
Abstract
The circadian clock governs body time and regulates many physiological functions including sleep and wake cycles, body temperature, feeding, and hormone secretion. Alignment of drug dosing time to body time can maximize the pharmacological effect and minimize untoward effects. Therefore, a simple and robust method for estimating body time is important for drug efficacy or “chronotherapy”. We previously reported that a metabolite timetable could estimate body time with good accuracy. A metabolite timetable was constructed by profiling metabolites in human and mouse with capillary electrophoresis-mass spectrometry (CE-MS) and liquid chromatography–mass spectrometry (LC-MS). In this chapter, we describe practical methods to profile and identify oscillating metabolites.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Chronotherapy
- Circadian
- Metabolomics
- Capillary electrophoresis–mass spectrometry
- Liquid chromatography–mass spectrometry
1 Introduction
Mammals, as well as other many organisms, have an internal 24 h period rhythm called a circadian rhythm, which is generated through cellular transcriptional and translational feedback loops of core-clock genes in the body [1–3]. Circadian rhythms control many physiological functions and metabolic processes [4]. For example, hormone secretion is under circadian rhythm control. The abundances of melatonin and cortisol in blood, for example, oscillate in a circadian manner to regulate the daily functions of peripheral organs [5–10].
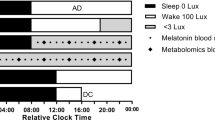
There is increasing evidence that dosing time affects drug efficacy and toxicity in humans [11–17]. Appropriately timed administration of two anticancer drugs in ovarian cancer patients (adriamycin at 6:00 AM and cisplatin at 6:00 PM) resulted in lower toxicity than arrhythmic administration [18]. However, it has been reported that individual internal body times can vary up to 5–6 h among healthy humans, and even 10–12 h for night-shift workers [19–22]. The conventional method to estimate body time is measure the blood level of melatonin or cortisol over 24 h because the abundance of these hormones fluctuates in a circadian manner [5, 8, 9]. However, this requires periodic blood sampling under a controlled environment, which severely restricts patients’ activity. A simpler method is needed to measure a patient’s body time in order to optimize drug efficacy [11–13, 15, 16]. We previously demonstrated that a molecular metabolite timetable could solve these issues [5, 23]. Our molecular timetable was inspired by Linnaeus’ flower clock. In Linnaeus’ flower clock, the opening or closing pattern of different flowers indicated the time of day. Likewise, we constructed a timetable of physiological molecules (e.g., metabolites) at particular times of day, and showed that the timetable could accurately estimate body time (Fig. 1).

The concept of body-time estimation based on a metabolite timetable method inspired by Linnaeus’ flower clock [23] reduces sampling costs. The conventional way is to monitor a single molecule over a few days (highlighted by a blue background). By contrast, the molecular timetable method measures multiple molecules at a few time points (highlighted by an orange background). Then, body-time is estimated by comparing the measured profile of multiple molecules to the timetable that was constructed in advance
For constructing the timetable, we used metabolomic techniques to profile hundreds to thousands of molecules in a sample. Metabolomic profiling by mass spectrometry has been used previously in other research fields including studies of bacteria, plants, and mammals [24–26]. The recent integration of capillary electrophoresis and mass spectrometry (CE-MS) has achieved high resolution with short measurement times [24, 27]. CE-MS is capable of identifying several hundred metabolites in a small sample volume (on the order of nanoliter). However, CE-MS, is not suited to measure neutral metabolites such as fatty acids and sugars because the technique is optimized for ionic molecules. Instead, liquid chromatography–MS (LC-MS) fills the gap left by CE-MS [24].
In this chapter, we describe how we constructed a metabolite timetable for mice and humans by using CE-MS and LC-MS and provide practical lab tips [5, 23].
2 Procedures
2.1 Construction of the Metabolite Timetable
Blood samples were periodically obtained from mice and humans. After pretreatment, samples were analyzed by CE-MS and LC-MS. We selected metabolites detected in the majority of time points, and found circadian oscillating metabolites by fitting to cosine curves. The fitness was statistically evaluated by a permutation test with a certain cutoff value for False Discovery Rate (FDR). Then, we constructed the metabolite timetable by using those oscillating metabolites as time-indicating metabolites [5, 23].
2.2 Application to Other Genetic Backgrounds
We confirmed that the metabolite timetable constructed by using CBA/N mice could also estimate internal body time of C57BL/6 mice. These two strains are genetically remote and are classified into two different clusters according to a single nucleotide polymorphism-based study [28]. This finding verified that this method can be applied to different populations with heterogeneous genetic backgrounds.
