
Abstract
SUMOylation is a widely used protein posttranslational mechanism capable of regulating substrates localization, stability, and/or activity. Identification and characterization of bona fide SUMO substrates is a laborious task but its discovery can shed light to exquisite and crucial regulatory signaling events occurring within the cell. Experiments performed in the SUMOylation field often demand a good understanding of the putative substrate’s function and necessitate a solid knowledge regarding both in vitro and in vivo approaches. This contribution offers a simplified view into some of the most common experiments performed in biochemical and cell biological research of the SUMO pathway in mammalian systems. It also summarizes and updates well established protocols and tricks in order to improve the likelihood to obtain reliable and reproducible results.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Small ubiquitin-like modifier (SUMO , originally named Sentrin) is a protein composed of about 97 residues (depending of the isoform considered) after proteolytic processing of its C-terminal tail mediated by SUMO-specific proteases. SUMO is present in eukaryotic genomes as several isoforms with, in many cases, overlapping functions [1].
SUMO-activating enzyme (SAE) mediates the attachment of SUMO into substrates in a similar fashion as ubiquitin-activating enzymes (E1s) in the ubiquitylation reaction activate ubiquitin. However, in the case of SUMO, SAE is not a single protein but a heterodimer composed of SAE1 and SAE2 [2]. Phylogenetic conservation analyses of the heterodimer show that regions of the ubiquitin E1s are split into SAE1 and SAE2 in the SUMO pathway . The mechanistic details concerning adenylation and thio-esther bond formation are conserved among both processes.
In the SUMOylation cascade, there is a unique SUMO-conjugating enzyme (E2 ) responsible for the transfer of SUMO into the substrate, termed Ubc9. The existence of a handful of SUMO ligases (E3s), directly responsible for mediating the conjugation of SUMO moieties into substrates, have been well established in the literature (such as the members of the PIAS family); however, it is also clear that Ubc9 can act at least in in vitro reactions as both E2 and E3 components during SUMO substrates’ modification [3, 4].
Upon SUMOylation , several substrates’ functions can be affected and there is no obvious rule to predict how SUMOylation will alter the structural and/or functional properties of a substrate. Remarkably, SUMOylation is a reversible mechanism since deSUMOylation can be achieved by SUMO proteases or SENtrin-specific proteases (SENPs) [5].
From many studies aimed to analyze the cellular SUMO proteome, it has become apparent that SUMOylation substrates often possess a SUMOylation consensus site in which a lysine (to which SUMO gets attached) is located in the middle of a linear motif defined by ΨXKE (where Ψ is a hydrophobic acid, X is any amino acid, and E is a glutamic acid or negatively charged residue). In addition, SUMOylation can also occur at different lysines which are not necessarily found within the context of the SUMOylation consensus sites of a given substrate.
The SUMOylation pathway also displays a great level of diversity and versatility. SUMOylated substrates can indeed be modified by mono-SUMOylation but also by poly- or multi-SUMOylation . Remarkably, SUMO isoforms have different preferences toward one or the other modifications, with SUMO1 being more prone to only conjugate once while SUMO2/3 rather generates poly-SUMOylated species, at least in mammals . Poly-SUMOylation occurs when the reaction employs another SUMO protein (which has been previously attached to a substrate) as the substrate. Among the three major SUMO isoforms in mammalians , only SUMO2 and 3 can form SUMO chains due to the presence of a SUMO consensus site in their primary sequences. SUMO1 lacks this site and therefore cannot form chains.
Finally, SUMOylated species can be recognized by other proteins via electrostatic interactions with SUMO. This process is mediated by the so-called SIMs or SUMO interacting motifs which are involved in the regulation of many different signaling pathways [6–8].
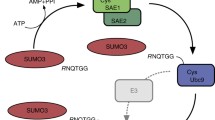
This chapter provides an updated insight into different protocols and experimental techniques that aim to identify and characterize SUMO substrates. Our contribution includes a standard research workflow (see Fig. 1) in which in vitro preliminary observations are subsequently confirmed and complemented with in vivo experiments.

