
Abstract
Anther culture is the most popular of the techniques used to induce microspore embryogenesis. This technique is well set up in a wide range of crops, including pepper. In this chapter, a protocol for anther culture in pepper is described. The protocol presented hereby includes the steps from the selection of buds from donor plants to the regeneration and acclimatization of doubled haploid plants derived from the embryos, as well as a description of how to analyze the ploidy level of the regenerated plants.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Androgenesis can be defined as the generation of an individual derived from a nucleus of male origin, usually a haploid microspore or young pollen grain [1]. Haploid embryos or calli are produced through the deviation of the microspore from its original gametophytic pathway towards a new sporophytic pathway. Haploid embryos may become doubled haploid individuals by themselves or through the application of treatments for genome doubling [2]. Doubled haploid individuals can be used as pure lines to produce hybrid seeds, which reduces considerably the time and resources needed to obtain pure lines when compared with conventional breeding methods [3].
For most of the studied species the optimal stage of male gametophyte development to induce embryogenesis is the transition between vacuolate microspores and young bicellular pollen [1, 4]. Technically, microspore embryogenesis can be induced through anther culture or isolated microspore culture . Isolated microspore culture is based on the isolation of the microspores in liquid medium. Since the maternal tissue is removed, microspores are directly in contact with the medium components. Therefore, the possible formation of somatic embryo s coming from the anther walls is avoided. Despite these advantages, isolated microspore culture is more complex than anther culture and therefore it is well set up just in a few species. Just tobacco ( Nicotiana tabacum ), rapeseed ( Brassica napus ), wheat ( Triticum aestivum ), and barley ( Hordeum vulgare ) can be considered as model systems for microspore culture [5]. For most crops of agronomic interest, the most used technique is still anther culture. Anther culture consists on the cultivation of the anthers in a solid or semisolid medium. It can be applied to a wide range of crops and it is the preferred technique used to produce doubled haploid s due to its simplicity [6], and to the possibility of culturing large amounts of anthers per isolation. In some species, the presence of the anther walls in the culture medium seems to provide a proper environment for microspore development, allowing for the induction of the microspores towards embryogenesis [7]. Anther culture in pepper ( Capsicum annuum L.) has been used to produce doubled haploid plants for breeding programs since the mid-1980s (see Chapter 9, this volume).
In this chapter, a protocol for anther culture of sweet pepper is explained according to Dumas de Vaulx et al. [8] with some modifications. The protocol was adapted for commercial F1 hybrids of sweet pepper [9] and the selection of donor flower buds was made according to Parra-Vega et al. [10]. In this protocol, the combination of two morphological markers (calix-bud length ratio and anther pigmentation) is used to select the optimal flower buds.
2 Materials
2.1 Plant Material
This stepwise protocol was developed with the following commercial F1 hybrids of pepper (C. annuum L.): ‘Herminio’ (Lamuyo type, from Syngenta Seeds), ‘Coyote’, ‘Quito’ (California type, both from Syngenta Seeds), and ‘Vélez’ (California type, from Enza Zaden).
2.2 Equipment
-
1.
Plastic tubes of 50 mL.
-
2.
Box with melting ice.
-
3.
Laminar flow hood.
-
4.
Sterile Whatman paper.
-
5.
Sterile forceps and scalpel.
-
6.
Sterile Petri dishes 90 × 25 mm (Ø × height).
-
7.
Parafilm.
-
8.
Inverted or light microscope.
-
9.
Microscope slides and cover slips.
-
10.
Aluminum paper.
-
11.
Incubator at 35 and 25 °C.
-
12.
Sterile baby food jars with plastic caps.
-
13.
Plastic plant pots 90 × 100 mm (width × height).
-
14.
Composite soil.
-
15.
Transparent plastic glass.
-
16.
Growth chamber at 25 °C.
-
17.
Pasteur pipettes, 3 mL.
-
18.
Razor blades.
-
19.
Filters of 30 μm pore (CellTricks, Partec).
-
20.
