
Abstract
The concept of Quality by Design (QbD) has been applied to the development of small-molecule pharmaceuticals and is increasingly being applied to the development of biopharmaceuticals. The successful implementation of a QbD approach requires a good understanding of its key elements. These include the quality target product profile (QTPP), critical quality attributes (CQAs), critical material attributes (CMAs), risk assessments, design space, critical process parameters (CPPs), a comprehensive control strategy, and lifecycle management. In this chapter, we have provided the basic definitions and considerations necessary to deliver these key elements for biopharmaceutical development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Quality target product profile
- Critical quality attributes
- Critical material attributes
- Critical process parameters
- QbD elements
- Risk assessment
- Control system
3.1 Introduction
The concept of Quality by Design (QbD) has provided both opportunities and challenges for the biopharmaceutical industry. A successful QbD approach can lead to a better understanding of products and more robust manufacturing processes, and offers the potential for timely and flexible regulatory approval (Stevenson and Cochrane 2011a, b). QbD uses a science and risk-based approach that emphasizes the importance of developing scientific knowledge and thorough understanding of both the product and the process. This concept has been successfully applied in many industries and is only recently being introduced in the pharmaceutical industry, first to small molecules and now to biologics (Elliott et al. 2013). The US Food and Drug Administration (FDA) and other health authorities are also engaged in applying QbD to pharmaceutical development and manufacturing. In the past several years, significant progress has been made to establish and implement the concepts of QbD to pharmaceutical development and manufacturing. A number of initiatives within the FDA have described their expectations and objectives and are encouraging pharmaceutical industry to utilize QbD concepts in their product development and manufacturing. Several International Conferences on Harmonization (ICH) guidance documents (ICH Q8, ICH Q9, ICH Q10, and ICH Q11), and the FDA Process Analytical Technology guidance have been published and laid the foundation for pharmaceutical companies to implement QbD in their operations and product development (FDA 2004; ICH 2009, 2005, 2008, 2012). In addition, a recently published mock case study on an antibody API and Drug Product by the CMC Biotech Working group, with FDA and other health authority feedback, has provided further useful and practical information about how to apply the key elements of QbD to process development (CMC Working Group 2009).
QbD requires a thorough understanding of the product and its manufacturing process. Additional time and resources are required to establish a company’s approach and framework to applying QbD for its first few products. But once established, such a program should streamline development by applying consistent approaches and tools, and leveraging data across the same class of products more easily. A successful QbD approach should provide a higher level of assurance of product quality and improved efficiency for industry and regulatory approval.
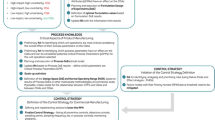
The key elements of the QbD approach include the quality target product profile (QTPP) , critical quality attributes (CQAs) , risk assessments, design space, critical material attributes (CMAs) , critical process parameters (CPPs), control strategy and product life cycle management which include continuous improvement. Figure 3.1 illustrates the QbD roadmap and how all of the elements of QbD are linked to each other.

Quality by design (QbD) roadmap
Despite much progress having been made in past several years, interpretation of QbD concepts and the scope of its key elements are still an ongoing process and will require further clarification and alignment within industry, particularly for more complex biopharmaceutical products and with regulators. In this chapter, we focus on the basic definition and scope of the key elements of QbD for biopharmaceutical drug development.
