
Abstract
Group II introns are large catalytic RNAs and mobile retroelements that encode a reverse transcriptase. Here, we provide methods for their identification in bacterial genomes and further analysis of their splicing and mobility capacities.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Group II introns are mobile metalloribozymes that self-splice from precursor RNA to generate excised intron lariat RNA that invade new DNA genomic locations by reverse splicing [1–3]. These retroelements also encode a reverse transcriptase that stabilizes the RNA structure for forward and reverse splicing and finally converts the inserted intron RNA back to DNA. Although not numerous, group II introns are found widely across the domains of life, being present in eubacteria, archaebacteria, and eukaryotic organelles.
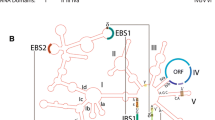
Typically, a complete group II intron consists of a conserved RNA structure of 500–800 nt organized in six domains and an intron-encoded protein (IEP; 400–700 amino acids) enclosed in one of the RNA domains (domain IV). The IEP is a multifunctional protein that has two conserved domains: an N-terminal RT domain and a Maturase/thumb domain (also named X-domain). Some IEPs additionally exhibit a C-terminal DNA-binding (D) region followed by a DNA endonuclease (En) domain (Fig. 1).

Group II intron structure. (a) Scheme of a group II intron. Three different group II introns representative of IIA, IIB, and IIC classes are depicted. The extension of the intron is defined by two exon edges (E1, E2) and contains six domains (I-VI) spanning a region of approximately 2 kb, which include an ORF region (IEP). In the IEP can be identified a reverse transcriptase domain (RT), maturase (X), and eventually DNA-binding motifs (D) followed by a canonical DNA endonuclease domain (En). Schematics of introns and ORFs are to scale (b) group II intron RNA secondary structure. Structure of the representative bacterial IIB1 intron RmInt1 (not to scale). Boxes indicate sequences involved in tertiary interactions (Greek letters, EBS, IBS). The loop of DIV (ORF), which encodes the IEP, is depicted by dashed lines. Specific nucleotides common in the intron boundaries as well as the conserved domain V are indicated
The retromobility mechanism of group II introns has been well studied biochemically and genetically elsewhere ([2], and references in). For identification of group II introns, it is relevant to know that most group II introns have very high sequence selectivity for a long DNA target insertion site known as a homing site (20–35 bp). Often, the homing site is in a conserved gene and the intron boundaries can be defined and confirmed based on a non-interrupted ORF. However, group II introns tend to evolve toward an inactive form by fragmentation, mainly, with loss of the 3′ terminus making it difficult to identify them [4, 5].
2 Materials
2.1 Primer Extension
-
1.
Total RNA preparations from at least three individual colonies harboring a plasmid which constitutively expresses the intron to be characterized (donor plasmid): This donor plasmid must contain the full-length group II intron flanked by at least 50 bp, which includes the intron-binding sites (IBSs). RNA extraction can be performed by a commercial kit or a suitable protocol described for your bacteria. A high yield protocol for bacterial RNA extraction is detailed in [6].
-
2.
20-mer oligonucleotide complementary to a sequence within the first 100 nt of the intron RNA sequence, that is, the extension has to proceed towards the 5′ end of the intron.
-
3.
T4 polynucleotide kinase supplied with its own concentrated buffer 10×.
-
4.
γ-[32P] ATP 3000 Ci/mmol; 10 mCi/μl.
-
5.
Illustra™ microspin G-25 columns (GE Healthcare).
-
6.
Annealing buffer: 10 mM PIPES pH 7.5, 400 mM NaCl.
-
7.
Extension mixture: 50 mM Tris pH 8.0, 60 mM NaCl, 10 mM DTT, 6 mM magnesium acetate, 1 mM each of all four dNTPs, 60 μg/ml actinomycin D, 2 U of RNase Inhibitor (Invitrogen), and 7 U AMV reverse transcriptase.
-
8.
3 M sodium acetate pH 4.8.
-
9.
100 % ethanol and 70 % ethanol (kept at −20 °C).
-
10.
TE 1×: 10 mM Tris pH 8.0, 1 mM EDTA.
-
11.
STOP solution: 0.3 % (w/v) each: Bromophenol blue and xylene cyanol FF, 10 mM EDTA, 97.5 % deionized formamide.
-
12.
