
Abstract
Mitosis which is a major step during plant development can also be observed in physiopathological conditions. During the compatible interaction between the root-knot nematode Meloidogyne incognita and its host Arabidopsis, the pathogen induce through repeated divisions without complete cytokinesis the formation of hypertrophied and multinucleate feeding cells, named giant cells. Due to the presence of hypertrophied plant cell material surrounding the giant cells, classical live cell imaging gave therefore very poor resolution. Here, we describe a protocol which allows the in vivo observation of the mitotic apparatus in developing giant cells using confocal imaging of vibrosliced tissues. This approach can also be used to visualize in vivo other cellular processes occurring in different steps of giant cells.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
The nematode infection provides an original model to explore key processes such as plant cell mitosis. Nematodes are obligate biotrophic pathogens which evolve the ability to induce “giant cells,” permanent feeding cells that constitute their unique source of nutrients [1]. Giant cells result from synchronous repeated karyokinesis without complete cell division [2]. Fully differentiated giant cells reach a final size about 400 times that of root vascular cells and contain more than a 100 polyploid nuclei, which have also undergone extensive endoreduplication [3]. Giant cell development is accompanied by division and hypertrophy of surrounding cells, leading to a typical root gall formation, the primary visible symptom of infection.
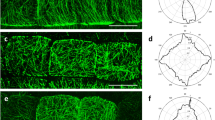
The distribution of the plant cell cytoskeleton in developing giant cells have been first determined using fixed tissues by immunostaining of α, β-, and γ-tubulins in butyl-methylmethacrylate embedded galls [4, 5]. New approach of live cell imaging was used to study the dynamic of cytoskeleton organization during giant cells ontogenesis using vibroslices through galls [6]. This technique allows the visualization in vivo of the mitotic apparatus in giant cells. Fluorescent markers were used―the MAP4 microtubule-binding domain (MBD)-GFP and H2B-YFP [7] as well as the microtubule-associated protein MAP65-3 [6]―to characterize accurately dynamic changes in the microtubules organization in mitotic giant cells. Imaging of these fluorescent-coupled proteins led to the confirmation of the presence of multiple microtubule spindles during the early steps of giant cell formation (Fig. 1a, b). Time-lapse in vivo also revealed the presence of early synchronous phragmoplast arrays with a restricted out-growth which lead to the formation of a novel cell plate structure—the giant cell mini cell plate—that does not extend across adjacent faces of the cell [6]. Here, we describe the protocol which allowed the observation in vivo of the mitotic apparatus in dividing giant cells using vibrosliced galls. This protocol would be compatible with study of any protein involved in nematode feeding site ontogenesis or physiology, and it make an ideal tool to study protein dynamics in this living tissue.

Microtubule organization in giant cells of Arabidopsis plant expressing (a) MBD:GFP (green) or (b) MBD:GFP (green) and H2B:YFP (red) using confocal microscope. Asterisks giant cells, N nematode; arrows mitotic structures in giant cells. Scale bar: 2.5 μm
2 Material
2.1 Equipment
-
1.
Plastic and glass Petri dishes (245 × 245 × 20 mm).
-
2.
Laminar-flow hood.
-
3.
100, 10 and 0.5 μm sieve.
-
4.
10 mL syringe.
-
5.
50 ml cornings.
-
6.
Syringe-driven filter unit and 3 μm membrane.
-
7.
Gelrite (phytagel, Sigma-Aldrich, St. Louis, United States of America).
-
8.
Fluorescent stereomicroscope.
-
9.
Tweezers, scalpel, scissors.
-
10.
1 ml tube.
-
11.
5 ml pipette tips and plastic Pasteur pipettes.
-
12.
HM650V vibrotome Microm (Walldorf, Germany).
-
13.
Glass slides and cover slips.
-
14.
Inverted confocal microscope (model LSM510 META; Zeiss, Germany).
2.2 Culture Media and Buffers
-
1.
Plant in vitro medium: Murashige and Skoog (MS) ½ medium, containing 1 % sucrose, 0.7 % plant cell culture-tested agar. Adjust pH (with potassium hydroxide, KOH) to 6.5 and autoclave at 121 °C for 15–20 min.
