
Abstract
Serine 216 constitutes a protein kinase C phosphorylation motif located within the DNA binding domain of estrogen receptor α (ERα). In this chapter we present experimental procedures confirming that mouse ERα is phosphorylated at serine 216 in peripheral blood neutrophils and in neutrophils that infiltrate the uterus, as well as the role of phosphoserine 216 in neutrophil migration. A phospho-peptide antibody (αP-S216) was utilized in Western blot, immunohistochemistry, and double immunofluorescence staining to detect this phosphorylation of an endogenous ERα. Both immunohistochemistry (with αP-S216 or neutrophil marker Ly6G antibody) and double immunofluorescence staining of mouse uterine sections prepared from C3H/HeNCrIBR females revealed that phosphorylated ERα was expressed in all infiltrating neutrophils during hormonal cycles but not in any other of the other uterine cells. Neutrophils infiltrate the uterus from the blood stream. White blood cells (WBC) were prepared from peripheral blood of C3H/HeNCrIBR females or males and double immunostained. Blood neutrophils also expressed phosphorylated ERα but in only about 20 % of cells in both sexes. Only the neutrophils expressing phosphorylated ERα spontaneously migrated in in vitro Transwell migration assays and infiltrated the uterus in mice.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Estrogen receptor α (ERα)
- Phosphorylation
- Neutrophils
- Migration
- Infiltration
- Mouse uterus
- Immunohistochemistry
- Immunofluorescence stain ing
1 Introduction
Serine 216 of mouse ERα is located on a loop between two zinc fingers and is conserved as serine 212 in human ERα. Mass spectroscopic analysis identified 15 different residues of human ERα that may be phosphorylated in a human breast cancer MCF7 cell line. One of the potential phosphorylation sites is serine 212 [1, 2]. Our cDNA microarray analysis of RNAs, prepared from human hepatoma-derived Huh7 cells, ectopically expressed phosphorylation mimicking ERα S212D or non-phosphorylation mimicking ERα S212A mutant, showed that S212D mutant regulated a distinct set of the genes [3]. While these observations suggest that this phosphorylation may impart a unique biological function to ERα, it has not been confirmed with endogenous ERα in normal tissues and/or cultured cells. Therefore, here we describe protocols that utilize an antibody specific to phosphorylated serine 216 of mouse ERα to detect endogenous phosphorylation. These methods demonstrate that ERα is phosphorylated at serine 216 in mouse neutrophils [4]. Furthermore, only a fraction of blood neutrophils were found to express phosphorylated ERα and only those neutrophils were able to migrate in an in vitro assay and also infiltrate the uterus in vivo.
Neutrophils infiltrate the mouse uterus during normal hormonal cycles or in inflammatory conditions. ERα plays an essential role in uterine cells to regulate normal infiltration in response to estrogen [5–7]. However, the presence of ERα and its biological function in infiltrating neutrophils is not well understood at the present time. Our present finding that ERα is phosphorylated at serine 216 in infiltrating neutrophils has provided new insight to investigate the role of neutrophil ERα in uterine functions as well as inflammation-associated development of estrogen-dependent diseases [8–10]. Although not covered in this chapter, knock-in (KI)/knockout (KO) mouse line (Esr1S216A) bearing an alanine mutation at residue serine 216 has now been generated. Utilizing these mice, the in vivo roles of phosphorylated ERα will be further investigated.
Serine 216 of mouse ERα is conserved as a phosphorylation motif not only in human ERα but also in 41 out of 48 human nuclear receptors. In fact, phosphorylation of this conserved motif was first confirmed with threonine 38 of nuclear receptor CAR (NR1I3) in hepatocytes and, moreover, its role in the activation of CAR by therapeutics such as phenobarbital [11, 12]. Thus, a unique opportunity to examine whether or not this conserved phosphorylation motif within the DNA binding domains engages a general regulatory mechanism common to the majority of nuclear receptors is presenting itself to us. Our work on serine 216 phosphorylation of ERα have strengthened this opportunity to be realized in future investigations. In this chapter we present experimental protocols, specifically western blot and double label immuno fluorescence staining immunohistochemistry, that confirm that mouse ERα is phosphorylated at serine 216 in peripheral blood neutrophils and in neutrophils that have infiltrated the uterus, as well as an in vitro migration assay protocol to investigate the role of phosphoserine 216 in neutrophil migration and infiltration.