3.2 Quality Target Product Profile
3.2.1 Definition
The QbD approach begins with the establishment of quality target product profile (QTPP) . The QTPP is a prospective summary of the quality characteristics of a Drug Product that ideally will be achieved to ensure the desired quality, taking into account safety and efficacy of the product (ICH 2009). The establishment of a good understanding of the target product profile (TPP) is an important step in determining QTPP. The TPP provides a statement of the overall intent of the drug development program and gives information about the drug at a particular time in its development lifecycle. Usually, it includes the specific studies (both planned and completed) that will supply the evidence for each conclusion that becomes part of the label (Lionberger et al. 2008). The QTPP is derived from an understanding of the mode of action of the product, patient profile, clinical indication, desired safety profile, and where appropriate, includes quality characteristics related to:
-
Route of administration and intended use (in a clinical setting or at home)
-
Dosage form, delivery system
-
Dosage strength
-
Container closure system
-
Therapeutic moiety release or delivery and attributes affecting pharmacokinetic characteristics (e.g., dissolution, aerodynamic performance) appropriate to the Drug Product dosage form being developed
-
Drug product quality criteria (e.g., sterility, purity, stability, and drug release) appropriate for the intended marketed product
3.2.2 Interpretation/Consideration for Biopharmaceuticals
The QTPP defines a target for product quality requirements. It forms the basis for the development of other key QbD elements, such as CQAs and control strategy , and drives formulation and process development decisions. The QTPP describes the product design criteria that will ensure the quality, safety, and efficacy of a specific product for patients. A series of critical considerations should be made for the QTPP of a biopharmaceutical product. This includes important information from the TPP or equivalent source that describes the use, safety and efficacy of the product. In addition, it also includes the understanding of scientific knowledge, health authority requirements and in case of the Drug Product- intrinsic API properties. The flow of inputs to and output of QTPP is shown in Fig. 3.2:

Inputs to and outputs from the quality target product profile (QTPP)
Establishment of the QTPP is a critical step for a QbD approach. The QTPP includes not only the relevant information from the product specification but also patient relevant product performance. For example, if the viscosity of a high concentration product is critical to the reconstitution or delivery of the drug product, then the QTPP should include viscosity information. The QTPP is a living document that can change as more information become available. When changes are made to the TPP or other key elements, a reevaluation must be performed to assess impact to the QTPP. The QTPP may be updated to reflect new knowledge about the product and changes in the clinical development program.
An example of a QTPP for a biopharmaceutical product is provided below. This example is taken from the A–MAb published mock case study on an antibody API and Drug Product (CMC Working Group 2009) . Detailed information in QTPP will vary from product to product based on the differences between indications, intended use, and the characteristics of the product itself. For example, in the A–MAb mock case study, Drug Product quality criteria, such as aggregate, fucose content, galactosylation, and host cell protein were listed in detail (Table 3.1).
3.3 Critical Quality Attributes
3.3.1 Definition
ICH Q8 defines a CQA as a physical, chemical, biological, or microbiological property or characteristic that should be within an appropriate limit, range, or distribution to ensure the desired product quality (ICH 2009). Product quality is typically interpreted as product safety and efficacy. A CQA is a product attribute and not an analytical test and is generally associated with the Drug Substance, excipients , intermediate and Drug Product. It should be noted that the intended safety, efficacy, stability, and performance of the product are generally not considered as CQAs. Safety and efficacy clearly fall under the domain of the TPP. CQAs can be further categorized as an obligatory CQA. An obligatory CQA is an attribute required by a health authority to be either monitored or controlled as part of the product’s control strategy .
CQAs are managed throughout the product lifecycle (see Fig. 3.2). During product development, potential CQAs (pCQAs) are identified based on an iterative application of risk-based tools. The list of pCQAs and their risk scores will be further modified as product knowledge increases through the various stages of product development. When changes are made to the QTPP, an evaluation must be performed to assess the impact on the pCQAs. At the time of filing for approval, the potential CQAs become CQAs and should reflect the current knowledge and understanding of the impact on patient safety and product efficacy. The CQAs must be described, justified, and documented. As more knowledge is gained about quality attributes post-licensure, the criticality of those attributes may change (increase or decrease) and the CQAs should be updated. When there is a change in a CQA, the impact on the design space and control strategy should be assessed and updated if necessary.
3.3.2 Interpretation/Considerations for Biopharmaceuticals
The approach to identifying CQAs should be dependent on the category of the quality attribute (QA) being assessed. Quality attributes for a biopharmaceutical product may be divided into the different assessment categories (see Table 3.2). Dividing quality attributes into categories enables distinction of QAs that are product or process specific and require a risk assessment, from those that are common across products and processes and can therefore be assessed generically, or those that must be controlled based on regulatory agency requirements or expectations.
In general, the risk assessment tools developed for evaluation of quality attributes are science and risk based, and designed to allow consistent CQA identification that is independent of process capability and applicable throughout the product lifecycle. This approach enables early identification of high-risk product variants and impurities that may be need to be studied further to lower the uncertainty and modify the impact based on the new information obtained. The risk assessment tool should be applied at defined stages of product development to incorporate new information and help guide development studies to better understand product quality attributes. Consistency among products and users determining QA criticality is assured through training, subject matter expert facilitation of assessments, standardized documentation, team and expert review, and management approval of CQA identification.