8 M Urea/polyacrylamide gel in TBE 1× (89 mM Tris pH 8.0, 89 mM boric acid, 2 mM EDTA).
-
13.
Imaging Plate 2040 or X-ray film.
2.2 RT-qPCR
-
1.
Total RNA preparations from at least three individual colonies containing the intron donor plasmid (see Subheading 2.1.).
-
2.
A primer pair spanning the splicing junction (Tm 60 °C): For instance, a 20-mer oligonucleotide which sequence corresponds to the 5′ end of the insertion site (designed into the 50 nt upstream of the 5′ intron boundary) and the other 20-mer oligonucleotide complementary to the 3′ end of the intron insertion site (designed into the 50 nt downstream of the intron end).
-
3.
qPCR mix: 20 mM Tris pH 8.4, 50 mM KCl, 3 mM MgCl2, 0.2 mM each of all four dNTPs, Sybr Green I mix, and 0.5 U Taq DNA polymerase. To prepare the Sybr Green I mix, combine 1 μl Sybr Green I 10,000×, 1 μl fluorescein, and 18 μl DMSO, and then dilute this mixture 1:250 (add 2.5 μl of the dilution to the 25 μl final volume reaction).
-
4.
The consumable material will depend on the technology used for amplification, but generally consist of see-through 96-well plates and cover films.
2.3 Plasmid to Plasmid Mobility Assay
-
1.
A donor plasmid (see above, Subheading 2.1).
-
2.
A receptor plasmid that contains the intron DNA target sequence. This DNA target must contain the sequences corresponding to the IBSs that interact with the exon-binding sites (EBSs) of the intron. Generally a region of 100 nucleotides spanning from −50 to +50 of the intron insertion site (+1) should include all the requirements as a homing site . Donor and receptor plasmids must be compatible.
-
3.
Plasmid DNA extraction kit adapted to your bacteria.
-
4.
Restriction enzymes and 10× buffers.
2.4 Southern Blot
-
1.
Agarose gel electrophoresis system: Generally 1 % agarose is used in TAE 1× (40 mM Tris-acetate, 2 mM EDTA; pH 8.0).
-
2.
DIG-labeled DNA molecular weight marker.
-
3.
Fluorescent nucleic acid gel stain (Gel Red, EtBr, …).
-
4.
Ultraviolet transilluminator.
-
5.
20× SSC: 3 M NaCl, 300 mM trisodium citrate (adjusted to pH 7.0 with HCl).
-
6.
VacuGene™ XL Vacuum Blotting System (GE Healthcare Amersham™).
-
7.
1 N NaOH solution.
-
8.
Positively charged nylon membranes.
-
9.
Whatman 3MM paper.
-
10.
Heated vacuum desiccator.
-
11.
Hybridization oven and tubes.
-
12.
Prehybridization solution: 50 % v/v formamide, 5× SSC, 2 % w/v blocking reagent (Roche), 0.1 % w/v N-lauroylsarcosine, 0.02 % w/v SDS, and deionized water. Heat until just about to boil to dissolve the blocking reagent. Store at −20 °C.
-
13.
DIG-labeled probe: 300–500 nt PCR DIG-labeled product which hybridizes at the 5′ or 3′ region of the insertion site. PCR amplification must be performed using the following dNTP mixture: 0.1 mM dATP, 0.1 mM dCTP, 0.1 mM dGTP, 0.065 mM dTTP, 0.035 mM DIG-dUTP.
-
14.
Digoxigenin-11-dUTP, alkali-stable.
-
15.
Thermocycler.
-
16.
Probe solution (5–10 ml): 60 ng of DIG-labeled probe/ml prehybridization solution.
-
17.
Solution 1: 2× SSC, 0.1 % (w/v) SDS and deionized water.
-
18.
Solution 2: 0.1× SSC, 0.1 % (w/v) SDS and deionized water.
-
19.
Solution 3: Buffer 1 (0.1 M malic acid, 0.15 M NaCl, deionized water; adjusted at pH 7.5 with NaOH) and 0.3 % (w/v) Tween 20.
-
20.
Solution 4: 1 % of Blocking Reagent (Roche) in Buffer 1. Heat the solution to dissolve the blocking reagent, but do not let it boil. Prepare 100 ml fresh.
-
21.