Graphical workflow for detection of SUMOylation substrates
2 Materials
2.1 Materials for In Vitro Experiments Aimed to Detect SUMOylation of Recombinantly Purified Substrates
-
1.
Recombinant E1 heterodimer or SUMO-activating enzyme (SAE) : It can be purchased from Enzo Life Sciences Cat No. BML-UW9330.
-
2.
Recombinant E2 or SUMO-conjugating enzyme : Enzo Life Sciences supplies an untagged Ubc9 (Cat No. ALX-201-046, see Note 1 ).
-
3.
SUMO isoforms: SUMO 1, 2, or 3: They can be purchased from several sources including Boston Biochem.
-
4.
SUMO-specific primary antibodies: anti-SUMO antibodies specific for each isoform are commercially available from different providers.
-
5.
ATP regenerating system: 1.25 mM ATP, 1.25 mM MgCl2, 1.9 mM Creatine Phosphate, and 6.25 μg/mL Creatine Phosphokinase (all components can be purchased from Sigma Aldrich).
-
6.
10× SUMO buffer (500 mM Tris–HCl pH = 7.5, 50 mM MgCl2, supplemented with 10 mM ATP).
-
7.
Purified substrate: produced and purified from recombinant organisms (such as E. coli). The substrate can be tagged in order to facilitate its detection by Western-blot ting analysis or non-tagged if primary antibodies are available.
-
8.
Temperature-controlled water bath.
-
9.
1.5 mL sterile test tubes, pipette tips, and pipettes.
-
10.
SDS-PAGE gradient gels (adjusted depending on the specific requirements), membranes for Western-blot , appropriate secondary antibodies, and all related consumables (running buffer, transfer buffer, filter paper, PBS-Tween solution, casein, etc.).
2.2 Materials for Semi-In Vivo Experiments Aimed to Detect SUMOylation of Substrates Produced in Rabbit Reticulocytes Extracts
-
1.
Rabbit reticulocytes in vitro transcription-translation kit (read carefully recommendations and detailed protocols provided from the manufacturer). It can be purchased from Promega.
-
2.
35S-Methionine or 35S-Cysteine (see Note 2 ).
-
3.
Plasmids containing the gene of interest under the control of a T7, T3, or SP6 promoter (see Note 2 ).
-
4.
N-ethylmaleimide, NEM (it acts as a SUMO protease (SENP) or deSUMOylase inhibitor) [9].
-
5.
1.5 mL sterile test tubes, pipette tips, and pipettes.
-
6.
15 % SDS-PAGE gels (the acrylamide concentration can be adjusted depending on the specific requirements), and all related consumables (running buffer, Western-blot detection reagents, etc.).
-
7.
All personal protecting equipment and devices needed in order to prevent and/or eliminate contaminations with radioactive material.
2.3 Materials for In Vivo Experiments Aimed to Detect SUMOylated Proteins in Cells
-
1.
Cell lines transfected with the gene(s) of interest.
-
2.
Plasmids for SUMO pathway proteins (SAE1/SAE2, Ubc9, and the SUMO isoforms). pCDNA3.1 plasmids for all components are available.
-
3.
Agarose beads with immobilized coating of protein A/G.
-
4.
Suitable primary anti-SUMO antibodies .
-
5.
Substrate-specific antibodies and/or tag-specific antibodies (these antibodies can be used either for detection or for co-immunoprecipitation experiments).
-
6.
Agarose glutathione beads.
-
7.
SUMO Interacting Motif traps (SIMtraps).
-
8.
N-ethylmaleimide, NEM (it acts as a SUMO protease or deSUMOylase inhibitor).
-
9.
DMEM media, fetal bovine serum, 6-well plates or 10 cm dishes, and all needed tissue culture reagents.
-
10.
1.5 mL sterile test tubes, pipette tips, and pipettes.
-
11.
