
Abstract
Adenosine (A)-to-inosine (I) RNA editing is a fundamental posttranscriptional modification that ensures the deamination of A-to-I in double-stranded (ds) RNA molecules. Intriguingly, the A-to-I RNA editing system is particularly active in the nervous system of higher eukaryotes, altering a plethora of noncoding and coding sequences. Abnormal RNA editing is highly associated with many neurological phenotypes and neurodevelopmental disorders. However, the molecular mechanisms underlying RNA editing-mediated pathogenesis still remain enigmatic and have attracted increasing attention from researchers. Over the last decade, methods available to perform genome-wide transcriptome analysis, have evolved rapidly. Within the RNA editing field researchers have adopted next-generation sequencing technologies to identify RNA-editing sites within genomes and to elucidate the underlying process. However, technical challenges associated with editing site discovery have hindered efforts to uncover comprehensive editing site datasets, resulting in the general perception that the collections of annotated editing sites represent only a small minority of the total number of sites in a given organism, tissue, or cell type of interest. Additionally to doubts about sensitivity, existing RNA-editing site lists often contain high percentages of false positives, leading to uncertainty about their validity and usefulness in downstream studies. An accurate investigation of A-to-I editing requires properly validated datasets of editing sites with demonstrated and transparent levels of sensitivity and specificity. Here, we describe a high signal-to-noise method for RNA-editing site detection using single-molecule sequencing (SMS). With this method, authentic RNA-editing sites may be differentiated from artifacts. Machine learning approaches provide a procedure to improve upon and experimentally validate sequencing outcomes through use of computationally predicted, iterative feedback loops. Subsequent use of extensive Sanger sequencing validations can generate accurate editing site lists. This approach has broad application and accurate genome-wide editing analysis of various tissues from clinical specimens or various experimental organisms is now a possibility.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Drosophila melanogaster
- RNA editing
- ADAR
- Double-stranded RNA
- Transcriptome
- Protein recoding
- Noncoding RNA s
- Neurological disorders
- Next-generation sequencing
- Single-molecule sequencing
- Inosinome
1 Introduction
1.1 A-to-I RNA Editing
Mature RNA molecules often vary substantially from their genomic origins via posttranscriptional RNA processing events such as alternative splicing. However, more subtle changes in mature RNAs can occur through RNA editing [1]. The most prevalent and evolutionarily conserved RNA-editing system is the deamination of adenosine-to-inosine (A-to-I). This phenomenon involves the conversion of adenosine nucleotides into inosine through hydrolytic deamination (Fig. 1a) mediated by adenosine deaminases acting on RNA (ADAR ) [2]. RNA editing enzymes consist of double-stranded RNA-binding domains (dsRBDs) as well as a catalytic domain in the C-terminal part of the protein [3]. ADAR targets duplex RNAs of various structural arrangements and lengths. The structural variability of RNA substrates confers two distinct types of editing specificities. For example, short imperfect dsRNA molecules containing mismatches, bulges, and loops are edited specifically while long perfectly base-paired dsRNAs are edited promiscuously [2]. Inosine nucleosides mimic the base pairing properties of guanosine through the formation of Watson-Crick bonds with cytosine (Fig. 1a). Therefore, the cellular machinery interprets inosines as guanosines [4]. Specific RNA editing in coding regions has the capacity to recode the genome via amino acid substitutions in highly conserved and functionally important residues within proteins [5]. For example, the rate of inactivation in potassium channels is regulated by specific RNA-editing events, that result in non-synonymous amino acid reassignments [6]. The A-to-I RNA editing system is highly active in the nervous system and edits transcripts encoding products which are involved in electrical and chemical neurotransmission, such as components of the synaptic release machinery as well as ligand-gated and voltage-gated ion channels [7]. Specific editing is also active in non-coding sequences and is associated with regulating RNA splicing through creation or elimination of splicing signals and with the regulation of biogenesis and function of microRNAs (miRNAs) [2]. Promiscuous ADAR editing activity occurs invariably in noncoding regions of the genome and this activity is typically observed in transposable element sequences embedded in introns, within untranslated regions (UTRs), and in long noncoding RNAs (lncRNAs) [8, 9]. This form of abundant editing is involved in nuclear retention of transcripts [10], in cellular defense against viral RNAs [11], and in regulation of RNA interference (RNAi) pathways [3]. Phenotypes of ADAR deficiencies in various model organisms provide evidence that appropriate nervous system function requires an adequate A-to-I RNA-editing activity. Loss of RNA editing in invertebrates results in severe neurological defects and diverse behavior abnormalities. In C. elegans, loss of RNA editing results in chemotaxis defects [12]. Furthermore, Drosophila editing mutants exhibit coordination defects, seizures, temperature-sensitive paralysis, defects in courtship display, and age-dependent neurodegeneration [13]. More severely, the loss of editing activity in mammals leads to lethality. Specifically, the deletion of ADAR1 editing enzyme is embryonic lethal due to hematopoiesis defects and elevated cellular apoptosis [14]. Similarly, ADAR2 deletion also results in postnatal lethality caused by severe seizure episodes [15]. These phenotypes highlight an important role for the posttranscriptional process of A-to-I RNA editing in metazoan physiology.