Solution 5: Prepare a 1:10,000 dilution of anti-digoxigenin-AP, Fab fragments (Roche) in 20 ml of Solution 4.
-
22.
Solution 6: 0.1 M Tris, 0.1 M NaCl, and deionized water. Adjust at pH 9.5 with HCl.
-
23.
Solution 7: Prepare a 1:100 dilution of CSPD (Roche) in Solution 6.
-
24.
X-ray film and exposure holder.
-
25.
X-ray developer and fixer.
3 Methods
3.1 How to Identify a Group II Intron
3.1.1 Identification of the Intron-Encoded Protein
Generally, a first indication of the presence of a group II intron is the identification of its reverse transcriptase domain (RT). Among others, group II intron RTs are characterized by a high conservation degree in all RT domains (0–7 domains). Examples of ORFs associated to group II introns can be identified (annotated) as (1) reverse transcriptase, (2) RNA-directed DNA polymerase, (3) retron-type reverse transcriptase, or even only (4) DNA polymerase. Alternatively, they can be searched by using representative group II intron-encoded proteins obtained from intron database (http://webapps2.ucalgary.ca/~groupii/) by BlastP searches on particular genomes or other sequence data. Finally, the query RT sequence could be aligned to the reported RT data set [7, 8], and performed phylogenetic analysis using FastTree so that it could be identified as an RT potentially associated to a group II intron RNA.
3.1.2 Identification of Intron RNA Component
In annotated genomes, frequently the RNA component of a group II intron has not been identified. In several genomes, ORFs overlapping the ribozyme sequence have been automatically annotated making it difficult to interpret if a particular genome sequence contains a canonical group II intron. In order to identify if a full-length group II intron is associated to a particular reverse transcriptase ORF, nucleotide sequences (4–5 kb) containing 2 kb upstream and 2 kb downstream of the ORF can be analyzed for the presence of characteristic group II intron RNA domains. In these cases, a conserved catalytic domain V (34–36 nt) can be easily identified, generally just downstream from the ORF sequence. As example, a local BlastN using the conserved domain V of RmInt1 (GAGCGGTGTGAATCGAGAGGTTTACGCACCGTTC) on these 6–8 kb can identify the potential domain V of the RNA structure. Additionally, the bioinformatics tool “intron boundaries” from the intron database (http://webapps2.ucalgary.ca/~Egroupii/primes.html) can be used to delimit the borders of the putative group II intron [9, 10]. In a canonical group II intron a “GUGYG” and “AY” as boundaries of 5′ and 3′ end, respectively, can be confirmed using this tool. A further study using Mfold (http://mfold.rna.albany.edu/?q = mfold;) for the detection of potential group II intron RNA structures can be performed in order to identify the remaining RNA domains (I, II, III, and VI). At this stage, the domain VI defining the 3′ end of the group II intron can be easily confirmed. In some cases, the identification of the domains I, II, and III (domains upstream of the ORF sequence) should require the uses of nucleotide-level comparisons with group II introns already described [10].
3.1.3 Identification of a Putative Exon Junction
The confirmation of the presence “in silico ” of a full-length intron can be finalized with an examination of putative exon junctions. In some cases the nucleotide corresponding with the IBSs (generally between −15 and +1) of the intron insertion site can be deduced as complementary stretches to some loop structures proposed for domain I of the RNA. Sometimes it is useful to investigate if this group II intron is interrupting a particular gene. A BlastX using the corresponding DNA fragment lacking the proposed group II intron sequence can reveal a new copy of the interrupted ORF but in this case without the group II intron. This fact is also indicative that our sequence contains a full-length mobile group II intron.
3.2 Group II Intron Splicing
The excision of group II introns can be detected by primer extension [11], and more accurately by RT-qPCR [12]. This protocol has been adapted to be carried out using a microfuge.
3.2.1 Primer Extension
-
1.
Prepare the (5′-32P)-labeled oligonucleotide. For that, mix 10 pmol of oligonucleotide with 10 μCi of γ-[32P] ATP, 8 U of T4 polynucleotide kinase, and the corresponding buffer. Incubate for 1 h at 37 °C. Remove the non-incorporate radioactive nucleotides using G-25 columns and measure 1 μl in a scintillation counter.
-
2.