15 % SDS-PAGE gels (acrylamide concentration can be adjusted depending on the specific requirements), membranes for Western-blot ting, appropriate secondary antibodies, and all related consumables (running buffer, transfer buffer, filter paper, PBS-Tween solution, casein, etc.).
2.4 Materials for Semi-In Vivo Experiments Aimed to Detect SUMOylated Proteins in E. coli
-
1.
Competent Escherichia coli BL21 (DE3).
-
2.
Suitable bicistronic or tricistronic plasmids containing SAE1, SAE2, E2 , and/or E3 enzymes as well as SUMO isoforms of interest (it is recommended to obtain these constructs from original sources reported in previous studies) [4, 10].
-
3.
Antibiotics, LB media, LB agar plates, small culture tubes (with loose caps) and 1.5 mL sterile test tubes, pipette tips, and pipettes.
-
4.
Plasmids containing the wild-type substrate of interest and also mutated (lysine-to-arginine) in its putative SUMOylation sites.
-
5.
Suitable anti-SUMO-specific primary antibodies.
-
6.
Substrate-specific antibodies and/or tag-specific antibodies for Western-blot applications.
-
7.
15 % SDS-PAGE gels (acrylamide concentration can be adjusted depending on the specific requirements), membranes for Western-blot , appropriate secondary antibodies, and all related consumables (running buffer, transfer buffer, filter paper, PBS-Tween solution, casein, etc.).
-
8.
Commassie-staining solution and distaining solution.
-
9.
Bradford reagent and suitable protein standards.
3 Methods
3.1 In Vitro Experiments Aimed to Detect SUMOylated Substrates
-
1.
Prepare and mix recombinant proteins (SAE1, SAE2, Ubc9, and SUMO isoforms) according to manufacturer’s instructions in a 1.5 mL test tube. Add ATP regenerating system if desired (see Note 3 ).
-
2.
Add the substrate protein and adjust the volume up to 30 μL with SUMO buffer and deionized water. Mix/homogenize properly.
-
3.
Incubate the sealed test tube at 37 °C for 1 h. It is recommended to prepare reactions in which the different components of the SUMO machinery are individually removed as well as preparing a reaction without substrate as negative controls.
-
4.
Upon completion, the reaction is stopped by addition of 10 μL of 4× SDS-PAGE sample buffer and boiled at 95 °C for 5 min.
-
5.
Spin down all samples and load 20 μL of each assay into two 15 % SDS-PAGE gels.
-
6.
Perform SDS-PAGE adjusting time and voltage. One of the gels will be subsequently used for Western-blot analysis while the other will be Coomassie-stained.
-
7.
Transfer the first gel onto a suitable membrane and block according to traditional methods for Western-blot. Incubate with primary antibodies against the specific substrate (or tag of the substrate). It is recommended to re-blot the membrane using antibodies against the specific SUMO isoform(s) employed. Develop the membrane using adequate secondary antibodies.
-
8.
A complete analysis of this experiment should reveal in first instance whether SUMOylation of the substrate takes place or not by evidencing a shift of ~15–20 kDa in the apparent weight of the substrate per SUMO moiety conjugated (see Note 4 ).
-
9.
In the second gel, it is also possible to visualize by Coomassie-staining the SUMOylation reactions although it is not always obvious to detect it given the substoichiometric amounts of substrates that are usually SUMOylated in the in vitro reactions. In addition, there is a higher risk of ambiguity related to the exact nature of putative SUMOylated bands observed.
3.2 Semi-In Vivo Experiments Aimed to Detect SUMOylated Proteins Using Rabbit Reticulocyte Extracts
3.2.1 Production of the Radiolabeled Substrate for SUMOylation
-
1.
Thaw the components of a rabbit reticulocyte lysates kit on ice 5–10 min prior to starting the assay. In parallel, place 1.5 mL test tubes on ice and set the temperature of a thermal bath at 37 °C.
-
2.
Organize all personal protective equipment according to radioactive material safety protocols.
