. Depending on the behavior to be tracked, it may be convenient to use a spotlight LED placed at an angle on the side of the fish that creates a clear light/dark contrast along the length of the tail: this contrast can enable easy automatic tail tracking.
-
4.
To combine high-speed tracking and visual stimulation from below, insert a 45° hot mirror, which transmits visible light and reflects infrared, into the optical path. Use a 900 nm short-pass filter to prevent the infrared laser light used for 2-photon excitation from reaching the camera sensor.
-
5.
Use a laser wavelength of 950 nm for GCaMP excitation. This ensures that there is minimal bleed-through into the camera that monitors behavior, as well as reducing absorption by melanin. We recommend using a power setting that delivers around 10 mW after the objective lens. Larvae can be imaged stably under these conditions for 12 or more hours.
-
6.
For a balanced trade-off of speed and spatial sampling, use an x–y pixel size just below 1 μm. Since a typical cell body diameter is about 5 μm, this ensures that each cell just spans sufficient pixels to allow subsequent automatic segmentation. We also suggest a similar distance between adjacent z planes. Needless to say, the higher the resolution the better, as long as the frame rate, and experimental duration remain compatible with the behavior to be studied (see Note 13 ).
-
7.
For quantitative analysis, behavioral variables such as eye angle and tail curvature should be extracted from the raw images captured by the high-speed camera [6, 10, 14]. This can be done in real-time, or alternatively the raw movies can be saved for later processing. To measure eye angle, first threshold the image to identify the eye regions, and then determine their angle relative to the body axis, for example by fitting their somewhat elongated outline with an ellipse and measuring the angle of the major axis of the ellipse. The tail can be tracked by selecting a point on the center of the tail where it enters the agarose, and computing the brightness of all points in an arc at a fixed radius from this point. The peak of brightness along this arc will indicate the tail direction (we are assuming that the illumination is such that the tail appears bright on a dark background). This process can be repeated, with each new point serving as the center for the next arc, in order to measure the curvature along the whole tail.
3.3 Data Analysis
The raw imaging data recorded in the experiments consists of the fluorescence time-series for each pixel in each plane. In addition, we have time-series for the behavioral variables monitored and for the stimulus shown. Can we understand the neuronal activity that gives rise to the former in terms of the latter? Here, present a workflow, based on that in ref. [6] which allows the experimenter to gain some insight into this question. This can represent a useful starting point for further analysis tailored to the particular experiment in question.
3.3.1 Elimination of Motion Artifacts
When imaging for prolonged periods of time, motion artifacts will arise either from slow drift or from movement of the brain as the fish behaves. It is important to eliminate these as far as possible from the data in order to perform the correlation analysis presented below. There are many software packages available to do this, for example [15]. In the protocol steps below, assume that the imaging experiment has resulted in NT fluorescence frames f n t , where t = 1,..,T labels the time points of the acquisition of the T frames per plane and z = 1,…,N labels the individual plane, out of a total of N planes.
-
1.
For each plane z create a total fluorescence image from several consecutive images, \( {f}_{\mathrm{sum}}^z=\kern0.28em {\displaystyle \sum}_{t=1}^M{f}_t^z \) that can serve as the initial template to align the individual fluorescence images to. Align all the individual images to this template and call the aligned images \( \overset{\sim }{f_t^z} \). It is typically sufficient to perform an affine transformation, i.e., one that maintains straight lines in the image. Using a measure of how well each individual frame aligns to the average (e.g., the normalized cross-correlation), it is possible to identify and discard frames that align very poorly due to a rapid and vigorous behavioral event such as a struggle. It may be beneficial to repeat this procedure once: reaverage all the aligned frames to a new template \( {\overset{\sim }{f}}_{\mathrm{sum}}^z=\kern0.28em {\displaystyle \sum}_{t=1}^T{\overset{\sim }{f}}_t^z \) and align the original frames to this new template to yield the final set of aligned frames \( {\hat{f}}_t^z \). This will improve the alignment and the crispness of the anatomical image, in cases where the larva moved during the period from which the template was taken.
-
2.
It is then important to register all planes to each other, to correct for the slow accumulation of drift across many planes. An anatomical image for each plane can be obtained by summing all the final aligned frames \( {f}_{\mathrm{anatomy}}^z=\kern0.28em {\displaystyle \sum}_{t=1}^T{\hat{f}}_t^z \). This z-stack can then be registered just as for the time-series in the previous step. The transformations should then be applied the raw frames from each plane.