-
2.
Arabidopsis seed sterilization:
-
2.63 % sodium hypochlorite (NaOCl) in water.
-
Ethanol 70 %.
-
-
3.
Nematode ultracleaning solutions:
-
4.
Embedding medium: 7 % agar (or agarose for molecular biology) dissolved in boiling distilled water.
2.3 Plant Material
-
1.
Arabidopsis line expressing the N-terminal domain of the microtubule-binding domain of MAP4 fused to the GFP (Pro 35S :MBD:GFP).
-
2.
Arabidopsis line expressing Histone H2B fused to YFP (Pro 35S :H2B:YFP).
-
3.
Plant expressing Pro 35S :MBD:GFP were crossed with plant expressing Pro 35S :H2B:YFP. Progeny expressing both constructions were selected for fluorescence using fluorescent stereo microscope. Homozygous progeny was used for microscopy analysis.
-
4.
Arabidopsis seed were surface sterilized by soaking for 5 min in ethanol 70 % and then for 5 min in 2.63 % sodium hypochlorite in a laminar flow hood. Seeds were extensively washed with ethanol 70 %, dried, and dispensed onto Petri dishes containing in vitro media (see Subheading 2.2). Plants were grown at 20 °C under a cycle of 8 h of light and 16 h of darkness for 5 days.
2.4 Pathogen Material
-
1.
M. incognita was grown on greenhouse-grown tomato (Solanum lycopersicum ‘St. Pierre’). The infected roots were collected, washed with tap water, and 5 cm-long root pieces grinded with 0.5% NaOCl in a blender.
-
2.
Eggs were collected by filtration through a strainer and two sieves (100 μm and 10 μm) and washes with tap water to get rid of all the bleach. The eggs were transferred on a 5 μm sieve and the sieve were placed in a tray containing sterile tap water for eclosion.
-
3.
J2 larvae were hatched in the water of the tray and were collected 7 days after the hatching on a 0.5 μm sieve. The number of larvae/mL was evaluated using cell counting chamber. Larvae attaching to the plastic, maximize the use of glass materials.
3 Methods
3.1 Ultra-Cleaning of M. incognita
-
1.
In a laminar-flow hood, put the freshly hatched larvae on a 3 μm membrane (maximum 50,000 larvae per membrane) placed inside a syringe-driven filter unit, using a 10 mL syringe (see Notes 3 and 4 ).
-
2.
Using 10 mL syringe, inject slowly the 0.01 % HgCl2 solution (see Note 1) through the syringe-driven filter unit containing the nematodes (see Note 5 ). The nematode larvae need to be in contact with the solution for 9–10 min.
-
3.
Using the 10 mL syringe, inject slowly the 0.7 % streptomycin sulfate solution (see Note 2) through the syringe-driven filter unit containing the nematode larvae. Wash three times by injecting through the syringe-driven filter unit with sterile water.
-
4.
Resuspend the larvae in 0.5 % gelrite in order to obtain 20 larvae per μL. To do so, pull out the membrane using sterile tweezers and place it in a glass Petri dish with the nematode larvae facing up. Take off the larvae from the membrane by gently pipeting up and down the gelrite solution on top of the membrane, using a cut 20 μl tip (see Note 6 ).
3.2 In Vitro Inoculation of Arabidopsis Seedlings with M. incognita
-
1.
Five days after sowing, screen Arabidopsis transgenic line expressing Pro 35S :MBD:GFP and/or Pro 35S :H2B:YFP for good level of fluorescent protein on the root meristem using fluorescent stereo microscope.
-
2.
In a laminar flow hood, prick out ten plants expressing a good level of fluorescent proteins into fresh MS (see Culture Media and Buffers) square Petri dishes.
-
3.
Let grow the seedlings for 10 days at 20 °C under a cycle of 8 h of light and 16 h of darkness. Petri dishes must be inclined at an angle of 60° to allow the roots to grow along the surface.
-
4.