-
3.
For a 10 μL reaction, add 5 μL of rabbit reticulocyte lysates into pre-chilled tubes and label them properly depending on the type of samples or controls to be tested.
-
4.
Add the T7, T3, or SP6 promoter-based DNA plasmid containing the gene of interest. Use concentrated and pure DNA (>500 ng/μL) in order to dilute as little as possible the reticulocyte extracts and mix gently, always keeping the samples on ice. The volume of DNA added should not exceed 2 μL.
-
5.
Extract enough 35S-Methionine or 35S-Cysteine from the shielded radioactive vial following safety protocols and transfer it to a designated tube kept on ice.
-
6.
Add 2 μL of radiolabeled amino acid (according to the manufacturer’s instructions) to the mixture still on ice, mix/homogenize and incubate the reaction for 90 min at 30 °C. Use a radiation-proof shield in front of the thermoblocker in order to contain radioactive emissions.
-
7.
Stop the reaction by cooling on ice and store the sample at −80 °C. Take an aliquot of the reaction (0.5–2 μL) and add SDS-PAGE sample buffer and heat at 95 °C for 5 min avoiding spills or that the lids will “pop” due to the boiling. Spin down each sample before further handling.
-
8.
Prepare an SDS-PAGE system in order to run the samples always behind a radiation-proof shield. Load samples and run using the appropriate settings for voltage and time.
-
9.
Stop the run before the migration front of the gel escapes from the SDS-PAGE system in order to minimize radioactivity in the contaminated liquid waste.
-
10.
Consider nonetheless the running buffer as radioactive waste and dispose it accordingly. Glass plates (if applicable), electrodes, and tank can be washed and reused only if they are decontaminated following protocols designed to handle contaminated radioactive materials (please check local regulations with the safety officer of your institution).
-
11.
The gel (eventually after Coomassie-staining) is vacuum- and heat-dried using an appropriate device and the correct protections: (a) plastic wrap to avoid contact of the gel with the surfaces inside the gel drier; and (b) radiation-proof shields.
-
12.
Expose the dried gel using a Phosphorimaging screen overnight at room temperature. Visualize and analyze the results with the help of a Phosphorimager and ad hoc software. The gel must be treated as radioactive solid waste once the analysis is done.
-
13.
Analysis of the position of the radioactive band should reveal efficient transcription-translation of the substrate in vitro.
3.2.2 SUMOylation Assay Using the Radiolabeled Substrate
-
1.
To 1–2 μL of rabbit reticulocyte lysates programmed to produce the radiolabeled substrate of interest, add a given amount (50–200 ng) of SUMO isoforms (mainly SUMO1, 2, or 3); 200 nM SAE1/SAE2; 500 nM Ubc9; and required volumes of 10× SUMO buffer and ATP regenerating system into a final volume reaction of 15–20 μL. Always keep in mind not to dilute too much the reactions.
-
2.
Incubate the reaction at 37 °C for the desired time. We recommend a kinetic including time zero, 15, 30, 45, 60, and 120 min.
-
3.
Analyze the reaction by SDS-PAGE as indicated above (Subheading 3.2.1). In this case, the radioactive bands detected by the Phosphorimager should show (usually) a major band corresponding to the molecular weight of the substrate while its SUMOylated form(s) should display apparent higher molecular weight(s) of 15–20 kDa per SUMO moiety conjugated.
-
4.
A complete analysis using this method should include reactions in which the lysine-to-arginine mutants (generated by DNA mutagenesis) are also included.
3.3 Methodology for In Vivo Experiments Aimed to Detect SUMOylated Substrates
-
1.
Obtain a cell line (transiently or stably) expressing or overexpressing the protein of interest. Protocols and suitable cell lines will depend on the nature of the given pathway that is scrutinized.
-
2.