Plastic tubes 3.5 mL, 55 × 12 mm (Ø × height).
-
21.
Flow cytometer Partec Ploidy Analyzer I.
2.3 Solutions and Culture Media
-
1.
70 % ethanol (v/v).
-
2.
4 g/L sodium hypochlorite with 0.05 % Tween (v/v).
-
3.
Sterile distilled water (three glass jars) autoclaved at 121 °C for 20 min.
-
4.
Induction medium: C medium (Table 1) supplemented with 0.01 mg/L kinetin and 0.01 mg/L 2,4-dichlorophenoxyacetic acid (2,4-D).
-
5.
Regeneration medium: R medium (Table 1), supplemented with 0.1 mg/L kinetin. Adjust the pH of media C and R to 5.9. Autoclave media at 121 °C for 20 min, and then pour it in 90 × 25 mm sterile Petri dishes.
-
6.
Rooting medium: V3 medium (Table 1). Adjust the pH to 5.9. Autoclave medium at 121 °C for 20 min and pour it in 90 × 25 mm sterile Petri dishes and sterile baby food jars (200 mL).
-
7.
Lysis buffer (LB01) [11]: 5 mM Tris(hydroxymethyl)aminomethane, 2 mM Na2EDTA, 0.5 mM espermine, 80 mM KCl, 20 mM NaCl, 15 mM β-mercaptoethanol, and 0.1 % (v/v) Triton X-100. The pH is adjusted at 7.5.
-
8.
Staining buffer: 4,6-Diamidino-2-phenylindole (DAPI ) (Partec CyStain UV precise P, PARTEC GmbH).
3 Methods
3.1 Donor Plant Growth Conditions
Donor plants are grown in a growth chamber at 25 °C, light intensity of 200 μmol/m2/s with a 16 h photoperiod and 60–65 % relative humidity.
3.2 In Vitro Culture of Anthers
-
1.
Select by eye the optimal buds for anther culture . In our genotypes, they are covered approximately the 80 % of them by the sepals (Fig. 1a), according to Parra-Vega et al. [10] (see Note 1 ). Excise the buds from the plant. Bring them to the lab in plastic tubes immersed on melting ice (see Note 2 ).
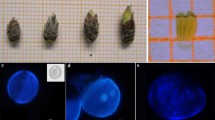
Fig. 1 
Process of anther culture in pepper . (a) Flower bud at the right stage for anther isolation. (b–d) Anther extraction out of the bud: transversal cut of the flower bud (b), longitudinal cut of the flower bud surface (c), and opening of the flower bud with scalpel and forceps to extract the anthers (d). (e) Anthers at the right stage for isolation. White arrow points the right position to culture the anthers in medium (concave part). (f) Anther culture d in vitro producing two microspore -derived embryos (white arrows) in c medium. (g) Microspore -derived embryo germinated in V3 medium. (h) Microspore-derived seedling cultured in vitro in V3 medium. (i) Acclimated seedling cultured ex vitro in a plastic plant pot. Bars: a–e, 2 mm; f and g, 5 mm; h, 1 cm; i, 2 cm
-
2.
Take the buds to the laminar flow hood.
-
3.
Surface sterilize the buds with 70 % ethanol for 30 s, and then with sodium hypochlorite 4 g/L for 5 min, and finally three washes of 4 min each with sterile distilled water (see Note 3 ).
-
4.
Place the buds over sterile Whatman paper and excise them to extract the anthers (see Note 4 ). At this step, make a second selection of the buds. Culture only buds containing anthers with purple distal tips (Fig. 1e), according to Parra-Vega et al. [10]. In case the optimal stage of anther development has not been well set up in advance for the genotype used, it is highly recommended, at this point, to check the microspores/pollen stage of every bud before culturing them (see Note 5 ).
-
5.
Place the selected anthers in Petri dishes with C medium. Place them with their concave part in contact with the medium. Seal the dishes with Parafilm and introduce them in the incubator at 35 °C in darkness for 4 days (see Note 6 ).