2.3 Application to Different Ages and Genders
We constructed the metabolite timetable by using young male mice, but it was reasonable to suppose that the abundance of metabolites changes with age or sex. To check the influence of age and sex on body time estimation, we also applied the metabolite timetable to aged male and young female mice. We demonstrated that the metabolite timetable method could accurately determine their internal body times [5] (see Note 1 ).
2.4 Jet Lag Experiment
To evaluate the accuracy of the molecular timetable method, we conducted a jet lag experiment in mice [5]. To mimic jet lag, we kept mice in a light–dark condition for 2 weeks, and then shifted the light-cycle forward by 8 h [29]. Blood was collected at two time points on three separate days after the light-cycle shift. The results confirmed that our metabolite timetable method could accurately estimate internal body time of jet-lagged mice.
3 Materials and Sampling
3.1 Subjects and Sampling
In the mice experiment, CBA/N and C57BL/6 were used. Young mice were 5–6 weeks old, and adult mice were about 6 months old. All mice were kept under light–dark condition (light: 12 h, dark: 12 h) for more than 2 weeks, and sampled under both light–dark and constant darkness conditions. We used blood plasma for metabolite profiling. The trunk blood was collected in tubes containing Novoheparin (Mochida Pharm) every 4 h for 2 days (12 samples in total). The blood from 4 to 10 mice per time point was mixed to obtain enough volume for constructing the metabolite timetable. The blood individual mice were used for body time measurement. Mice were fasted by removing food pellets from their cages at the light-off point one prior day to sampling to cancel the effect of food intake. Collected blood samples were quenched on ice (see Note 2 ) and centrifuged to remove cell products. For LC-MS samples, acetonitrile was added and strongly shaken, then supernatants were collected and lyophilized. The dried samples were stored in the freezer (−80 °C) until use (see Note 3 ). For CE-MS samples, 1.8 ml of methanol containing 55 μM each methionine sulfone and 2-Morpholinoethanesulfonic acid were mixed with plasma (100 μl) as internal standard and strongly shaken. Next, 800 μl of deionized water and 2 ml of chloroform were added. They were centrifuged at 2500 × g for 5 min at 4 °C. The supernatant aqueous layer (800 μl) was filtered using 5-kDa cutoff filter (Millipore) to remove cell products and proteins. Solvents were evaporated and stored in the freezer (−80 °C) until mass spectrometry analysis.
In the human experiment, six healthy volunteers (male, 20–23 years old, average 21.5 years old) participated in this study. They had no previous history of psychiatric or severe physical disease. They stayed in controlled laboratory conditions without time cues (i.e., no clock, radio, newspaper, mobile phone, or Internet access). Meals (200 kcal) were provided every 2 h and blood was collected every 2 h, using an i.v. catheter placed in a forearm vein of the subjects (see Note 4 ). To separate plasma, blood was centrifuged twice at 1000 × g for 5 min at 4 °C. The supernatant was stored at −80 °C until mass spectrometry analysis.
4 Reagents, Instruments, and Settings for Metabolomics
4.1 Metabolite Standards
We used a mixture of about 500 chemicals as internal standards. All chemical standards were purchased from commercial sources. They were dissolved in Milli-Q water, 0.1 N NaOH and stocked as 10 or 100 mM. Stock solutions were diluted to 20 or 50 μM.
4.2 Instruments for Metabolome Analysis
All CE-MS experiments were performed by using Agilent CE capillary electrophoresis system (Agilent technologies), Agilent G3250AA LC/MSD TOF system, Agilent1100 series binary HPLC pump, G1603A Agilent CE-MS adapter, and G1607A Agilent CE-ESI-MS sprayer kit. LC-MS experiments of mice were performed by using Agilent 1100 series HPLC loaded with the ZORBAX SB-C18 RRHT (Agilent Technologies). Mass spectral data was acquired by using Qstar XL mass spectrometry (Applied Biosynthesis). LC-MS human experiments were performed by using Agilent 1290 infinity HPLC loaded with Acquity ultra performance liquid chromatography BEH C18 column (Waters) and 6530 Accurate-Mass Q TOF LC/MS using the dual spray ESI of G-3521A (Agilent technologies) (see Notes 5 and 6 ).