Prepare a lysis buffer according to the specific experimental requirements of the substrate and cell line utilized. We suggest a buffer with the following composition: salts 0–1 M, ionic detergent (if applicable): 0.01–0.5 %, non-ionic detergent (if applicable): 0.1–1 %, divalent cations: 0–10 mM, EDTA: 0–5 mM, pH = 7–8. As an example, we propose the following buffer: 100 mM Tris–HCl, 5 mM EDTA, 1 mM TCEP, 0.5 % NP-40, supplemented with Complete tablets (Roche Diagnostics) and 1 mM NEM .
-
3.
Select a plate or set of wells containing at least 106 cells (transfected and non-transfected as negative controls), strip them out using trypsin-EDTA in the case of adherent cells. Wash the cells with PBS once and resuspend them in 500 μL of lysis buffer and incubate on ice for 20 min.
-
4.
Centrifuge the lysates at 16,000 × g at 4 °C for 20 min. The pellet contains cellular debris and insoluble material. Save the supernatant and collect it into pre-chilled 1.5 mL test tubes kept on ice.
-
5.
Measure the total protein concentration of the supernatant by performing a Bradford assay and incubate identical amounts (usually at least 1 mg) of lysates produced from transfected and non-transfected cells with 1 μg of an antibody directed against the substrate of interest (tag or protein substrate itself). Incubate at 4 °C for 4–6 h with constant rotation.
-
6.
Prepare agarose-protein A and/or G beads (according to the specific antibody used) by washing and equilibrating them in the appropriate buffer used for co-immunoprecipitation (see details above). Add protein A and/or G beads (15 μL of slurry) to the lysate previously incubated with the immunoprecipitating antibody and incubate again for 1–2 h at 4 °C with constant rotation.
-
7.
Centrifuge for 5 min at 1,600 × g at 4 °C to separate supernatant from pellet (containing the beads). Wash the pellet (beads + antibodies + immunoprecipitated proteins) three times with 1 mL of buffer, always performing very mild centrifugation steps (1,600 × g at 4 °C for 5 min).
-
8.
Add 15 μL 2× of SDS-PAGE loading buffer into the tube and boil for 5 min at 95 °C. Load samples onto an SDS-acrylamide gel. Set the proper voltage and time and perform SDS-PAGE.
-
9.
Transfer the gel into a suitable membrane (usually nitrocellulose or PVDF) and block the membrane according to traditional methods for Western-blot ting. Incubate with primary antibodies directed against different SUMO isoforms (mainly SUMO1, 2, or 3). Develop the membrane using appropriate secondary antibody taking into consideration that IgGs from the antibody used for immunoprecipitation were also transferred to the same membrane. A careful choice of antibodies can avoid cross-reactions and interference between antibodies (see Note 5 ).
-
10.
The result of this Western-blot will complement any in vitro assay previously done with the same substrate. This experiment provides evidence that the endogenous cellular machinery can SUMOylate the substrate of interest. In the same way as indicated before, the SUMOylation site can be confirmed or identified if several conditions using candidate lysine-to-arginine mutants are employed.
-
11.
SUMOylation efficiency in cells can be “boosted” by co-overexpressing Ubc9 together with the substrate of interest. On the other hand, a dominant-negative (or activity-dead) mutant of Ubc9 (mutated in the Cysteine used for SUMO esterification) should reduce the efficiency of SUMOylation of the substrate by the cellular endogenous machinery.
3.3.1 Optional Protocol
-
1.
From step 5, it is possible to perform a pull-down experiment rather than an immunoprecipitation . This protocol takes advantage of the ability of some short linear motifs to bind SUMO isoforms [6]. SUMO-interacting motifs (SIM) are conserved linear elements that can be placed in tandem along an engineered sequence to create an artificial polypeptide called SUMO-trap [11]. In this particular protocol, SUMO-traps are tagged with GST (but other tags such as V5 also exist).
-
2.
Wash and equilibrate GSH beads.
-
3.