2 Materials
-
1.
Antibody directed against phospho-Ser216 ERα (seeNote 1 ).
-
2.
Protein kinase C (PKC) (Promega, V5261, seeNote 2 ): store at −80 °C.
-
3.
Glutathione S Transferase (GST)-mouse ERα and its mutant (Ser216Ala) (seeNote 3 ): purified ERα proteins purified from bacteria transformed with plasmids carrying these cloned receptors.
-
4.
Tris-buffered saline, pH 7.4 (TBS).
-
5.
Kinase buffer: 41.5 mM TBS, pH 7.4, 6.67 mM CaCl2, 3.3 mM dithiothreitol, 1.67 mM MgCl2, 1.0 mg/mL phosphatidylserine, and 330 μM ATP.
-
6.
4× SDS-sample buffer: 314 mM Tris–HCl, pH 6.8, 8 % SDS, 50 % Glycerol and 0.02 % Bromophenol Blue.
-
7.
Polyvinylidene difluoride (PVDF) membrane.
-
8.
TBS/Tween 20 (TBS-T) buffer: 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.1 % Tween-20.
-
9.
Bovine serum albumin (BSA).
-
10.
Skim milk (nonfat dry milk).
-
11.
Horseradish peroxidase conjugated goat anti-rabbit antibody (Goat anti-rabbit IgG-HRP). Store at 4 °C.
-
12.
Luminol reagent (Advansta).
-
13.
Xylene.
-
14.
Ethanol: 100, 95, and 70 %.
-
15.
Antigen retrieval reagent and system such as Antigen Decloaker (Biocare Medical).
-
16.
3 % hydrogen peroxide.
-
17.
Normal goat serum.
-
18.
Biotinylated goat anti-rabbit antibody (ABC kit, Vector Laboratories): store at 4 °C.
-
19.
ExtrAvidin-peroxidase (Sigma-Aldrich).
-
20.
3,3′-diaminobenzidine solution (DAB+ Chromogen, Dako Cytomation).
-
21.
Hematoxylin solution.
-
22.
Coverslips and mounting solution such as Permount.
-
23.
3.8 % sodium citrate (seeNote 4 ).
-
24.
6 % dextran (seeNote 5 ).
-
25.
Sterile saline (0.9 % sodium chloride).
-
26.
Phosphate buffered saline (PBS).
-
27.
Cytospin.
-
28.
Glass microscope slides treated for adherence of tissue sections (such as Superfrost Plus, Thermo Scientific).
-
29.
4 or 10 % formaldehyde in PBS.
-
30.
Avidin/biotin blocking kit (Vector Laboratories).
-
31.
Fluorescein Avidin D at a cell sorter grade (DCS) (Vector Laboratories).
-
32.
Texas Red® Avidin D at a cell sorter grade (DCS) (Vector Laboratories).
-
33.
Anti-Ly6G (Lymphocyte antigen 6G) antibody (Clone 1A8, BD Pharmingen) as a neutrophil marker.
-
34.
Biotinylated rabbit anti-rat antibody (ABC kit, Vector Laboratories). Store at 4 °C.
-
35.
Alexa Fluor® 488 Goat anti-rabbit IgG (H + L) antibody (Life Technologies).
-
36.
Alexa Fluor® 594 Goat anti-rat IgG (H + L) antibody (Life Technologies).
-
37.
Mounting medium/DAPI (Vector Laboratories).
-
38.
24-well transwell plate (3.0 μm pore) (Corning Life Sciences).
-
39.
RPMI conditional medium: RPMI1640 medium (no-phenol red), 2 mM l-glutamine, penicillin (100 U/mL), streptomycin (100 μg/mL). Store at 4 °C.
-
40.
Charcoal–dextran stripped fetal bovine serum (FBS).
-
41.
0.1 M glycine–HCl, pH 2.0.
-
42.
3 % hydrogen peroxide.
-
43.
Trypan blue.
-
44.
Hemocytometer for cell counting.
3 Methods
3.1 In Vitro PhosphorylationERαProteins by PKC
-
1.
Add 0.5 μg of GST-mERα protein or its S216A mutant protein to 30 μL of kinase buffer in a 1.5 mL centrifuge tube on ice.
-
2.