3.3.2 Unbiased Identification of Functionally Active Units
A common approach to analyzing imaging data is to group pixels in regions of interest (ROIs) and ask: What is the activity in this ROI during the experiment? ROIs can be selected manually, or chosen using automatic segmentation methods. These methods may identify neurons using morphological criteria, pixel correlations, or both together (see refs. [3, 16]). Uneven partitioning of the GCaMP protein between the nucleus and cytoplasm can be used to identify cell bodies (e.g., [17]), or the reverse effect can be achieved, at the expense of some temporal resolution, by specific nuclear targeting of the indicator [18]. Such approaches can identify on the order of 90,000 neurons in the brain of a 6dpf larval zebrafish. Here, we describe a method introduced in [6] that can identify, in an unbiased way, groups of voxels in the brain, corresponding either to cell bodies, or neuropil structures, that are active during an experiment, based on spatial clustering of correlated pixels.
-
1.
Low-pass filter the fluorescence time-series of all individual pixels. The filter should take into account the dynamics of the calcium indicator in question: for GCaMP5G, which has a half-decay time of 0.667 s [19], one can use a filter cut-off of about 2 Hz. This step makes the assumption that high frequency components in the fluorescence signal arise predominantly from photon shot noise and not from changes in indicator fluorescence.
-
2.
Consider a specific pixel (x,y) in a given plane. If it is part of an active neuron, its fluorescence will correlate well with its neighboring pixels, some of which will also be part of this neuron. Calculate the correlation between this pixel and its neighboring pixels. Figure 3b shows the case where the neighboring pixels considered are 24 pixels that together with the pixel in question make up a 5 by 5 square. Each pixel can then be represented by its average correlation with surrounding pixels to create an anatomical correlation map, such as the one shown in Fig. 3c (see Note 14 ).
Fig. 3 
Unbiased identification of functional units. (a) Anatomical image of a single plane created by summing all the fluorescence frames collected for this plane. The field of view corresponds to that shown in Fig. 1a. (b) Every pixel in the plane has an associated fluorescence time-series. For each pixel, one can ask how well it’s fluorescence time-series correlates with that of its neighboring pixels. Here, we show a pixel in red and its 24 neighboring pixels in a 5 by 5 square. The time-series of all 25 pixels are shown as a color map below. (c) Anatomical correlation map obtained by performing the procedure depicted in (b) for all the pixels in plane shown in (a). (d) Starting with the pixels with highest correlation value (seeds), pixels can be aggregated to form functionally active units if their correlation with the seed exceeds a threshold value. One such functionally identified unit is overlaid on the correlation map
-
3.
Generate a control map, to estimate the probability of observing particular correlation values by chance. Partition each pixel’s time-series into a number of equally sized segments lasting ~10 s. Randomly permute these segments and concatenate them to create a shuffled time-series for the pixel. Do this for all the pixels, performing each time a new random permutation for each. Create a control anatomical correlation map by repeating the procedure outlined in the point above. Take care that the duration of each segment is not meaningful in the context of the task (i.e., 10 s segments should not be used if the stimulus repeats itself every 10 s).
-
4.
Threshold the correlation map to identify putative active regions. The threshold value can be determined either by inspection, or by using the control performed above as follows: create a histogram of the distribution of correlation values for the true anatomical correlation map. Repeat this for the control map. Divide the former by the later and choose the threshold value to be that appears 20 times more often in the true versus the control correlation map.
-
5.
Use this correlation map, together with the raw data to iteratively locate and segment ROIs. Here, we give the procedure for 3-D data, but the same approach is applicable to 2-D. Determine the location of the first ROI by choosing the voxel with the highest local average correlation and using this as a “seed.” This voxel has six closest neighbors. If the correlation of the activity in any of these neighboring voxels with the activity in the “seed” voxel exceeds a predefined threshold (see below), incorporate the voxel into the ROI. Calculate the fluorescence time-series for the new ROI, and repeat the previous steps for the new set of nearest neighbors. On each iteration, perform a morphological close operation (e.g., using the Matlab imclose function) to remove holes within ROIs. Once no more voxels can be added, the ROI is considered segmented. Then, find the remaining voxel with the highest local correlation value and use this as the “seed” for the next ROI. The choice of threshold value determines how readily pixels are incorporated, and can be based on the distributions of true and control correlations as outlined above. This algorithm is extremely effective when the signals across planes are expected to be very stereotypical.