Mix SUMO-trap and lysates prior to incubation with GSH beads. Incubation of SUMO-traps with the lysate should be adjusted depending on the substrate since selective attachment of SUMO1, 2, or 3 may affect interaction with SUMO-traps (see Note 6 ). In general, incubation should be carried out at 4 °C for several hours.
-
4.
Wash beads with lysis buffer three times and recover the beads (and save the supernatants that can also be analyzed by SDS-PAGE to confirm depletion of SUMOylated species by the SUMO-traps) after centrifuging the mixture at 4 °C for 5 min at 1,600 × g.
-
5.
Elute bound material to SUMO-traps using a small volume of lysis buffer supplemented with 25 μM freshly dissolved Glutathione (check pH of the buffer when preparing it). Centrifuge at 3000 × g at 4 °C for 3 min and recover supernatant in a new pre-chilled test tube.
-
6.
Proceed with the regular (previous) protocol from step 8.
3.4 Methodology for Semi-In Vivo Experiments Aimed to Detect SUMOylated Proteins in E. coli
-
1.
Transform SAE1/SAE2 or fusion construct into E. coli BL21 (DE3) cells. Obtain positive colonies and proceed sequentially with another transformation using Ubc9 DNA plasmid [4, 10]. Plate and select using antibiotics after each transformation.
-
2.
Generate competent cells from the positive colonies above and split into three different new transformations, one for each SUMO isoform. Prepare competent cells of all the bacterial lines.
-
3.
Finally, transform the gene of interest or its lysine-to-arginine mutants in different reactions, plate and select them using the appropriate antibiotics. Keep in mind that a negative control should be included in which an empty plasmid (the backbone plasmid of the substrate) should be transformed in parallel.
-
4.
Select a single colony and grow it on 3 mL LB media supplemented with the adequate antibiotics overnight at 37 °C with shaking.
-
5.
Transfer 250 μL of each culture into fresh media up to a final volume of 5 mL. Supplement with the proper antibiotics and grow until the O.D. at 600 nm reaches between 0.6 and 1.0.
-
6.
Induce using the corresponding compounds (IPTG, arabinose, etc.).
-
7.
Collect the pellet after 2 h of expression and lyse the cells by sonication using the lysis buffer described in Subheading 3.3, step 2 but without detergents.
-
8.
Measure total protein concentration by Bradford and prepare samples of ~30 μL of each lysate with the same amount of proteins (60–80 μg).
-
9.
Add 10 μL of 4× SDS-PAGE sample buffer into each tube and boil for 5 min at 95 °C. Split samples in two.
-
10.
Load samples onto two different SDS-PAGE gels. Set the proper voltage and time and perform SDS-PAGE.
-
11.
Transfer each gel into a suitable membrane and block according to traditional methods for Western-blot ting. Incubate one membrane with primary antibodies against different SUMO isoforms (mainly SUMO1, 2, or 3). The second membrane could be blotted using a substrate-specific antibody.
-
12.
Process the membranes using the corresponding secondary antibodies (see Note 7 ).
4 Notes
-
1.
It is also possible to use a fusion protein containing the functional units of both SAE1 and SAE2 [12].
-
2.
Check how many methionines or cysteines are encoded within the sequence of the substrate as it can determine the feasibility of detecting a proper signal and which radiolabeled amino acid(s) to employ.
-
3.
Protein concentrations can be adapted but is recommended to use a molar ratio of 1:2:20 of E1 heterodimer:Ubc9:SUMO isoforms at the nM–μM range. Substrates can be added as a 10× molar excess compared to the total amount of SUMO in the test tube.
-
4.
Consensus SUMOylation sites can be inferred using different SUMO predictors available on the Internet. Based on this information, it is also possible to produce lysine-to-arginine mutants at particular locations on the substrates. Quikchange mutagenesis can be performed to disrupt the SUMOylation site(s). Including this control in the protocol will offer an invaluable test of the specificity of the SUMOylation reaction, in addition to allowing the identification of the modification site(s) in the substrate.
-
5.