Add 1 μL (1.6 units) of PKC to this solution and incubate it at 30 °C for 30 min.
-
3.
Stop the kinase reaction by boiling the reaction solution for 15 min in 4× SDS sample buffer containing β-mercaptoethanol.
-
4.
Separate the phosphorylated mouse ERα proteins in SDS sample buffer (5–15 μL/lane from step 3 of Subheading 3.1) by electrophoresis on a 10 % SDS polyacrylamide gel. Electro-transfer the proteins from the gel to a PVDF membrane.
-
5.
Incubate the PVDF membrane in TBS-T/1 % BSA for 1 h at room temperature using a rotating shaker.
-
6.
Incubate the membrane with anti-P-S216 antibody (1:1000) in TBS-T/1 % BSA overnight at 4 °C.
-
7.
Wash with TBS-T buffer for 10 min three times.
-
8.
Incubate the membrane with a horseradish peroxidase conjugated goat anti-rabbit antibody (1:10,000) in TBS-T/5 % skim-milk for 1 h at room temperature.
-
9.
Wash with TBS-T buffer for 15 min three times.
-
10.
Develop membrane with Luminol reagent and expose the membrane to X-ray film.
-
11.
After detection of phosphorylated ERα, reuse the same membrane for Western blot with an anti-GST antibody. Shake the membrane in 0.1 M glycine-HCl for 10 min at room temperature.
-
12.
Wash the membrane with TBS-T buffer for 15 min three times.
-
13.
Incubate the membrane in TBS-T/5 % skim-milk/anti-GST antibody overnight at 4 °C or for 1 h at room temperature. Hereafter, continue procedures with steps 7–10 of Subheading 3.1 to develop the anti-GST bands (Fig. 1).
Fig. 1 
Western blot analysis to show the specificity of an anti-P-S216 antibody aP-S216. Purified glutathione S transferase (GST)-tagged mERα wild type (WT) and its S216A mutant were incubated with or without protein kinase C (PKC). Subsequent Western blots were performed with αP-S216 or an anti-GST antibody as described in Subheading 3.1
-
14.
Quantitate the P-S216 and GST bands by densitometry.


3.2 Immunohistochemistry of Uterine Sections
-
1.
Fix mouse uterus in 10 % formalin, embed in paraffin, cut 6 μm thick sections, and place them on glass slides.
-
2.
De-paraffinize the sections by submerging the glass slides in xylene for 5 min twice and by sequentially washing with 100, 95 and 70 % ethanol for 3 min at each step and finally with H2O.
-
3.
Submerge de-paraffinized sections in antigen retrieval solution (decloaker buffer) and heat in the decloaking chamber (seeNote 6 ).
-
4.
Cool down to room temperature and replace decloaker buffer with H2O.
-
5.
Incubate with 3 % hydrogen peroxide solution for 15 min to inactivate endogenous peroxidases.
-
6.
Wash twice with TBS-T buffer for 5 min.
-
7.
Incubate with blocking buffer (PBS/1.5 % normal goat serum) for 1 h at room temperature.
-
8.
Incubate with anti-P-S216 antibody (1:25–50 dilution) in blocking buffer for 40 min at room temperature and wash with TBS-T buffer twice for 5 min.
-
9.
Incubate with biotinylated rabbit secondary antibody (1:500 dilution) in blocking buffer for 15 min at room temperature.
-
10.
Wash with TBS-T buffer twice for 5 min.
-
11.
Treat with ExtrAvidin-peroxidase (50-fold dilution in PBS buffer) for 20 min and wash with TBS-T buffer twice for 5 min.
-
12.
React with 3,3′-diaminobenzidine solution for 6 min and wash by flowing water onto the back of glass slides for 3 min.
-
13.
Dip in hematoxylin solution for 10–30 s to counter-stain and wash by flowing water until color disappears from the solution.
-
14.
Wash with TBS-T buffer for 1 min and sequentially dehydrate in 70, 95 and 100 % ethanol for 30 min and finally clear in xylene for 15 min or more.
-
15.
Mount surface of the section with mounting solution and place a cover slip.
-
16.
Observe staining using microscopy.
3.3 Competitive Immunohistochemistry of Uterine Sections
-
1.
To examine the specificity of the anti-P-S216 antibody, add phosphorylated antigen peptide or the non-phosphorylated peptide counterpart to an anti-P-S216 antibody solution (seeNote 7 ).