4.3 CE-MS Conditions
Details of CE-MS conditions have been described previously [30, 31]. Cationic metabolites were separated using fused-silica capillary (50 μm inner diameter × 100 cm length) filled with 1 M formic acid [32]. Sheath liquid containing 0.1 μM Hexakis (2,2-difluoroethoxy) phosphazene for reference electrolyte in MeOH/water (50 % v/v) was delivered at 10 μl/min. Sample solution was injected at 50 mbar for 3 s (less than 3 nl) and 30 kV was applied. The capillary temperature was maintained at 20 °C and sample tray was kept at 5 °C. Time-of-flight mass spectrometry (TOF-MS) was controlled in the positive ion mode. An automatic recalibration function was performed by using reference masses of standards ([2MeOH + H2O + H]+, m/z 65.0597) and reserpine ([M+H]+, m/z 609.2806). Exact mass data were acquired at a rate of 1.5 cycles/s over the range of 50–1000 m/z.
4.4 LC-MS Conditions for the Mice Experiments
The column was kept at 60 °C. The mobile phase consisted of 0.1 % acetic acid/water and methanol. The gradient went from 40 % of methanol at 0 min, 99 % at 20 min, 99 % at 30 min, 40 % at 30.01 min and then kept at 40 % until 40 min. The mobile phase flow rate was 0.2 ml/min. Both positive ion mode and negative ion mode were used for sample analyses. The detail MS conditions for positive ions were as follows: spray voltage was 5.5 kV, scan range was 250–700 m/z, declustering potential 1/2 was 50 V/15 V. The settings for negative ion mode was almost the same as positive ion conditions, but the spray voltage was −4.5 kV and declustering potential 1/2 was −50 V/−15 V.
4.5 LC-MS Conditions for the Human Experiments
The settings for the human experiments were almost same as the mice experimental conditions, but the column was maintained at 50 °C, mobile phase consisted of 0.5 % acetic acid/water and isopropanol, the gradient was linearly changed from 1 to 99 % of isopropanol for 12 min and held for 5 min and the flow rate was 0.3 ml/min. Detailed MS conditions were as follows: gas temperature was 350 °C, drying gas was 10 l/min, nebulizer was 30 psig, fragmentor was 200 V, skimmer was 90 V, and scan range was 100–1600 m/z.
5 Data Analysis and Statistics
5.1 Identification and Quantification of Metabolomics Data
Data from CE-MS was analyzed by in-house software KEIO MasterHands (available on a contract basis) (see Note 7 ). The data analysis was performed in the following five steps: (1) Noise-filtering, (2) Baseline-correction, (3) Migration time alignment, (4) Peak detection, (5) Integration of peak area. Migration time was normalized by an algorithm based on dynamic-programming [33]. Metabolites were identified by matching both m/z value and migration time to standard reagents, and all peaks were manually checked. Peak areas were normalized according to internal standards spiked into samples.
5.2 Screening of Time-Indicating Metabolites
First of all, we chose candidate metabolites that were constantly detected (more than 10 time points in both light–dark condition and constant darkness condition). Next, we selected metabolites that have high Pearson’s correlation with a cosine curve (see Note 8 ). We calculated p-values and False Discovery Rate (FDR) of the correlation by using permutation tests. By applying the thresholds of FDR < 0.1 for CE-MS data and FDR < 0.01 for LC-MS data, we selected significantly oscillating metabolites as components of the metabolite timetable (Fig. 2).

The circadian oscillation patterns of selected metabolites [5]. Yellow indicates a high level of metabolite concentration and blue indicates a low level in blood plasma. Metabolites were sorted by their peak time of concentration. Blood samples were collected under both light–dark (labeled with Zeitgeber time) and constant darkness (labeled with circadian time) condition. The bar above the graph indicates day (white), subjective day (gray ), and night or subjective night (black )
5.3 Accuracy Test of Metabolite Timetable
To check the validity of the constructed metabolite timetable, we tried the jet-lag experiment. Young male CBA/N mice were first kept under light–dark cycle (light 12 h, dark 12 h) for 2 weeks, and then exposed to 8 h advanced light–dark cycle. They were sacrificed and blood was collected on 1, 5, and 14 days after the phase advance. Their behaviors were recorded by an infrared monitoring system NS-AS01 (Neuroscience) and data were visualized with Clock-Lab software (Actimetrix).
6 Conclusion
In this chapter, we have described how a metabolite timetable was constructed for mice and humans by using CE-MS and LC-MS. We showed that our timetable method could be used in different populations, such as gender, age, and species of mice.
Our metabolite timetable method provides a robust way for minimally invasive internal body time estimation and may contribute to personalized medicine in the near future.