It is also possible to overexpress, in the cells transfected with the putative SUMOylatable substrate, the different (tagged) SUMO isoforms using mammalian plasmids suitable for transfection.
-
6.
SIM-traps can be produced in-house, requested to other laboratories under MTAs, or purchased from providers such as Ubiquitin-Proteasome Biotechnologies (Cat. No. J4410) or Boston Biochem (Cat. No. AM-200).
-
7.
The results from these blots will provide evidence that the SUMO machinery can SUMOylate the substrate under physiological conditions in a heterologous system. As suggested, the SUMO site can be confirmed if several bacterial clones are created using candidate lysine-to-arginine mutants. The system is advantageous in the sense that large amounts of SUMOylated substrate can be produced and purified in order to perform further biophysical characterizations. There is also a well-described way of performing this protocol using the plant-specific SUMO machinery [13].
References
Hay RT (2013) Decoding the SUMO signal. Biochem Soc Trans 41:463–473
Tang Z, Hecker CM, Scheschonka A et al (2008) Protein interactions in the sumoylation cascade—lessons from X-ray structures. FEBS J 275:3003–3015
Wang J, Taherbhoy AM, Hunt HW et al (2010) Crystal structure of UBA2ufd-Ubc9: insights into E1-E2 interactions in sumo pathways. PLoS One 5:e15805
O’Brien SP, DeLisa MP (2012) Functional reconstitution of a tunable E3-dependent sumoylation pathway in Escherichia coli. PLoS One 7:e38671
Hickey CM, Wilson NR, Hochstrasser M (2012) Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13:755–766
Song J, Durrin LK, Wilkinson TA et al (2004) Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc Natl Acad Sci U S A 101:14373–14378
Namanja AT, Li YJ, Su Y et al (2012) Insights into high affinity small ubiquitin-like modifier (SUMO) recognition by SUMO-interacting motifs (SIMs) revealed by a combination of NMR and peptide array analysis. J Biol Chem 287:3231–3240
Kerscher O (2007) SUMO junction-what’s your function? New insights through SUMO-interacting motifs. EMBO Rep 8:550–555
Da Silva-Ferrada E, Lopitz-Otsoa F, Lang V et al (2012) Strategies to identify recognition signals and targets of SUMOylation. Biochem Res Int 2012:875148
Mencía M, De Lorenzo V (2004) Functional transplantation of the sumoylation machinery into Escherichia coli. Protein Expr Purif 37:409–418
Da Silva-Ferrada E, Xolalpa W, Lang V et al (2013) Analysis of SUMOylated proteins using SUMO-traps. Sci Rep 3:1690
Uchimura Y, Nakao M, Saitoh H (2004) Generation of SUMO-1 modified proteins in E. coli: towards understanding the biochemistry/structural biology of the SUMO-1 pathway. FEBS Lett 564:85–90
Okada S, Nagabuchi M, Takamura Y et al (2009) Reconstitution of Arabidopsis thaliana SUMO pathways in E. coli: functional evaluation of SUMO machinery proteins and mapping of SUMOylation sites by mass spectrometry. Plant Cell Physiol 50:1049–1061
Acknowledgements
Research performed at the laboratory of Pathophysiological Cell Signaling is funded by the following bodies: FWO (G0C7514N grant), BELSPO (IAP-VII/07 program), VUB Research Council (new PI grant), and Innoviris (Brains Back to Brussels program to GJG). CC thanks financial support from the European Union’s Seventh Framework Program (FP7) 2007–2013 under grant agreement no. 264257. The authors would also like to acknowledge networking support by the Proteostasis COST Action (BM1307).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Cedeño, C., La Monaca, E., Esposito, M., Gutierrez, G.J. (2016). Detection and Analysis of SUMOylation Substrates In Vitro and In Vivo. In: Matthiesen, R. (eds) Proteostasis. Methods in Molecular Biology, vol 1449. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3756-1_16
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3756-1_16
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3754-7
Online ISBN: 978-1-4939-3756-1
eBook Packages: Springer Protocols