-
2.
Gently shake the mixture for 1 h at room temperature.
-
3.
Incubate uterine sections in one or the other mixture for 40 min at room temperature, then continue the immunohistochemistry procedure from steps 9 to 16 of Subheading 3.2. If the anti-P-S216 antibody is specific, this step should demonstrate that phosphorylated antigen peptide, but not the non-phosphorylated counterpart, inhibits staining by the anti-P-S216 antibody.
3.4 Preparation of Peripheral White Blood Cell (WBC) Fractions
-
1.
Collect blood from mouse postcaval veins.
-
2.
Add 44 μL of 3.8 % sodium citrate to 0.4 mL of blood in a swing rotor tube and shake gently.
-
3.
Centrifuge the tube at 350 × g for 20 min at room temperature (turn off the centrifuge brake).
-
4.
Remove upper layer (platelet rich plasma) from the tube.
-
5.
Add a quarter volume of 6 % dextran solution to the cell pellet and gently mix with the lower layer by pipeting several times (seeNote 8 ).
-
6.
Add one volume of 0.9 % sodium chloride to the above mixture and gently pipet up and down several times (seeNote8).
-
7.
Let the tube stand at room temperature for 20–30 min to allow RBC to sediment (seeNote 9 ).
-
8.
Collect the upper layer (WBC) and place in a new tube.
-
9.
Centrifuge at 220 × g for 6 min at room temperature, with centrifuge brake set low.
-
10.
Collect the resulting pellet and resuspend the WBC in less than 0.5 mL of PBS.
-
11.
Dilute an aliquot of the WBC suspension in trypan blue (1/100) and count cells using a hemocytometer (seeNote 10 ).
3.5 Double Fluorescence Staining with Fluorescein and Texas Red
This section introduces two staining methods using Fluorescein and Texas Red or Alexa.
-
1.
Cytospin mouse WBC (1 × 105 cells per 100 μL) from step 10 of Subheading 3.4 onto a glass slide. Allow the cells to dry overnight at room temperature.
-
2.
Fix the cells by submerging the slide in 4 % formalin solution for 20 min at room temperature.
-
3.
Incubate with Avidin solution for 15 min at room temperature to block for avidin/biotin.
-
4.
Rinse briefly with PBS and incubate with a Biotin solution for 15 min at room temperature.
-
5.
Wash with PBS twice for 5 min.
-
6.
Incubate with blocking buffer (PBS/1.5 % normal goat serum) for 20 min at room temperature.
-
7.
Incubate with anti-P-S216 antibody (a 1:25–50 dilution) in blocking buffer for 40 min at room temperature and wash with PBS twice for 5 min (Fig. 2).
Fig. 2 
Flowchart of double immunofluorescent labeling using two primary antibodies from different species
-
8.
Incubate with a biotinylated goat anti-rabbit antibody (5–10 μg/mL dilution) in blocking buffer for 30 min at room temperature and wash with PBS for 5 min twice.
-
9.
Incubate with Fluorescein Avidin DCS (1:100) in PBS for 8 min and wash with PBS twice for 5 min.
-
10.
To stain the neutrophils in the WBC sample, use Ly6G as a neutrophil marker. Repeat the blocking reactions in steps 3 and 4 of Subheading 3.5, then continue with step 11 of Subheading 3.5.
-
11.
Incubate with blocking buffer (PBS/1.5 % normal rabbit serum) for 20 min at room temperature.
-
12.
Incubate with anti-Ly6G antibody (1:70 dilution) in blocking buffer for 30 min at room temperature.
-
13.
Wash with PBS twice for 5 min.
-
14.
Incubate with a biotinylated rabbit anti-rat antibody (5–10 μg/mL dilution) in blocking buffer for 30 min at room temperature and wash with PBS twice for 5 min.
-
15.
Incubate with Texas Red® Avidin DCS (1:100) in PBS for 8 min and wash with PBS buffer twice for 5 min.
-
16.
Coverslip with mounting medium/DAPI.
-
17.
Observe fluorescence staining using a confocal microscopy.


3.6 Double Fluorescence Staining with Alexa 488 and Alexa 594
-
1.
Fix cells as described in Subheading 3.5, steps 1 and 2.