-
6.
The segmentation algorithms presented above may yield ROIs that span many adjacent cells with similar functional properties or large areas of neuropil with graded responses. In order to uncover spatial variations in activity, where no clear morphological markers exist, it is useful to split large ROIs into smaller ones by repeated bisection along their longest axis until the constituent ROIs have the size of about one cell body, although, in this case, the ROI boundaries will have no particular significance.

3.3.3 Analysis of Behavior-Activity Correlations
Once we have identified ROIs that are active in our experiment we want to know what feature of the stimulus or behavior the activity reflects. A simple approach is to identify the behavioral parameters which best explain the pattern of neural activity (see ref. [14]). The method we present here aims to identify linear correlations between such parameters and observed activity (see Note 15 ). The workflow is schematized in Fig. 4.

Correlation analysis of neuronal activity and behavioral data. (a) The data collected during the experiment consists of the fluorescence traces of the units detected as described above, the monitored behavior, which in this case consists of left eye position, right eye position and tail deflection, and the stimulus presented. The trace of the unit shown in Fig. 3 is shown here in magenta as an example. (b) From the behavioral and stimulus variables directly measured, derived variables are defined as described in the text (black traces). (c) Each variable to be considered is convolved with the kernel of the calcium indicator (in this case GCaMP5G shown top right) to yield the fluorescence expected from a unit that would linearly code for this variable. Examples of these predictors from the variables in (b) are shown here in blue. For this particular unit (magenta trace) the predictor with the best correlation (0.864) is shown in red
-
1.
First identify the set of behavioral parameters you wish to test. In this approach, neural activity is compared just with a fixed set of predictors, which reflect prior assumptions about the possible signals carried by single neurons. These could be the “raw” variables measured or controlled in the experiment, such as stimulus velocity or eye position, or “derived” variables, which involve simple combinations, or nonlinear transformations of these raw signals. For example, while we measure absolute eye position over the whole range, we can include predictors that consist only of nasal or temporal deviations from the resting position of the eye, or represent the degree of convergence between the two eyes (see Fig. 4b). Similarly, we can generate “derived” predictors from the stimulus velocity, such as binocular rotational or translational motion.
-
2.
The question we ask is: is the neuronal signal we observe linearly related to one of the behavior or stimulus variables we have defined? In order to answer this question, we use correlation analysis. The first step is to convolve all the behavioral and stimulus variables with the estimated spike response function of the calcium indicator we used in our experiment. In the top of Fig. 4c we show the example of an exponential kernel with decay of 0.667 s, consistent with measured responses of GCaMP5G [19]. The resulting convolved traces are the fluorescence signals we would expect to observe if the underlying neural activity in an ROI was linearly related to the associated behavioral variable.
-
3.
For each ROI, correlate its fluorescence trace with all the regressors. This will provide a vector of correlation values for each ROI. As a first pass classification, each ROI can be assigned to the predictor with the maximum correlation value.
-
4.
The simple approach above may lead to some incorrect assignments, as regressors themselves can be correlated, ROIs may have similar correlation values with several different parameters or the appropriate predictors were not included in the tested set. As an alternative, the data can be clustered according to the complete pattern of behavioral correlations, and each ROI assigned to a cluster. In order to perform cluster analysis using a method such as k-means, it is helpful to reduce the dimensionality of the data. Perform principal component analysis on the correlation vectors calculated for all the ROIs. Choose the first n principal components that explain most of the variance in the data, for example 95 %. To reduce the dimensionality of the data, project the correlation vector of each ROI onto this subset of principal components, which are necessarily orthogonal to each other: each ROI will now be assigned an n-dimensional vector of coefficients. Following cluster analysis, the spatial distribution of ROIs belonging to each cluster can be compared, and traces from each cluster can be averaged within or across different trials.
4 Notes
-
1.
It is straightforward to adjust the field of view and working distance of fixed focal length camera objectives by using extension tubes to increase their distance from the camera sensor. Handy web tools for calculating camera field of view and extension tube choices can be found at http://www.1stvision.com.
-
2.