In a laminar flow hood, add 100 surface-sterilized M. incognita J2s on each seedling by deposing droplet of 10 μl close to the root apex (see Note 7 ).
-
5.
Seal the Petri dishes with parafilm to prevent evaporation and keep them on the bench at RT for 5–6 days.
3.3 Preparation of the Infected Material Prior to Observation
-
1.
5–6 days after inoculation of the seedlings with the nematode larvae, extract from the Petri dish root systems infected with the nematodes using tweezers.
-
2.
Wash carefully the root system of each plant with distilled water to remove all the pieces of plant medium and place them in an empty Petri dish (see Note 8 ).
-
3.
Excise galls under the stereo microscope using tweezers and scalpel and place them in a 1 ml tube filed with distilled water (see Note 9 ).
-
4.
To prepare the mould, cut the last 1 cm of a 5 ml pipette tip using scissors and place it in an open small Petri dish (Fig. 2a).
Fig. 2 
Scheme of the preparation of the infected material prior to observation. (a) Cut the last 1 cm of a 5 ml pipette tip using scissors and place it in an open small Petri dish. Using tweezers place on it two to three galls. (b) Pour on top cooled embedding medium using plastic Pasteur pipette. (c) After solidification, extract the preparation from the plastic mould using pencil back. (d) With razor blade, cut the top of the preparation to flatten it and fix the preparation with glue on the vibratome receptacle putting the galls in the top part. (e) Vibroslice the block into sections of about 100 μm thick, using a vibratome. (f) Dispose the sections in a glass slide and add distilled water with a plastic Pasteur pipette before covering them with a cover slip until observation with confocal microscope
-
5.
Using tweezers place two to three galls in the bottom part of the mould and pour on top cooled embedding medium (see Culture Media and Buffers) using plastic Pasteur pipette (Fig. 2b).
-
6.
After solidification, extract the preparation from the plastic mould using pencil back.
-
7.
With razor blade, cut the top of the preparation to flatten it and fix the preparation with glue on the vibratome receptacle putting the galls in the top part of the block (Fig. 2c).
-
8.
Vibroslice the block into sections of about 100 μm thick using a vibratome (Fig. 2d).
-
9.
Dispose the sections in a glass slide and add distilled water with a plastic Pasteur pipette before covering them with a cover slip (see Note 10 , Fig. 2e).
-
10.
Prepare a humid box by placing a soaked-paper roll on the bottom of a square Petri dish and display the slides on it before closing the lid.
-
11.
Let the preparation in the humid box until observation with confocal microscope.

3.4 Imaging of the Mitosis in Giant Cells
-
1.
For imaging of the dividing giant cells, observe the vibroslice with a ×63 water immersion apochromat objective using confocal microscope.
-
2.
In order to image both dividing nucleus and microtubule mitotic structures in giant cells of Arabidopsis transgenic line expressing Pro 35S :MBD:GFP and Pro 35S :H2B:YFP, use the Lambda acquisition mode (see Note 11 ) with a 499–550 nm beam path (488 nm excitation line).
-
3.
Select two region of interest (ROI) which correspond to the YFP signal of H2B:YFP in the dividing nucleus and the GFP signal of MBD:GFP in the microtubule mitotic structure.
-
4.
In the graph plotting the pixel intensity versus the mean wavelength of each band, verify that the fluorescence observed matches the desired fluorophore or represents nonspecific emission.
-
5.
Choose two different colors to distinguish YFP signal of H2B:YFP in the dividing nucleus and the GFP signal of MBD:GFP in the microtubule mitotic structure.
-
6.
In order to reconstruct in 3D the structures observed in giant cells, use Z-stack mode.
4 Notes
-
1.
Mercuric chloride is Very toxic (T+), Corrosive (C), Dangerous for the environment (N). Wear two pairs of gloves and discard it in the appropriate chemical bin (ICSC 0979; R-phase: R28, R34, R48/24/25, R50/53; S-phrases: (S1/2), S36/37/39, S45, S60, S61.
-
2.