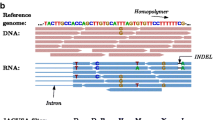
The hydrolytic deamination of adenosine to inosine. (a) An adenosine is converted to inosine via the hydrolytic deamination of an adenine base. Inosine shares the binding properties of guanosine, and thus forms bonds with cytidine. (b) Sequences generated through deep sequencing technologies contain a mixture of edited (G) and unedited (A) reads after proper alignments. Contrary, sequences generated from ADAR -deficient and wild-type DNA samples invariably contain unedited (A) reads. (c) The signature of A-to-I RNA editing. Example electropherograms generated by Sanger sequencing of cDNA molecules exhibit mixed A/G peaks in wild-type RNA sample. In contrast, ADAR-deficient and wild-type DNA electropherograms contain only the genomic encoded version in sequences
1.2 A-to-I RNA Editing and Neurological Disorders
Analysis of transcriptional landscapes within 15 human cell lines via deep sequencing technologies revealed that the majority of the genome is transcribed [16]. Indeed, pervasive transcription produces vast numbers of RNA molecules originating from noncoding portions of the genome [17]. These lncRNAs participate in diverse cellular functions during mammalian development, such as dosage compensation, genomic imprinting, and cell differentiation [18], through the formation of intricate secondary and tertiary structures that act as gene regulatory elements [19]. Not surprisingly, surveys of RNA folding in various eukaryotic genomes suggest that the transcriptome occupies a highly complex structural configuration [20], providing an additional informational layer analogous to the genetic code [21]. Moreover, these discoveries suggest that proper cellular function depends on the accurate expression of noncoding and coding RNA molecules, whose orchestrated processing allows for functional specificities. Thus, it is not surprising that mutations within proteins involved in almost all aspects of RNA metabolism lead to cellular catastrophes and various human diseases [22]. In particular, several studies have linked abnormal RNA editing with various neurological disorders [23]. For example, aberrant RNA editing of glutamate receptor is strongly linked to Amyotrophic lateral sclerosis and epilepsy. Likewise, serotonin receptor editing has been implicated in depression, schizophrenia and Prader-Willi syndrome. Alterations in ADAR expression occur in glioblastoma, a brain-specific cancer, and misregulation of dsRNA metabolism mediated by the ADAR1 editing enzyme is linked to Aicardi-Goutieres syndrome, a neurodevelopmental disorder [24].
Despite these links between RNA editing and cellular disease, appropriate tools remain largely unavailable for the measurement of changes in editing levels. Additionally, relatively little data exists on stress and activity induced changes in editing levels. Current research in the field implements next generation sequencing in order to identify authentic RNA-editing sites across a broad range of phyla. Understanding the dynamics of ADAR -mediated editing in a variety of environmental and physiological contexts represents an important avenue of current research.