While arbitrary stimuli can be generated directly using low-level graphics libraries such as OpenGL (http://www.opengl.org), a number of freely available packages are specifically tailored for generating visual stimuli for neuroscience, such as Psychophysics Toolbox (http://psychtoolbox.org, Matlab-based) and VisionEgg (http://visionegg.org, Python-based) [20, 21].
-
3.
Larvae may become stuck on the plastic surface of disposable transfer pipettes, and should only be handled using a fire-polished glass pasteur pipette. Twirl a pipette in your fingers while holding the tip in a bunsen flame for a few seconds. After cooling, the pipette tip should feel smooth to the touch, but not be closed.
-
4.
While nacre fish are particularly appropriate for visual experiments, since they have normal eye pigmentation, it is possible to suppress all pigment formation in any fish larva by treatment with 1-phenyl 2-thiourea (PTU). However it should be noted that PTU may directly affect visual processing in the retina [22].
-
5.
Using enough agarose so that it extends to the sides of the Petri dish, can help prevent the agarose from getting unstuck from the Sylgard. However, be careful to limit the depth of agarose, since this will minimize the deviation in pitch of the larva and also ensure that the objective can get close enough to have the appropriate working distance.
-
6.
If the larva is on its side, tap the rim of the dish with the forceps. This will typically induce it to right itself up and swim forward slightly. It is therefore useful to position the larva radially on a side of the dish facing the center and tap the edge slightly. Try to touch the larva as little as possible and avoid touching its head.
-
7.
Make sure the larva is close to the center of the Petri dish, so that the objective does not risk touching the rim.
-
8.
Judging that the fish is not tilted to the left or right can be tricky when using stereo microscopes that offer different views through the left and right eyepieces. To avoid this problem, turn the dish so that the long axis of the fish is oriented left to right.
-
9.
Pass the E3 through a syringe filter, to remove any dust or other particles floating in the water that may interfere with imaging.
-
10.
It is important to cut all the way to the Sylgard to make sure that the agarose will not flake away in pieces. Make sure a closed loop is cut around every section of agarose to be removed, and then pull the whole piece carefully away.
-
11.
Larvae should have vigorous blood flow and a heartbeat at around 2–3 Hz. Edema around the eyes or a cloudy appearance in the brain or spinal cord are indicators of poor health.
-
12.
Due to the very high intensity of the visual stimulus, compared with fluorescence emission, it is usually necessary to filter the projector output to remove any stray light in the emission wavelengths. It is important to consider, when choosing and placing the filters, that the properties of interference filters are usually only specified for a small range of incidence angles, and that sometimes an absorptive filter may be a better choice.
-
13.
It is advisable to image every plane for a period of time that ensures the occurrence of about three events of interest, to clearly distinguish relevant activity from background. These events may be stimulus presentations or particular behavioral actions. It is also important to include a sufficient “baseline” period. In order to cover most of the brain in a single experiment, we suggest using a stimulus protocol that lasts around 2 min per plane. This will ensure that 250 planes can be imaged in 500 min, just over 8 h. The frame rate should be matched to the indicator kinetics to avoid missing fast signals, at least 2 Hz for GCaMP5G.
-
14.
There may be certain cases when the activity that we are interested in identifying is temporally reproducible from plane to plane. This is the case when the same stimulus sequence is presented in each plane and we are looking at averaged and/or highly reproducible patterns of activity. In this case, the set of neighboring pixels can be extended to include those in adjoining planes and a cube can be considered in Fig. 3b instead of a square. This is however not possible if we are seeking activity related to temporally variable features of behavior: in this case, this analysis must be performed within an individual plane.
-
15.
In many cases, the relationship of neuronal firing to behavioral parameters will be nonlinear. For example, responses may only occur above a threshold, show saturation, or depend on conjunction of multiple variables. In cases where a particular nonlinearity is expected, e.g., threshold-linear responses to eye position, this can be explicitly included in the set of predictors [6, 23]. Alternatively, nonlinear regression methods can be used [24], including some that allow arbitrary relationships between behavioral variables and activity (e.g., see ref. [25]), although there will typically be a trade-off between model flexibility and interpretability.