For the annealing mixture, combine 10 μg of total RNA with 300,000 cpm (~0.2 pmol) of (5′-32P)-labeled primer in annealing buffer (10 μl final volume). Heat the mixture at 85 °C for 5 min in a heater block, then cool the block fast to 60 °C in water bath, and, finally, leave it at room temperature for slowly cooling until 44–45 °C.
-
3.
Prepare the extension mixture and preincubate it at 42 °C for 2–5 min. When the annealing is completed, add 40 μl of preheated extension mixture and incubate at 42 °C for 60 min.
-
4.
Stop the reverse transcription reaction by adding 15 μl 3 M sodium acetate pH 4.8 and 150 μl of cold 100 % ethanol. Keep for at least 60 min at −80 °C. Alternatively, sample precipitation could be placed at −20 °C overnight.
-
5.
Centrifuge at 16,000 g for 15 min, and remove the supernatant. Wash samples with 200 μl of cold 70 % ethanol. Centrifuge at 16,000 g for 5–10 min, remove supernatant with a tip, and let the pellets to dry at room temperature.
-
6.
Resuspend reactions in 4 μl of TE1x + 4 μl of STOP solution.
-
7.
Load 4 μl of the sample from step 6 on a denaturing, polyacrylamide gel. Pre-run the gel for 30 min at 45 W. Denature the samples by heating for 3 min at 95 °C, and keep them on ice until loading. Run the gel at 50 W until the bromophenol blue reaches the bottom of the gel. Dry the gel and expose to an imaging plate or an X-ray film.
-
8.
Acquire images after 24–48 h of exposure, with a laser scanning system (for the imaging plates), or by conventional developing (developer/fixer, for the X-ray films).
3.2.2 RT-qPCR
-
1.
Prepare cDNA from total RNA samples extracted from cells harboring the intron whose splicing efficiency will be quantified. For this purpose, we reverse transcribe 10 μg of the RNA using random hexamers (5 ng/μl final concentration) and AMV RT (see above in the primer extension section, but omitting actinomycin D in the extension mixture).
-
2.
The qPCR experiments can be conducted with a real-time PCR detection system. Each reaction must contain l μg of cDNA template and 5 pmol of each primer in 25 μl of qPCR mix final volume.
-
3.
The PCR cycling conditions are as follows: 5-min hot start at 95 °C, followed by 45 cycles of denaturation at 95 °C for 10 s, annealing at 63–65 °C for 2 s, and extension at 72 °C for 2 s.
-
4.
Relative or absolute quantification can be performed. Relative quantification requires a parallel reaction with a primer pair amplifying a reference gene; any housekeeping gene with constitutive expression is suitable. Thus, the mRNA levels will be calculated according to the 2-ΔΔCT method [13], where ΔΔCT = ΔCT (calibrator or WT) − ΔCT (test or mutant). For absolute quantification, a standard curve is needed preferably from a plasmid containing the exon junction.
3.3 Group II Intron Mobility Assay
An outline of the double-plasmid retrohoming assay [14] is shown in Fig. 2.

Plasmid-to-plasmid group II intron mobility assay. (a) Outline of the double-plasmid retrohoming assay: the target on the recipient plasmid could be invaded by an intron from the donor, resulting in the homing product. (b) Southern hybridization with a DNA probe specific to the target. Negative control is indicated as a minus sign in a circle above the blot. Target invasion rate was calculated as described in Subheading 3 and is plotted in the histogram shown beside the blot
-
1.
Transform bacteria with both donor and recipient plasmids.
-
2.
Grow bacteria in liquid media supplemented with the corresponding antibiotics and extract the plasmid DNA according to a suitable protocol or kit.
-
3.
Digest DNA samples with one or a combination of restriction enzymes which release the DNA fragment containing the invaded or non-invaded target site from the recipient plasmid.
-
4.
Run equal amount of digested DNA samples in a preparative agarose gel using a DIG-labeled DNA molecular weight marker.
-
5.
Reveal the bands using a fluorescent nucleic acid gel dye. Expose the gel to the UV during 15–20 min to facilitate the DNA break and transfer to the membrane.
-
6.
Cut the nylon membrane and wet it with distilled water. Note that the membrane dimensions must exceed 1–1.5 cm from the gel size. Equilibrate by immersion in 20× SSC for 5 min with occasional swinging.
-
7.
Assemble the transfer system. Place first the nylon membrane on the foam bed followed by the perforated, plastic mask in such a way that the membrane overlaps with the open gap boundaries in the mask. Close the tab to fix the components and finally put the gel on the top covering the mask window to ensure the sealing of the system.