1.3 Next-Generation Sequencing to Study RNA Editing in Drosophila
Since inosine forms base pairs with cytosine, the cellular machinery recognize inosine as guanosine. This is observed as A-to-G substitutions in RNAseq reads obtained from deep sequencing platforms (Fig. 1b). More importantly, these A-to-G substitutions in reads can be validated using Sanger sequencing. Electropherograms generated from cDNA libraries exhibit a mixed A/G peak at the edited adenosine (Fig. 1c). During the last decade several studies attempted to identify the exact genomic locations of RNA editing sites in various cell lines, tissues and model systems. Although identification of A-to-G substitutions in RNAseq experiments sounds simple, an inherent number of technical and biological factors can lead to variations and errors in the detection and measurement of RNA-editing sites. For example, a recent study reported that RNA-editing events are mechanistically more widespread than previously thought, leading to all possible nucleotide substitutions in human B cells, thereby expanding the range of RNA editing types [25]. Yet, most of these RNA editing events were attributed to artifacts generated from common sources of errors by next generation sequencing technologies [26–28]. Additionally, editing site discovery studies in Drosophila showed relatively poor overlap between sites and uncovered a large number of editing sites that are specific to individual wild type lab stocks [29–31]. Although different lab stocks may carry specific RNA editing events, stock-specific RNA editing alone cannot explain the unprecedented variation observed in these studies (Fig. 2a). To different degrees, the discrepancies seen between the three independent Drosophila datasets serve as an important reminder of the quality-control issues that may arise from the use of high throughput next generation sequencing experiments. Additionally, they highlight the need for more rigorous methodologies for the assessment of experimental reproducibility [32].

Comparison of RNA-editing site datasets from recent studies in Drosophila. (a). Venn diagram showing the relations between three independent Drosophila editing datasets identified by next-generation sequencing technologies. RNA-editing sites reported by modENCODE, Rodriguez et al., and Ramaswami et al. exhibit relatively poor overlap. (b) Venn diagram showing the relations between RNA-editing sites reported through the method described here (St. Laurent et al.), compared to the other three published RNA-editing lists
Here we describe our editing site discovery pipeline protocol, in Drosophila, using single-molecule sequencing. This method identified 3581 editing sites (Fig. 2b) and achieved a measure of success in both specificity (false-positive rate) and sensitivity (False Negative Rate) [33]. Coupled with extensive Sanger sequencing validations, the method generated the most accurate editing site discovery dataset in Drosophila to date. Our method provides a benchmark necessary to observe meaningful biological patterns resulting from the process of A-to-I RNA editing. Most available datasets do not include sufficient information or validation experiments for quality metrics, such as sensitivity, and specificity. Comparison of our results with other recently published Drosophila datasets (Table 1) demonstrates the effectiveness of the protocol. Our pipeline achieves three goals: (a) the identification of putative RNA-editing sites with high validation rate, (b) the successful capture of the majority of editing sites in any given experimental sample, and (c) the usage of this dataset to increase the visibility of ADAR -mediated editing in the context of transcriptome systems biology.
2 Materials
2.1 DNA Preparation
-
1.
Maxwell® 16 Tissue DNA Purification Kit (Promega).
2.2 RNA Preparation
-
1.
TRIzol® reagent (Invitrogen).
-
2.
TurboDNase Buffer (Applied Biosystems).
-
3.
RNaseOut (Invitrogen).
-
4.
TurboDNase (Applied Biosystems).
-
5.
RNeasy MinElute kit (Qiagen).
2.3 Ribosomal RNA Depletion
-
1.
Oligos complementary to the Drosophila 18S and 28S rRNA.
-
2.
DEPC water.
-
3.
10 mM ddNTP mixture (Roche).
-
4.
2.5 mM CoCl2.
-
5.
10× TdT buffer (NEB).
-
6.
Terminal Transferase (NEB).
-
7.
Performa DTR cartridges (EdgeBio).
-
8.
RiboMinus Eukaryote Kit for RNA-Seq (Invitrogen).
2.4 Synthesis of cDNA
-
1.
Superscript III kit (Invitrogen).
-
2.
RNAseIf (NEB).
-
3.
Performa Gel Filtration Columns (EdgeBio).
2.5 PolyA Tailing and 3′ Blocking
-
1.
PolyA Control Oligo (Helicos).