The streptomycin sulfate is used as standard antibiotic to prevent bacterial infection in the culture. Wear gloves.
-
3.
Autoclave the syringe-driven filter unit with the membrane in place.
-
4.
To prevent contamination, the number of larvae/membrane must be inferior at 50,000.
-
5.
Do not push too strongly and verify using stereomicroscope that there is no leak: you shouldn’t see nematode in the solution placed in a Petri dish.
-
6.
The homogenized solution must become more and more opaque, due to the presence of the nematodes.
-
7.
Before inoculating the plant, observe with the stereomicroscope the larvae to be sure that they are numerous and still alive.
-
8.
Be careful to let the root system infected with nematodes always immerged in water to not let it dry.
-
9.
When you excise galls under the stereo microscope using tweezers and scalpel, let 1 mm on each side of the gall: this will allow you to manipulate them with tweezers without damaging the Giant cells.
-
10.
When you start to cut through the block containing the giant cells, observe some of the section with the stereomicroscope to see if you are close to the giant cells. When you will manage to remove enough tissue around them, increase the size of the sections in order to not cut through the giant cells and then empty them. A young giant cell is about 100–150 μm in diameter.
-
11.
The lambda acquisition mode collects the fluorescence in a stack that sorts the emission output into a series of wavelength bands.
References
Caillaud MC, Dubreuil G et al (2008) Root-knot nematodes manipulate plant cell functions during a compatible interaction. J Plant Physiol 165(1):104–113
Huang CS (1985) Anatomy and physiology of giant cells induced by root-knot nematodes. In: Sasser JN, Carter CC (eds) An advanced treatise on Meloidogyne, vol II. North Carolina State University Graphics, Raleigh, pp 155–164
de Almeida Engler J, Gheysen G (2013) Nematode-induced endoreduplication in plant host cells: why and how? Mol Plant Microbe Interact 26(1):17–24
de Almeida Engler J, Van Poucke K et al (2004) Dynamic cytoskeleton rearrangements in giant cells and syncytia of nematode-infected roots. Plant J 38(1):12–26
Banora MY, Rodiuc N et al (2011) Feeding cells induced by phytoparasitic nematodes require gamma-tubulin ring complex for microtubule reorganization. PLoS Pathog 7(12):e1002343
Caillaud MC, Lecomte P et al (2008) MAP65-3 microtubule-associated protein is essential for nematode-induced giant cell ontogenesis in Arabidopsis. Plant Cell 20(2):423–437
Caillaud MC, Abad P et al (2008) Cytoskeleton reorganization, a key process in root-knot nematode-induced giant cell ontogenesis. Plant Signal Behav 3(10):816–818
Acknowledgements
We thank Michaël Quentin (Université de Nice Sophia Antipolis, France) for the careful reading of the manuscript, Richard Cyr (Pennsylvania State University, USA) for the generous gift of Pro35S:MBD:GFP seeds and Frederic Berger (Gregor Mendel Institute of Molecular Plant Biology GmbH, Vienna Austria) for Pro35S:H2B:YFP seeds. We thank Janice de Almeida Engler (INRA Sophia Antipolis, France) for her training at the vibratome and Philippe Lecomte (INRA Clermont-Ferrand, France) for his help with the writing of the protocol. We thank Nathalie Marteu (INRA Sophia Antipolis, France) for technical help and support. This work was supported by INRA and by the French Government (National Research Agency, ANR) through GENOPLANTE contracts AF2001032, ANR05GPLA020 AFINDIS and the ‘Investments for the Future’ LABEX SIGNALIFE: program reference # ANR-11-LABX-0028-01. M.-C.C. was supported by a fellowship from the Ministère de la Recherche et l’Enseignement Supérieure.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Caillaud, MC., Favery, B. (2016). In Vivo Imaging of Microtubule Organization in Dividing Giant Cell. In: Caillaud, MC. (eds) Plant Cell Division. Methods in Molecular Biology, vol 1370. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3142-2_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3142-2_11
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3141-5
Online ISBN: 978-1-4939-3142-2
eBook Packages: Springer Protocols