-
2.
Wash cells with PBS twice for 5 min.
-
3.
Incubate with blocking buffer (PBS/1.5 % normal goat serum) for 20 min at room temperature.
-
4.
Incubate the cells with anti-P-S216 antibody (1:25–50 dilution) in blocking buffer for 40 min at room temperature.
-
5.
Wash with PBS twice for 5 min.
-
6.
React with a neutrophil marker anti-Ly6G antibody (1:70 dilution) in blocking buffer for 30 min at room temperature.
-
7.
Wash with PBS twice for 5 min.
-
8.
Incubate with a mixture of Alexa Fluor® 488 Goat anti-rabbit secondary antibody and Alexa Fluor® 594 Goat anti-rat secondary antibody (each 1:500 dilution) in blocking buffer for 2 h at room temperature in the dark.
-
9.
Wash with PBS twice for 5 min.
-
10.
Coverslip with mounting medium/DAPI.
-
11.
Observe fluorescence staining of the cells that are attached to the insert using confocal microscopy.
3.7 In Vitro Migration Assay
-
1.
Add 600 μL of RPMI conditional medium/3 % FBS to each well of a 24-well plate.
-
2.
Dilute WBC (from step 10 of Subheading 3.4) to 2 × 106 cells per 100 μL in RPMI conditional medium/3 % FBS.
-
3.
Gently add the diluted WBC to a Transwell insert (Fig. 3).
Fig. 3 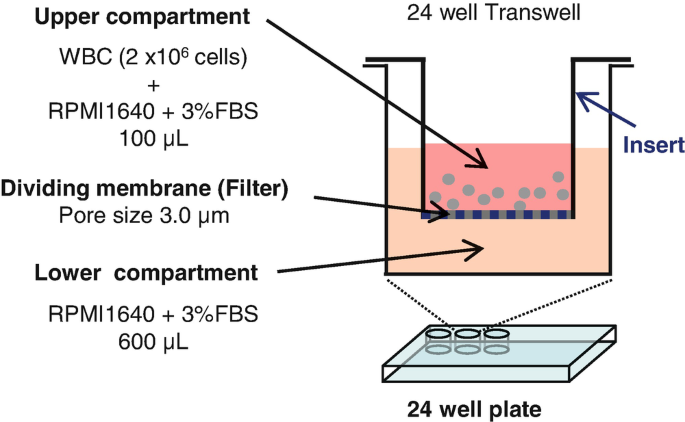
Schematic of the in vitro Transwell cell migration system
-
4.
Set insert into the well and incubate for 90 min at 37 °C.
-
5.
Collect media from the upper and lower compartments.
-
6.
Attach cells from both compartments on separate glass slides using a Cytospin and dry overnight at room temperature for double fluorescence staining (see Subheading 3.5 or 3.6) (seeNote 11 ).
-
7.
Some neutrophils will migrate through the filter that divides the upper from lower compartments but do not move into the media of the lower compartment. To stain the neutrophils that remain attached to the membrane, employ the following procedures.
-
8.
Wash the insert with PBS to remove cells on the upper surface of the filter.
-
9.
Place the insert in a new well filled with 1 mL of 4 % formalin for 20 min at room temperature to fix cells on the bottom surface of the filter.
-
10.
Perform procedures for double fluorescence staining (see Subheading 3.5 or 3.6).
-
11.
Remove the stained filter from the insert and place it on a glass slide with its lower surface facing upwards.
-
12.
Mount this filter with a coverslip and mounting medium/DAPI.
-
13.
Observe staining using confocal microscopy as in Fig. 4.
Fig. 4 
Migration of phosphorylated ERα-expressing neutrophils in Transwell system. The WBC fraction prepared from peritoneal blood of C3H/HeNCrIBR females was subjected to an in vitro migration assay as described in Subheading 3.7. Panel a illustrates the total WBC cell fraction that was added to the upper compartment. Panel b shows the cells that remained in the upper compartment after the migration assay. Panel c illustrates the cells that migrated into the lower compartment. Panel d illustrates the cells that migrated to the lower surface of the dividing filter. These cells were double stained by anti-Ly6G (in red) and aP-S216 (in green) antibodies. DAPI stains nuclei in blue. Neutrophils stained by both antibodies are in yellow. The data demonstrate that only those blood neutrophils that expressed phosphorylated ERα were able to migrate in this in vitro Transwell assay




4 Notes
-
1.