-
2.
2.5 mM CoCl2.
-
3.
10× TdT buffer (NEB).
-
4.
PolyA tailing dATP (Helicos).
-
5.
Biotinylated ddATP (Perkin Elmer).
-
6.
USER enzyme (NEB).
-
7.
DEPC water.
-
8.
AMPure beads (Beckman Coulter).
-
9.
70 % Ethanol.
-
10.
TE buffer.
2.6 Sequencing
-
1.
Helicos Single Molecular Sequencer.
2.7 Validation
-
1.
PCR primers.
-
2.
Sequencing primers.
-
3.
Phusion High-Fidelity PCR Kit (NEB).
-
4.
ExoSAP-IT (Affymetrix).
3 Methods
Highly complex genomic datasets produced with the implementation of next-generation sequencing technologies have relatively high error rates and present challenges in distinguishing patterns of biological knowledge from sources of noise and variation. These sources include unannotated SNPs, alternative splicing events, sequencing platform errors, sequence read misalignments [34], and potentially non-ADAR -mediated RNA sequence alterations [25]. Recently published RNA editing lists were shown to contain numerous false positives [33], likely because minimal Sanger validation was performed. Similarly, several other recent editing site datasets provided only sparse validation through Sanger sequencing [29, 35, 36]. In order to generate authentic information of sufficient quality to measure both the sensitivity and specificity of RNA editing sites in a whole organism we chose the Helicos single-molecule sequencing platform due to its advantages in transcript detection and reproducibility when compared to other sequencing platforms. Other advantages include unbiased coverage of rarely expressed transcripts [37], minimal sample preparation and avoidance of PCR amplification and ligation [38], and a very low A-to-G substitution error rate [33]. As a model organism, Drosophila bestows an ideal system for RNA editing profiling for the following reasons: Firstly, the presence of a well characterized collection of known editing sites exists for Drosophila [5]. Secondly, the existence of an ADAR deficiency model [13], and the availability of 15 sequenced genomes from various Drosophila species [39] further provide a well-established platform for the study of RNA editing. The method described here couples the depth of SMS with the accuracy of Sanger sequencing to determine bona fide RNA-editing events at the genome-wide scale. Specifically, our method uses a three-way comparison between the transcriptomes of wild-type (WT) and ADAR-deficient Drosophila and with the resequencing of our WT lab stock genome to comprehensively uncover the inosinome of an adult metazoan organism (Fig. 3).

A schematic diagram of the discovery pipeline for the Drosophila inosinome. Total (rRNA-depleted) RNA from adult wild-type flies served as the starting material for the discovery of novel A-to-I RNA-editing events. To account for single-nucleotide polymorphisms (SNPs) and various other artifacts not related to the RNA-editing process, we additionally sequenced DNA from wild-type flies and RNA from ADAR -deficient samples. Single-molecule sequencing reads were examined for the highest alignment quality, using an alignment pipeline designed to select the highest quality alignments while completely avoiding penalties for A-to-G substitutions, the signature of A-to-I RNA editing. Subsequent analysis generated a database of possible novel RNA-editing sites. With the implementation of Basic Filters and Machine Learning Algorithms the editing database was filtered further to distinguish between real editing sites and the many different kinds of false positives. One of the key features of this discovery pipeline is a strong reliance on validation of randomly selected sites through Sanger sequencing to generate True Positives and True Negative sites, and further train the Machine Learning Algorithms at each iteration. Using this repetitive computational approach the final version of the Machine Learning Algorithm was used to partition possible RNA editing sites to establish the Tier 1 list (conservative thresholds), and the Tier 2 list (medium thresholds). Finally, with the implementation of Sanger sequencing the validation rates for the two Tier lists were confirmed by sequencing random selected sites
3.1 DNA Preparation
-
1.
Isolate DNA from whole male Canton-S (wild type) and ADAR null flies using the Maxwell® 16 Tissue DNA Purification Kit following the manufacturer’s protocol. Place 10–20 flies of the same genotype in well #1 of the DNA cartridge. Place a plunger in well #7 of the DNA cartridge. Add 500 μl of the elution buffer to the elution tube. Place the DNA cartridge and elution tube in a Maxwell 16 robot to isolate genomic DNA from Drosophila tissue.