-
8.
Connect the vacuum pump and regulate the power to 50–60 mbar.
-
9.
Add 1 N NaOH until the gel gets covered. Let the transfer during 2 h.
-
10.
Disassemble the transfer system and incubate the nylon membrane on a tray containing 2× SSC during 15 min. Swing occasionally.
-
11.
Let the membrane dry on Whatman 3MM paper. Introduce the membrane in a Whatman 3MM paper envelope and fix the DNA at 120 °C on a heated vacuum desiccator during 35 min.
-
12.
Introduce the nylon membrane in a hybridization tube in such a way that the DNA side remains opposite to the glass walls. Add 50 ml of prehybridization solution. Incubate in a preheated hybridization oven at 42 °C for at least 2 h.
-
13.
Denaturalize the probe solution in boiling water during 10 min and cool quickly on ice for 10 min.
-
14.
Remove the prehybridization solution from the hybridization tube. Add the denatured probe solution in the membrane container. Incubate at 42 °C for 12–16 h (overnight).
-
15.
Collect the probe solution from the hybridization tube and store it at −20 °C. The probe solution can be reused at least for four to five additional hybridizations.
-
16.
Wash the membrane for 5 min twice with 100 ml of Solution 1.
-
17.
Wash with 100 ml of Solution 2 for 15 min at 68 °C twice. These two washing steps will remove all non-hybridized probe.
-
18.
Chill the hybridization tube at room temperature (RT). Incubate the membrane with 100 ml of Solution 3 for 5 min at RT. Hereafter, all the incubation steps will be performed at RT.
-
19.
Block the membrane by adding 80 ml of Solution 4 to the tube and incubate for 30 min.
-
20.
Drain the Solution 4 and add 20 ml of Solution 5. Incubate for 30 min to allow the antibody to recognize the DIG molecules.
-
21.
Wash with 100 ml of Solution 3 for 15 min. Repeat this washing step to remove the unbounded antibody.
-
22.
Equilibrate the membrane by incubating in 50 ml of Solution 6 for 5 min at room temperature.
-
23.
Finally, incubate in 5 ml of Solution 7 for 5 min.
-
24.
Place the membrane on Whatman 3MM paper and let it dry for at least 5 min.
-
25.
Wrap the membrane in plastic wrap, avoiding wrinkles. Incubate at 37 °C for 15 min in darkness to activate the alkaline phosphatase.
-
26.
Expose the wrapped membrane to X-ray film during 4–12 h (overnight) in an X-ray exposure holder.
-
27.
Reveal the X-ray film using developer and fixer as indicated by the manufacturer.
-
28.
Digitalize the image obtained from the exposure and quantify the band intensities using proper software.
-
29.
The homing efficiency is calculated as the percentage of the ratio of homing product (H) to the addition of homing product and non-invaded recipient plasmid (H + R), (H/(H + R) × 100) [14].
4 Notes
4.1 Group II Intron Splicing
-
1.
For RNA handling precautions are essential: keep all solutions and material free of RNase contamination, wear gloves during all the steps, and preserve the RNA and cDNA samples at 4 °C during all the processes.
-
2.
Radioactivity protocols require supervising and properly conducting.
-
3.
Do not dry the primer extension samples using high temperature because it should be difficult to resuspend them.
-
4.
The size and the percentage of the polyacrylamide gel to run the primer extension samples depend on the cDNA length. For instance, a 100 nt cDNA could be separated in a denaturing, 6 % polyacrylamide gel of 40 cm length/0.2 mm thick.
-
5.
In the qPCR, the same cDNA has to be amplified in triplicates, and three cDNAs are recommended to be synthesized from each RNA sample. Use fine calibrated pipettes.
-
6.
To obtain a better reproducibility in the RT-qPCR experiments use the master mixes available from several commercial suppliers either for first-strand cDNA synthesis and/or for qPCR amplification.
-
7.
Depending on the size of the PCR product, it would be necessary to suppress the extension step in order to minimize the plausible amplification of the unspliced RNA precursor.
4.2 Group II Intron Mobility
-
1.
Since it is required to release the fragment containing the target DNA by restriction enzymes, design your recipient plasmid suitably. It is also recommended that the enzyme linearize the donor plasmid.