References
Fetcho JR, O’Malley DM (1997) Imaging neuronal networks in behaving animals. Curr Opin Neurobiol 7:832–838
Kettunen P (2012) Calcium imaging in the zebrafish. Adv Exp Med Biol 740:1039–1071
Ahrens MB, Li JM, Orger MB et al (2012) Brain-wide neuronal dynamics during motor adaptation in zebrafish. Nature 485:471–477
Ahrens MB, Orger MB, Robson DN et al (2013) Whole-brain functional imaging at cellular resolution using light-sheet microscopy. Nat Methods 10:413–420
Wolf S, Supatto W, Debrégeas G et al (2015) Whole-brain functional imaging with two-photon light-sheet microscopy. Nat Methods 12:379–380
Portugues R, Feierstein CE, Engert F et al (2014) Whole-brain activity maps reveal stereotyped, distributed networks for visuomotor behavior. Neuron 81:1328–1343
Renninger SL, Orger MB (2013) Two-photon imaging of neural population activity in zebrafish. Methods 62:255–267
Niell CM, Smith SJ (2005) Functional imaging reveals rapid development of visual response properties in the zebrafish tectum. Neuron 45:941–951
O'Malley DM, Sankrithi NS, Borla MA et al (2004) Optical physiology and locomotor behaviors of wild-type and nacre zebrafish. Methods Cell Biol 76:261–284
Portugues R, Engert F (2011) Adaptive locomotor behavior in larval zebrafish. Front Syst Neurosci 5:72
Lister JA, Robertson CP, Lepage T et al (1999) Nacre encodes a zebrafish microphthalmia-related protein that regulates neural-crest-derived pigment cell fate. Development 126:3757–3767
Severi K, Portugues R, Marques J et al (2014) Neural control and modulation of swimming speed in the larval zebrafish. Neuron 83(3):692–707
Orger MB, Kampff AR, Severi KE et al (2008) Control of visually guided behavior by distinct populations of spinal projection neurons. Nat Neurosci 11:327–333
Miri A, Daie K, Burdine RD et al (2011) Regression-based identification of behavior-encoding neurons during large-scale optical imaging of neural activity at cellular resolution. J Neurophysiol 105:964–980
Nestares O, Heeger DJ (2000) Robust multiresolution alignment of MRI brain volumes. Magn Reson Med 43:705–715
Mukamel EA, Nimmerjahn A, Schnitzer MJ (2009) Automated analysis of cellular signals from large-scale calcium imaging data. Neuron 63:747–760
Romano SA, Pietri T, Pérez-Schuster V et al (2015) Spontaneous neuronal network dynamics reveal circuit’s functional adaptations for behavior. Neuron 85:1070–1085
Kim CK, Miri A, Leung LC et al (2014) Prolonged, brain-wide expression of nuclear-localized GCaMP3 for functional circuit mapping. Front Neural Circ 8:138
Akerboom J, Chen TW, Wardill TJ et al (2012) Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci 32:13819–13840
Straw AD (2008) Vision egg: an open-source library for realtime visual stimulus generation. Front Neuroinformatics 2
Brainard DH (1997) The psychophysics toolbox. Spat Vis 10:433–436
Page-Mccaw PS, Chung SC, Muto A et al (2004) Retinal network adaptation to bright light requires tyrosinase. Nat Neurosci 7:1329–1336
Kubo F, Hablitzel B, Maschio MD et al (2014) Functional architecture of an optic flow-responsive area that drives horizontal eye movements in zebrafish. Neuron 81:1344–1359
Bianco IH, Engert F (2015) Visuomotor transformations underlying hunting behavior in zebrafish. Curr Biol 25:831–846
Huber D, Gutnisky DA, Peron S et al (2012) Multiple dynamic representations in the motor cortex during sensorimotor learning. Nature 484:473–478
Acknowledgments
The authors would like to acknowledge the contribution of colleagues Claudia Feierstein and Florian Engert to the development of the methods described in this protocol. MBO was supported by a Marie Curie Career Integration Grant, PCIG09-GA-2011-294049. RP was supported by the Max Planck Society.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Orger, M.B., Portugues, R. (2016). Correlating Whole Brain Neural Activity with Behavior in Head-Fixed Larval Zebrafish. In: Kawakami, K., Patton, E., Orger, M. (eds) Zebrafish. Methods in Molecular Biology, vol 1451. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3771-4_21
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3771-4_21
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3769-1
Online ISBN: 978-1-4939-3771-4
eBook Packages: Springer Protocols