3.2 RNA Preparation
-
1.
Isolate total RNA from whole male Canton-S (wild type) flies using TRIzol® reagent (Invitrogen) following the manufacturer’s protocol.
-
2.
Mix 20 μg of total RNA with 10 μl TurboDNase Buffer; 1 μl RNaseOut; and 2 μl TurboDNase. Incubate the reaction for 30 min at 37 °C.
-
3.
Purify the RNA using the RNeasy MinElute Kit following the manufacturer’s protocol.
3.3 Ribosomal RNA Depletion
-
1.
Deplete rRNA from total RNA samples using the RiboMinus Eukaryote Kit for RNA-Seq following the manufacturer’s protocol (see Note 1 ).
3.4 Synthesis of cDNA
-
1.
Use between 100 and 200 ng of rRNA-depleted RNA for cDNA synthesis using the Superscript III Kit (see Note 2 ).
3.5 PolyA Tailing and 3′ Blocking
-
1.
Use 100 ng of cDNA in 28 μl water and add 5 μl of Helicos PolyA Control Oligo. Incubate the reaction for 5 min at 95 °C and rapid cool on ice.
-
2.
Add 5 μl 2.5 mM of CoCl2, 5 μl of Helicos PolyA tailing dATP, and 5 μl of 10× terminal deoxynucleotide transfer (TdT) buffer. Incubate the reaction for 1 h at 42 °C and then at 70 °C for 10 min.
-
3.
Denature the reaction at 95 °C for 5 min and then rapidly cool the reaction on ice.
-
4.
Add 0.4 μl of biotinylated ddATP and then 2 μl of TdT buffer. Incubate the reaction for 1 h at 37 °C and then at 70 °C for 10 min.
-
5.
Digest the reaction with 1 μl of USER enzyme and incubate at 37 °C for 30 min.
-
6.
Use DEPC water to bring the volume of the reaction to 60 μl and then add 72 μl of AMPure beads. Incubate the reaction for 30 min at room temperature with intermittent agitation.
-
7.
Collect the beads using a magnetic stand and wash them twice with 500 μl of 70 % ethanol. Air-dry the beads for 5–10 min.
-
8.
Resuspend the beads in 20 μl of TE buffer. Place the samples on the magnetic stand for 5 min and then remove the supernatant (see Note 3 ).
3.6 Sequencing
-
1.
Sequence samples with the Helicos Single Molecule Sequencer (see Note 4 ).
3.7 Validation
-
1.
Perform PCR reactions with Phusion High-Fidelity PCR Kit following the manufacturer’s protocol.
-
2.
Clean 7.5 μl of PCR sample with 3 μl of ExoSAP-IT following the manufacturer’s protocol.
-
3.
Sanger sequence the samples (see Note 5 ).
3.8 Bioinformatics and Machine Learning Methods
-
1.
Align standard sequences.
Align SMS reads from WT and ADAR deficient flies to DM3 reference genome. Perform alignments with the indexDP genomic aligner. Realign RNAseq reads that do not match the genome by setting the score for A-to-G or T-to-C substitutions (reference → read) to be the same as for the matching bases. Combine these alignments with the standard alignments for downstream processing. Capture all possible alignments for each RNAseq read and filter them with a minimum normalized score. Remove all reads mapping to rDNA or chrM (see Note 6 ).
-
2.
Use basic filters for editing site discovery (Table 2) (see Note 6 ).
Table 2 Basic filters for editing site discovery -
3.
Use machine learning algorithms.
Train the machine learning models on true positive (TP) and true negative (TN) sites obtained from Sanger sequencing validation. Assess the quality of the models using the following criteria: specificity (TN/N), sensitivity (TP/P), AUC (the area under receiver operating characteristic curve), and positive predictive value (TP/(TP + FP)). Furthermore, assess the performance of generated models through the ROCR package. Generate subsequent predictive models using the R environment for statistical computing. Finally, partition the data through extensive training and tuning of the models using the classification and regression training (caret) package for R (see Note 7 ).