7 Notes
-
1.
Our mouse metabolite timetable can be applied regardless of gender and age. However, some reports show that subject backgrounds, such as gender, age, and clinical history, have the potential to affect metabolomic data [34, 35]. Therefore, experimental design should be carefully considered depending on the purpose.
-
2.
Samples should be kept cold and analyzed as quickly as possible to prevent metabolites from being degraded or modified. We recommend bringing materials such as reagents, and equipments such as a centrifuge and pipettes needed for sampling into the animal room or near the sampling space. For example, red blood cells are easily broken and can confound measurements by releasing their contents, in particular, amino acids. Samples become stable after separating plasma from red blood cells, white blood cells, and blood platelets.
-
3.
When samples are stored before analysis over a long period of time (more than 1 week), it is advisable to dry up liquid and store in a deep freezer at −80 °C or in liquid nitrogen to prevent oxidation and degradation of metabolites.
-
4.
Food intake is a primary cause of bias in blood metabolomic experiments in humans and mice, because diet directly affects body fluids [36]. The easiest and the most effective way to minimize the problem is fasting during time-course sampling. If fasting raises an ethical issue, we recommend providing a defined caloric amount of food at a constant time interval. In our human experiments, we served food to subjects every 2 h during sampling, and blood samples were collected just before having food [23]. In this way, we could select metabolites that oscillated independently of food intake.
-
5.
We recommend using not only MS but also a focused approach to measure well-known oscillating metabolites. For example, measurements of melatonin or corticosterone with radioimmunoassay or enzyme-linked immunosorbent assay are beneficial to assess sample quality (Fig. 3).
Fig. 3 
Circadian change of corticosterone level in mice plasma under light–dark (left) and constant darkness (right) conditions [5]. The bar above the graph indicates day (white), subjective day (gray), and night or subjective night (black). The gray shadow in the graph indicates the dark condition
-
6.
There are a variety of systems, such as CE-MS, LC-MS, gas chromatography, and nuclear magnetic resonance to measure metabolites. Among them, we recommend CE-MS or LC-MS, because they are the current primary platforms for metabolomics. CE-MS is particularly appropriate for ionic metabolites and can detect a wide range of metabolites. LC-MS is appropriate for neutral metabolites such as fatty acids and sugars. We recommend using a combined platform of CE-MS and LC-MS.
-
7.
There can be multiple causes of error in MS analysis. For example, capillary, column, and sheath liquid may deteriorate, and even detector sensitivity may change over the course of many measurements. It is important to occasionally, for example after every set of analysis, measure quality control samples to keep the data consistent over many samples (12 times/day × 2 days × 3 subjects = 72 samples in the case of our metabolite timetable construction). The quality control samples usually contain some reagents such as amino acids or organic acids with predefined concentrations. This allows for canceling errors and checking sample degradation. It is also important to keep the sample tray cold (e.g., 5 °C) during analysis.
-
8.
We selected only metabolites which showed a gradual change in their abundance over 24 h. This is because gradual changes fit to a cosine curve provide more information about the time of day than discrete or peaky 24 h rhythms.

References
Mohawk JA, Green CB, Takahashi JS (2012) Central and peripheral circadian clocks in mammals. Annu Rev Neurosci 35:445–462
Reppert SM, Weaver DR (2002) Coordination of circadian timing in mammals. Nature 418:935–941
Ueda HR (2007) Systems biology of mammalian circadian clocks. Cold Spring Harb Symp Quant Biol 72:365–380
Takahashi JS, Hong H-K, Ko CH et al (2008) The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet 9:764–775
Minami Y, Kasukawa T, Kakazu Y et al (2009) Measurement of internal body time by blood metabolomics. Proc Natl Acad Sci U S A 106:9890–9895
Eckel-Mahan KL, Patel VR, Mohney RP et al (2012) Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci U S A 109:5541–5546
Dallmann R, Viola AU, Tarokh L et al (2012) The human circadian metabolome. Proc Natl Acad Sci U S A 109:2625–2629
Weitzman ED, Fukushima D, Nogeire C et al (1971) Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J Clin Endocrinol Metab 33:14–22