3.3.2.1 Product Variants and Process-related Impurities
Product variants and certain process-related impurities should be evaluated carefully using a risk assessment approach that assesses the impact of each QA on safety and efficacy. Product variants are assessed on a product-specific basis to account for the unique modifications, mechanism of action, indication, route of administration, preclinical and clinical experience, in vitro studies, and other factors that influence risk assessment. General product and platform knowledge from similar molecules can also help this process. Process-related impurities are often common among similar products and process. Therefore, prior knowledge is often applied to assess risk for these attributes in products manufactured using similar processes. Criticality of each QA is assessed independent of actual levels present or the ability of the process to control the QA. Process capability should be considered later, during development of the control strategy and any post-approval lifecycle management plan. An exception to this is the assessment done with raw materials and leachables (see 4.3.2.3).
3.3.2.2 Risk Assessment Approach
Many biopharmaceutical companies, with input from the regulators, have adopted the use of a risk ranking and filtering (RRF) approach to assess criticality of QAs (Martin-Moe et al. 2011). The risk-ranking approach typically incorporates two factors: impact and the uncertainty of that impact. Impact is the potential affect a variant or impurity may have on patient safety and product efficacy (together these constitute “harm”). Uncertainty is related to the degree of confidence that the impact is correctly assigned for the QA of interest. Also, the impact and uncertainty rankings may have different scales to reflect the relative importance of the two factors, with impact outweighing uncertainty. Numerical values are assigned to impact and uncertainty and multiplied to generate a relative risk score, which is used in ranking. Filters , in the form of cut-offs for risk scores, are then used to identify attributes that are high risk (classified as CQAs) and low risk (classified as non-CQAs).
Application of risk assessments to identify the criticality of QAs should not take the place of the need for review by subject matter experts and technical management before final CQA classifications are endorsed. Business practices should ensure review of QA classification by technical experts and management. In the event that a QA is categorized incorrectly with a risk assessment tool, this practice would enable reclassification, with appropriate justification. Moreover, the justification for the assigned classifications will also be presented to health authority reviewers as a part of the summary of the outcomes of the risk assessment . As a result of this internal and external oversight, there is assurance that no high-risk QAs are inadvertently classified as low risk due to strict application of a risk assessment.
The criticalities of composition/strength and adventitious agents are assessed in a different approach. Regulatory requirements specify that certain attributes in the composition/strength and adventitious agent categories must always be controlled due to their potentially significant impact on safety and efficacy of products. Therefore, these attributes have been classified as obligatory CQAs and do not require using a risk-ranking tool for further evaluation of criticality. For these attributes, appropriate process and analytical controls should be implemented. Examples of such attributes are summarized in Table 3.3.
3.3.2.3 Raw Materials and Leachables
The assessment of the criticality of raw materials is a challenging task. One approach is to consider the toxicity of the raw material assuming no clearance in the manufacturing process as a worse case. This approach evaluates and expresses theoretical risk to patients related to the direct impact of the presence of these materials on the Drug Product. In practice, many of the raw materials used in the manufacturing process have been studied extensively in animal or clinical studies. For example, extensive data are available for culture additives such as insulin (Smith et al. 1980), and process chemicals such as phosphate and acetate (Haut et al. 1980).
Raw materials are evaluated for criticality by assessing the potential toxicity of the compound itself. This approach evaluates the theoretical risk to patients related to the direct impact of the presence of these materials on the Drug Product. Those raw materials that pose a potential toxicity risk are considered potential CQAs (pCQAs). Those pCQAs are then assessed for clearance and the potential toxicity is reassessed based on the levels determined in those studies. Not all compounds identified as pCQAs can be measured directly. In those cases, clearance was supported by removal of detectable compounds with similar physicochemical properties . For example, clearance of a subset of ionic salts may be used to demonstrate clearance of all ionic salts (e.g., magnesium, chlorides, and sodium). Raw materials that are still potentially a toxicity risk after considering that clearance through the process becomes CQAs, and a control strategy will need to be developed for them. The impurity of a raw material that affects a CQA is also known as a CMA . A CMA is defined as a physical, chemical, biological, or microbiological property or the characteristic of a raw material whose variability has an impact on CQAs. Therefore, they need to be monitored or controlled to ensure the desired product quality. CMAs may include purity of raw materials, physical properties of excipient, and chemical and/or microbiological purity of API or excipient. Acceptable ranges for CMAs must be specified to ensure that the CQAs of the final product will be within the acceptable ranges.