4 Notes
-
1.
For ribosomal depletion the manufacturer’s protocol was modified as follows: Primers complementary to the Drosophila 18S and 28S rRNA transcript were designed to have a 5′-biotin. First, the primers were resuspended at 1000 μM and an equimolar mastermix prepared. A total of 2000 pmol oligo was added to 19 μl DEPC water and the mixture incubated at 95 °C for 5 min and then rapidly cooled on ice. A total of 8 μl of 10 mM ddNTPs, 4 μl 2.5 mM CoCl2, 10× TdT buffer, and 3 μl Terminal Transferase were added to the primers and incubated at 37 °C for 1 h, followed by an additional incubation at 70 °C for 10 min. The primers were then cleaned twice on Performa DTR cartridges following the manufacturer’s protocols. 2.5 μl of the prepared primer mixture was added to the total RNA samples prior to hybridization, when the RiboMinus probe was added. The exact sequence of the Drosophila 18S and 28S rRNA complementary primers can be found in St. Laurent et al. [33].
-
2.
Upon completion of cDNA synthesis RNA was eliminated by adding 1 μl RNAseH. We modified the manufacturer’s protocol to include the addition of 1 μl RNAseIf as well and incubated the mixture at 37 °C for 30 min. Furthermore, the resulting cDNA was then purified by the serial use of two Performa Gel Filtration Columns and quantified.
-
3.
An additional 20 μl elution was performed and pooled with the first sample.
-
4.
20 μl of samples were hybridized to the HeliScope flow cell at a loading concentration of 100–350 pM.
-
5.
PCR and Sanger sequencing primers were designed with BatchPrimer 3 (BatchPrimer 3: a high-throughput Web application for PCR and sequencing primer design).
-
6.
More detailed descriptions of computational methods implemented for sequencing alignments as well as information on the basic filters used for editing site discovery can be found in St. Laurent et al. [33].
-
7.
Data used for the testing and training of machine learning models, description of variables, and additional details of the machine learning algorithms implemented for editing site discovery can be found in St. Laurent et al. [33].
References
Bass BL (2002) RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 71:817–846
Nishikura K (2010) Functions and regulation of RNA editing by adar deaminases. Annu Rev Biochem 79:321–349
Savva Y, Rieder LE, Reenan R (2012) The adar protein family. Genome Biol 13:252
Basilio C, Wahba AJ, Lengyel P, Speyer JF, Ochoa S (1962) Synthetic polynucleotides and the amino acid code, v. Proc Natl Acad Sci U S A 48:613–616
Hoopengardner B, Bhalla T, Staber C, Reenan R (2003) Nervous system targets of RNA editing identified by comparative genomics. Science 301:832–836
Bhalla T, Rosenthal JJC, Holmgren M, Reenan R (2004) Control of human potassium channel inactivation by editing of a small mRNA hairpin. Nat Struct Mol Biol 11:950–956
Rosenthal JJC, Seeburg PH (2012) A-to-I RNA editing: effects on proteins key to neural excitability. Neuron 74:432–439
Lehmann KA, Bass BL (2000) Double-stranded RNA adenosine deaminases adar1 and adar2 have overlapping specificities. Biochemistry 39:12875–12884
Savva YA, Jepson JEC, Chang Y-J, Whitaker R, Jones BC et al (2013) RNA editing regulates transposon-mediated heterochromatic gene silencing. Nat Commun 4:2745
Kumar M, Carmichael GG (1997) Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts. Proc Natl Acad Sci U S A 94:3542–3547
Bass BL, Weintraub H, Cattaneo R, Billeter MA (1989) Biased hypermutation of viral RNA genomes could be due to unwinding/modification of double-stranded RNA. Cell 56:331
Tonkin LA, Saccomanno L, Morse DP, Brodigan T, Krause M, Bass BL (2002) RNA editing by adars is important for normal behavior in Caenorhabditis elegans. EMBO J 21:6025–6035
Palladino MJ, Keegan LP, O’Connell MA, Reenan RA (2000) A-to-I pre-mRNA editing in Drosophila is primarily involved in adult nervous system function and integrity. Cell 102:437–449
Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL et al (2004) Stress-induced apoptosis associated with null mutation of adar1 RNA editing deaminase gene. J Biol Chem 279:4952–4961
Higuchi M, Maas S, Single FN, Hartner J, Rozov A et al (2000) Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme adar2. Nature 406:78–81