The purified antibody used here (0.29 mg protein/mL) was produced in rabbits by AnaSpec, Inc [4]. This antibody is not commercially available at the present time.
-
2.
Purified fraction from rat brain that consists primarily of α, β and γ isoforms with lesser amount of δ and ζ isoforms.
-
3.
GST-mouse ERα and its mutant (Ser216Ala) were constructed in our laboratory.
-
4.
Weigh directly into bottle (without a spatula) and dissolve in sterile water. Store at 4 °C.
-
5.
Weigh directly into bottle (without a spatula) and dissolve in sterile water by sonicating, warming and shaking. After filtration, store the solution at 4 °C.
-
6.
Set timer of the decloaker apparatus (a type of pressure cooker) for 3 min. However, the entire decloaking process takes a total of 30 min because of the time needed for increasing and decreasing the internal pressure.
-
7.
Dilute anti-P-S216 antibody with PBS/1.5 % normal goat serum to the concentration of 10 μg/mL and add either phospho-peptide or non-phospho-peptide in PBS to the same final concentration as that of the antibody.
-
8.
Pre-warm 6 % dextran solution and 0.9 % sodium chloride in 37 °C water bath just before use.
-
9.
Pipet out bubbles from solution before allowing the tube to stand for 20–30 min.
-
10.
Count only granulocytes based on their smaller size.
-
11.
Centrifuge medium at 220 × g for 6 min at room temperature. Remove the supernatant and resuspend the pellet in 100 μL PBS in preparation for the Cytospin.
References
Murphy LC, Seekallu SV, Watson PH (2011) Clinical significance of estrogen receptor phosphorylation. Endocr Relat Cancer 18:R1–R14
Atsriku C, Britton DJ, Held JM et al (2009) Systematic mapping of posttranslational modifications in human estrogen receptor-alpha with emphasis on novel phosphorylation sites. Mol Cell Proteomics 8:467–480
Shindo S, Sakuma T, Negishi M, Squires J (2012) Phosphorylation of serine 212 confers novel activity to human estrogen receptor alpha. Steroids 77:448–453
Shindo S, Moore R, Flake G, Negishi M (2013) Serine 216 phosphorylation of estrogen receptor alpha in neutrophils: migration and infiltration into the mouse uterus. PLoS One 8, e84462
Tibbetts TA, Conneely OM, O'Malley BW (1999) Progesterone via its receptor antagonizes the pro-inflammatory activity of estrogen in the mouse uterus. Biol Reprod 60:1158–1165
Daimon E, Wada Y (2005) Role of neutrophils in matrix metalloproteinase activity in the preimplantation mouse uterus. Biol Reprod 73:163–171
Wood GA, Fata JE, Watson KL, Khokha R (2007) Circulating hormones and estrous stage predict cellular and stromal remodeling in murine uterus. Reproduction 133:1035–1044
Cunningham M, Gilkeson G (2011) Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol 40:66–73
Baumgarten SC, Frasor J (2012) Minireview: Inflammation: an instigator of more aggressive estrogen receptor (ER) positive breast cancers. Mol Endocrinol 26:360–371
Lang TJ (2004) Estrogen as an immunomodulator. Clin Immunol 113:224–230
Mutoh S, Osabe M, Inoue K et al (2009) Dephosphorylation of threonine 38 is required for nuclear translocation and activation of human xenobiotic receptor CAR (NR1I3). J Biol Chem 284:34785–34792
Mutoh S, Sobhany M, Moore R et al (2013) Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by Inhibition of epidermal growth factor receptor signaling. Sci Signal 6(274):ra31
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences: Z01ES1005-01. We would like to acknowledge Dr. Kenneth Korach’s lab members and also Drs. Gordon Flake and Hideki Nakano for various experimental protocols.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Shindo, S., Moore, R., Negishi, M. (2016). Detection and Functional Analysis of Estrogen Receptor α Phosphorylated at Serine 216 in Mouse Neutrophils. In: Eyster, K.M. (eds) Estrogen Receptors. Methods in Molecular Biology, vol 1366. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3127-9_32
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3127-9_32
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3126-2
Online ISBN: 978-1-4939-3127-9
eBook Packages: Springer Protocols