-
2.
Plasmid-to-plasmid assay must be performed in an intronless strain to obtain an accurate quantification of the homing rates.
-
3.
It is important to design a negative control of invasion, like a receptor plasmid lacking the insertion site.
-
4.
In order to seal the transfer system, you can cover the edges of the gel with 2 % agarose.
-
5.
Use gloves to manipulate both the Whatman paper and the nylon membrane.
-
6.
Before using the anti-digoxigenin-AP antibody, vortex and centrifuge the stock for 3 min at full speed. Take the necessary volume near the surface, not from the bottom of the tube.
-
7.
If you have no vacuum transfer system, you can opt for the traditional capillary transfer protocol.
-
8.
There are several devices to acquire chemiluminescent signal that can be used in these analyses.
References
Toro N, Jiménez-Zurdo JI, García-Rodríguez FM (2007) Bacterial group II introns: not just splicing. FEMS Microbiol Rev 31:342–358
Lambowitz AM, Zimmerly S (2010) Group II introns: mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a003616
Enyeart PJ, Mohr G, Ellington AD, Lambowitz AM (2014) Biotechnological applications of mobile group II introns and their reverse transcriptases: gene targeting, RNA-seq, and non-coding RNA analysis. Mob DNA. doi:10.1186/1759-8753-5-2
Dai L, Zimmerly S (2002) Compilation and analysis of group II intron insertions in bacterial genomes: evidence for retroelement behavior. Nucleic Acids Res 30(5):1091–1102
Toro N, Martínez-Rodríguez L, Martínez-Abarca F (2014) Insights into the history of a bacterial group II intron remnant from the genomes of the nitrogen-fixing symbionts Sinorhizobium meliloti and Sinorhizobium medicae. Heredity (Edinb). doi:10.1038/hdy.2014.32
Molina-Sánchez MD, Martínez-Abarca F, Toro N (2006) Excision of the Sinorhizobium meliloti group II intron RmInt1 as circles in vivo. J Biol Chem 281:28737–28744
Toro N, Martínez-Abarca F (2013) Comprehensive phylogenetic analysis of bacterial group II intron-encoded ORFs lacking the DNA endonuclease domain reveals new varieties. PLoS One. doi:10.1371/journal.pone.0055102
Toro N, Nisa-Martínez R (2014) Comprehensive phylogenetic analysis of bacterial reverse transcriptases. PLoS One. doi:10.1371/journal.pone.0114083
Candales MA, Duong A, Hood KS et al (2012) Database for bacterial group II introns. Nucleic Acids Res. doi:10.1186/1759-8753-4-28
Abebe M, Candales MA, Duong A et al (2013) A pipeline of programs for collecting and analyzing group II intron retroelement sequences from GenBank. Mob DNA. doi:10.1186/1759-8753-4-28
Muñoz-Adelantado E, San Filippo J, Martínez-Abarca F et al (2003) Mobility of the Sinorhizobium meliloti group II intron RmInt1 occurs by reverse splicing into DNA, but requires an unknown reverse transcriptase priming mechanism. J Mol Biol 327:931–943
Chillón I, Martínez-Abarca F, Toro N (2011) Splicing of the Sinorhizobium meliloti RmInt1 group II intron provides evidence of retroelement behavior. Nucleic Acids Res 39:1095–1104
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C (T)) Method. Methods 25:402–408
Nisa-Martinez R, Jiménez-Zurdo JI, Martínez-Abarca F et al (2007) Dispersion of the RmIntI group II intron in the Sinorhizobium meliloti genome upon acquisition by conjugative transfer. Nucleic Acids Res 35:214–222
Acknowledgments
This work was supported by research grants CSD 2009-0006 from the Consolider-Ingenio program, and BIO2011-24401 from the currently Ministerio de Economía y Competitividad, including ERDF (European Regional Development Funds).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Toro, N., Molina-Sánchez, M.D., Nisa-Martínez, R., Martínez-Abarca, F., García-Rodríguez, F.M. (2016). Bacterial Group II Introns: Identification and Mobility Assay. In: Garcia-Pérez, J. (eds) Transposons and Retrotransposons. Methods in Molecular Biology, vol 1400. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3372-3_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3372-3_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3370-9
Online ISBN: 978-1-4939-3372-3
eBook Packages: Springer Protocols