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T et al (2012) Landscape of transcription in human cells. Nature 489:101–108
Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC et al (2005) The transcriptional landscape of the mammalian genome. Science 309:1559–1563
Fatica A, Bozzoni I (2014) Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15:7–21
Serganov A, Serganov A, Patel DJ, Patel DJ (2007) Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat Rev Genet 8:776–790
Li F, Zheng Q, Ryvkin P, Dragomir I, Desai Y et al (2012) Global analysis of RNA secondary structure in two metazoans. Cell Rep 1:69–82
Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY (2011) Understanding the transcriptome through RNA structure. Nat Rev Genet 12:641–655
Lukong KE, Chang K, Khandjian EW, Richard S (2008) RNA-binding proteins in human genetic disease. Trends Genet 24:416–425
Slotkin W, Nishikura K (2013) Adenosine-to-inosine RNA editing and human disease. Genome Med 5:105
Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM et al (2012) Mutations in adar1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat Genet 44:1243–1248
Li M, Wang IX, Li Y, Bruzel A, Richards AL et al (2011) Widespread RNA and DNA sequence differences in the human transcriptome. Science 333:53–58
Kleinman CL, Majewski J (2012) Comment on “widespread RNA and DNA sequence differences in the human transcriptome”. Science 335:1302
Lin W, Piskol R, Tan MH, Li JB (2012) Comment on “widespread RNA and DNA sequence differences in the human transcriptome”. Science 335:1302
Pickrell JK, Gilad Y, Pritchard JK (2012) Response to comment on “widespread RNA and DNA sequence differences in the human transcriptome”. Science 335:1302
Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM et al (2011) The developmental transcriptome of Drosophila melanogaster. Nature 471:473–479
Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P et al (2013) Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10:128–132
Rodriguez J, Menet JS, Rosbash M (2012) Nascent-seq indicates widespread cotranscriptional RNA editing in Drosophila. Mol Cell 47:27–37
Sugden LA, Tackett MR, Savva YA, Thompson WA, Lawrence CE (2013) Assessing the validity and reproducibility of genome-scale predictions. Bioinformatics 29:2844–2851
St Laurent G, Tackett MR, Nechkin S, Shtokalo D, Antonets D et al (2013) Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nat Struct Mol Biol 20:1333–1339
Qu W, Shen Z, Zhao D, Yang Y, Zhang C (2009) Mfeprimer: multiple factor evaluation of the specificity of PCR primers. Bioinformatics 25:276–278
Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, Li JB (2012) Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods 9:579–581
Peng Z, Cheng Y, Tan BC-M, Kang L, Tian Z et al (2012) Comprehensive analysis of RNA-seq data reveals extensive RNA editing in a human transcriptome. Nat Biotechnol 30:253–260
Sam LT, Lipson D, Raz T, Cao X, Thompson J et al (2011) A comparison of single molecule and amplification based sequencing of cancer transcriptomes. PLoS One 6:e17305
Raz T, Kapranov P, Lipson D, Letovsky S, Milos PM, Thompson JF (2011) Protocol dependence of sequencing-based gene expression measurements. PLoS One 6:e19287
Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L et al (2007) Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450:219–232
Acknowledgements
The authors wish to thank members of the Reenan lab for helpful discussions and suggestions, especially Michael Fuchs, Natalie Palaychuk, and Ali Rezaei. The authors are grateful to Cindi Staber, Alexander Leydon, Mustafa Talay, and Alissa Trepman for useful comments.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Savva, Y.A., Laurent, G.S., Reenan, R.A. (2016). Genome-Wide Analysis of A-to-I RNA Editing. In: Dassi, E. (eds) Post-Transcriptional Gene Regulation. Methods in Molecular Biology, vol 1358. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3067-8_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3067-8_15
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3066-1
Online ISBN: 978-1-4939-3067-8
eBook Packages: Springer Protocols