-
6.
At day 4, place the dishes in the incubator at 25 °C with a 12-h photoperiod for 4 days more.
-
7.
At day 8, transfer the anthers to R medium and incubate them at 25 °C, light intensity of 32 μmol/m2/s and a 12 h photoperiod. Every 2 months, change the anthers to fresh R medium.
-
8.
As soon as the embryos pop out of the anthers, pick them with forceps and transfer them to V3 medium in 90 × 25 mm Petri dishes, incubate them at 25 °C, light intensity of 32 μmol/m2/s and a 12 h photoperiod. Transfer the embryos that germinate correctly to sterile baby food jars with V3 medium (see Note 7 ).
-
9.
When seedlings develop a proper root system (one or two primary roots and some secondary roots), transfer them to plastic plant pots with wet soil.
-
10.
Acclimate the seedlings in the growth chamber at 25 °C and a 16 h photoperiod (see Note 8 ).

3.3 Analysis of the Ploidy Level
-
1.
Analyze the nuclear DNA content with a flow cytometer (Partec Ploidy Analyzer I) according to its commercial specifications. Use DAPI as the fluorescent stain.
-
2.
Use donor plants as control for 2C DNA content. Plants derived from embryos will be analyzed in order to know the ploidy level (see Note 9 ).
-
3.
Excise young leafs from the plant and place them in a box with ice (see Note 10 ).
-
4.
Chop with a razor blade a piece of 1 cm2 of a young leaf in a plastic Petri dish containing 0.5 mL of lysis buffer (see Note 11 ).
-
5.
Filter the extracted nuclei with a 30 μm pore filter into a 3.5 mL plastic tube.
-
6.
Add 1.5 mL of DAPI staining buffer with a 3 mL Pasteur pipette.
-
7.
Keep the tubes on ice for 2 min prior to analyze the samples using the flow cytometer. Count a minimum of 10,000 cells per sample.
4 Notes
-
1.
The selection of anthers is one of the critical steps of anther culture . The anthers must contain vacuolate microspores and young bicellular pollen grains to efficiently induce embryogenesis. As this parameter determination is highly genotype dependent, it is recommended to study previously, in each genotype, the right size and appearance of anthers containing the appropriate stage of microspore /pollen development to be induced towards embryogenesis.
-
2.
Once the buds are excised from the plant, keep them on ice in order to slow down the development of the microspores/pollen . Also, keep the sterilized solutions at 4 °C before using them to reduce the degradation process of anthers.
-
3.
Pour the sterilized solutions into the plastic tube, close the lid and shake the solutions during the corresponding time for each solution. After that, open the lid and remove the liquid keeping the buds. Pour the next solution into the tube and repeat the process. An alternative to the plastic tubes is to use tea filter sieves.
-
4.
Excise the anthers with a scalpel avoiding breaking them. First, make a transversal cut at the basal part of the bud (near to the pedicel), removing the basal part of the floral bud (Fig. 1b). Second, make a longitudinal cut, only at the surface of the bud (Fig. 1c), to open the sepals. Later, take away the sepals and petals with forceps, and extract the anthers (Fig. 1d). It is important to remove the anther filament as much as possible, just to avoid callus formation from this tissue, which is especially prone to proliferate.
-
5.
After extracting the anthers from the bud, take one anther to observe it under the microscope and keep the remaining anthers waiting in the laminar flow hood. Place the anther onto a microscope slide with a drop of water, chop the anther with a razor blade in order to extract the microspores/pollen and cover it with a standard cover slip. Observe the preparation under a light or inverted microscope checking the stages of microspores contained. If the anther contains mostly vacuolate microspores and young bicellular pollen, the rest of anthers from the same bud will be used for anther culture .
-
6.
Cover the Petri dishes with aluminum paper to create a darkness environment inside the incubator.
-
7.
Transfer the germinated embryos to baby jars in order to increase the space to develop the roots and aerial parts of the new plant.
-
8.