Kennaway DJ, Voultsios A, Varcoe TJ et al (2002) Melatonin in mice: rhythms, response to light, adrenergic stimulation, and metabolism. Am J Physiol Regul Integr Comp Physiol 282:R358–R365
Fustin J-MM, Doi M, Yamada H et al (2012) Rhythmic nucleotide synthesis in the liver: temporal segregation of metabolites. Cell Rep 1:341–349
Halberg F (1969) Chronobiology. Annu Rev Physiol 31:675–725
Reinberg A, Smolensky M, Levi F (1983) Aspects of clinical chronopharmacology. Cephalalgia 3(Suppl 1):69–78
Reinberg A, Halberg F (1971) Circadian chronopharmacology. Annu Rev Pharmacol 11:455–492
Bocci V (1985) Administration of interferon at night may increase its therapeutic index. Cancer Drug Deliv 2:313–318
Labrecque G, Bélanger PM (1991) Biological rhythms in the absorption, distribution, metabolism and excretion of drugs. Pharmacol Ther 52:95–107
Lemmer B, Scheidel B, Behne S (1991) Chronopharmacokinetics and chronopharmacodynamics of cardiovascular active drugs. Propranolol, organic nitrates, nifedipine. Ann N Y Acad Sci 618:166–181
Ohdo S, Koyanagi S, Suyama H et al (2001) Changing the dosing schedule minimizes the disruptive effects of interferon on clock function. Nat Med 7:356–360
Hrushesky WJ (1985) Circadian timing of cancer chemotherapy. Science 228:73–75
Hasan S, Santhi N, Lazar AS et al (2012) Assessment of circadian rhythms in humans: comparison of real-time fibroblast reporter imaging with plasma melatonin. FASEB J 26:2414–2423
Smith MR, Eastman CI (2008) Night shift performance is improved by a compromise circadian phase position: study 3. Circadian phase after 7 night shifts with an intervening weekend off. Sleep 31:1639–1645
Wright KP, Gronfier C, Duffy JF et al (2005) Intrinsic period and light intensity determine the phase relationship between melatonin and sleep in humans. J Biol Rhythms 20:168–177
Horowitz TS, Cade BE, Wolfe JM et al (2001) Efficacy of bright light and sleep/darkness scheduling in alleviating circadian maladaptation to night work. Am J Physiol Endocrinol Metab 281:E384–E391
Kasukawa T, Sugimoto M, Hida A et al (2012) Human blood metabolite timetable indicates internal body time. Proc Natl Acad Sci U S A 109:15036–15041
Hirayama A, Kami K, Sugimoto M et al (2009) Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res 69:4918
Ishii N, Nakahigashi K, Baba T et al (2007) Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 316:593–597
Timm S, Florian A, Wittmiß M et al (2013) Serine acts as metabolic signal for the transcriptional control of photorespiration-related genes in Arabidopsis thaliana. Plant Physiol 162:379–389
Sugimoto M, Wong DT, Hirayama A et al (2010) Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics 6:78–95
Tsang S, Sun Z, Luke B et al (2005) A comprehensive SNP-based genetic analysis of inbred mouse strains. Mamm Genome 16:476–480
Moriya T, Yoshinobu Y, Ikeda M et al (1998) Potentiating action of MKC-242, a selective 5-HT1A receptor agonist, on the photic entrainment of the circadian activity rhythm in hamsters. Br J Pharmacol 125:1281–1287
Soga T, Ohashi Y, Ueno Y et al (2003) Quantitative metabolome analysis using capillary electrophoresis mass spectrometry research articles. J Proteome Res 2:488–494
Soga T, Baran R, Suematsu M et al (2006) Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J Biol Chem 281:16768–16776
Soga T, Heiger DN (2000) Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem 72:1236–1241
Baran R, Kochi H, Saito N et al (2006) MathDAMP: a package for differential analysis of metabolite profiles. BMC Bioinform 7:530
Saito K, Maekawa K, Pappan KL et al (2013) Differences in metabolite profiles between blood matrices, ages, and sexes among Caucasian individuals and their inter-individual variations. Metabolomics 10:402–413
Ishikawa M, Maekawa K, Saito K et al (2014) Plasma and serum lipidomics of healthy white adults shows characteristic profiles by subjects’ gender and age. PLoS One 9:e91806
Froy O, Miskin R (2007) The interrelations among feeding, circadian rhythms and ageing. Prog Neurobiol 82:142–150
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Iuchi, H., Yamada, R.G., Ueda, H.R. (2016). Metabolites as Clock Hands: Estimation of Internal Body Time Using Blood Metabolomics. In: Karpova, N. (eds) Epigenetic Methods in Neuroscience Research. Neuromethods, vol 105. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2754-8_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2754-8_15
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2753-1
Online ISBN: 978-1-4939-2754-8
eBook Packages: Springer Protocols