In order to avoid drastic change in humidity conditions, use a transparent plastic cup to protect the seedlings. Pinch holes in the cup every 2 days, to gradually reduce the humidity inside the cup down to the levels of the growth chamber. Then remove the glass.
-
9.
The flow cytometer is used to analyze the ploidy level, but when a 2C individual appears, molecular analysis marker (preferentially SSRs) has to be performed in order to clarify whether this individual has a somatic or an embryogenic origin. For donor plants polymorphic for the SSR used, if the regenerated samples analyzed are homozygous for the used molecular markers, the origin of these plants will be gametophytic. However, if the samples are heterozygous for the SSRs used, their origin will likely be somatic (most likely coming from anther wall tissues).
-
10.
Young tissues are used to analyze the ploidy level because these tissues present more cells in G2 phase; therefore the second peak of the histogram appears clearer.
-
11.
The nucleic extraction buffer from Partec (CyStain UV precise P, PARTEC GmbH) may be used at this step. However, with pepper is recommended to use the lysis buffer in order to slow down the oxidizing process of pepper samples.
References
Seguí-Simarro JM (2010) Androgenesis revisited. Bot Rev 76:377–404
Seguí-Simarro JM, Nuez F (2008) Pathways to doubled haploidy: chromosome doubling during androgenesis. Cytogenet Genome Res 120:358–369
Wedzony M, Forster BP, Zur I, Golemiec E, Szechynska-Hebda M, Dubas E, Gotebiowska G (2009) Progress in doubled haploid technology in higher plants. In: Touraev A, Forster BP, Jain SM (eds) Advances in haploid production in higher plants. Springer, Dordrecht, Netherlands, pp 1–33
Soriano M, Li H, Boutilier K (2013) Microspore embryogenesis: establishment of embryo identity and pattern in culture. Plant Reprod 26:181–196
Forster BP, Heberle-Bors E, Kasha KJ, Touraev A (2007) The resurgence of haploids in higher plants. Trends Plant Sci 12:368–375
Germanà MA (2011) Anther culture for haploid and doubled haploid production. Plant Cell Tissue Organ Cult 104:283–300
Seguí-Simarro JM, Nuez F (2008) How microspores transform into haploid embryos: changes associated with embryogenesis induction and microspore-derived embryogenesis. Physiol Plant 134:1–12
Dumas de Vaulx R, Chambonnet D, Pochard E (1981) Culture in vitro d’anthères de piment (Capsicum annuum L.): amèlioration des taux d'obtenction de plantes chez différents génotypes par des traitments à +35°C. Agronomie 1:859–864
Parra-Vega V, Renau-Morata B, Sifres A, Seguí-Simarro JM (2013) Stress treatments and in vitro culture conditions influence microspore embryogenesis and growth of callus from anther walls of sweet pepper (Capsicum annuum L.). Plant Cell Tissue Organ Cult 112:353–360
Parra-Vega V, González-García B, Seguí-Simarro JM (2013) Morphological markers to correlate bud and anther development with microsporogenesis and microgametogenesis in pepper (Capsicum annuum L.). Acta Physiol Plant 35:627–633
Dolezel J, Binarova P, Lucretti S (1989) Analysis of nuclear DNA content in plant cells by flow cytometry. Biol Plant 31:113–120
Chambonnet D (1988) Production of haploid eggplant plants. Bulletin interne de la Station d'Amelioration des Plantes Maraicheres d'Avignon-Montfavet, France, 1-10
Acknowledgments
This work was supported by the grant AGL2014-55177 from Spanish MINECO to J.M.S.S.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Parra-Vega, V., Seguí-Simarro, J.M. (2016). Anther Culture in Pepper (Capsicum annuum L.). In: Germana, M., Lambardi, M. (eds) In Vitro Embryogenesis in Higher Plants. Methods in Molecular Biology, vol 1359. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3061-6_26
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3061-6_26
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3060-9
Online ISBN: 978-1-4939-3061-6
eBook Packages: Springer Protocols