Identification of specific leachables as CQAs is much dependent on whether a specific compound or its impact can be detected. Leachables are compounds that leach into the drug or biological product from elastomeric or plastic components or coatings of the primary container and closure system. If development and stability data show evidence that leachables are consistently below levels that are demonstrated to be acceptable and safe, no leachables will be classified as CQAs. Typically, no leachables are classified as CQAs. However, if leachables are shown to have significant impacts on CQAs of the final product (e.g., glue, tungsten, or silicone oil from a prefilled syringe) by a stability study, they can be classified as CQAs.
Examples of identifying critical quality attributes have been presented recently (CMC Working Group 2009) . These examples have demonstrated the importance of using prior product knowledge, laboratory data, nonclinical data, and clinical data for the criticality assessment.
3.4 Critical Process Parameters (CPPs) and Design Space
3.4.1 Definition
Once pCQAs are identified, the next important step in the QbD process is to define CPPs and design space. This work is usually done in parallel with the identification and characterization of CQAs. A CPP is defined as a process parameter whose variability has an impact on a critical quality attribute and therefore should be monitored and controlled to ensure the process produces the desired quality (ICH 2006a).
The concept of design space has been defined in ICH Q8 (R2) as “The multidimensional combination and interaction of input variables (e.g., material attributes and process parameters) that have been demonstrated to provide assurance of quality within an acceptable range.” A design space can be applied to a single unit operation, multiple unit operations, or the entire process. The establishment of a design space for a manufacturing process is based on a good understanding of how the process impacts the CQAs. The limits of the design space should correspond to the acceptable ranges for the CQAs. In general, a change within the established design space for a manufacturing process is not considered as a significant change, while moving beyond the established design space is considered to be a significant change and would require pre-approval by the health authorities. In addition, more extensive preclinical or clinical data may also be required to support such a change.
3.4.2 Interpretation/Considerations for Biopharmaceuticals
The concepts of CPPs and design space are used in the manufacturing process development studies to define the acceptable ranges for the manufacturing process parameters and formulation conditions for biopharmaceutical industry (Jameel and Khan 2009; Martin-Moe et al. 2011). Key steps for establishing CPPs and design space include performing risk assessments to identify which process parameters should be studied; designing those studies using design of experiments (DOE) and using qualified scale-down models; and executing the studies and analyzing the results to determine the importance of the process parameters, as well as define the design space.
Prior knowledge from early manufacturing development experience with the molecule in question and knowledge from similar molecules is used to assess initial criticality. The initial criticality of the process parameters is usually assessed based on likelihood that a parameter can impact a pCQA on its own or in combination with other parameters. For a product with limited prior knowledge, small-scale pilot studies are often conducted before doing more thorough DOE studies.
Process characterization and validation studies are the final steps for establishing CPPs and design space. Process characterization is a systematic investigation to understand the relationship between key operating parameters and critical quality attributes. Objectives of process characterization include identification of key operational and performance parameters, establishment of acceptable range for key parameters, and demonstrating process robustness (Li et al. 2006). Process validation is establishing documented evidence that provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-defined specifications and quality attributes. Process validation often includes results of full-scale runs under target manufacturing conditions, and the collection of data on an appropriate number of production batches. Cumulative impact of multiple unit operations on pCQAs may be assessed by doing multi-step DOE studies or by assessing the impact on a given pCQA by running the various process steps that impact that pCQA at a worse case (process step linkage study). DOE is used during process characterization studies to establish CPPs and design space for the manufacturing process. DOE is a systematic and rigorous approach to determine the multidimensional relationship among input variables and their influences on outputs of a process. The input variable can be a process parameter (e.g., process time and/or temperature) and formulation attributes (e.g., concentration and excipients), while outputs are the product quality and impurity levels which usually are defined by CQAs.
For a DOE study, it should be emphasized that a combination of acceptable ranges based on univariate experiments can provide supporting data but may not be sufficient to establish a design space. The acceptable ranges may need to be based on multivariate experiments that take into account the main effects, as well as interactions of the process parameters and formulation attributes. For many of biopharmaceutical products , site- and scale-independent characterization studies that support identification of CPPs and define the design space are conducted using scale-down models of the manufacturing-scale unit operations. Site-specific studies include characterization and validation studies conducted at manufacturing scale in the intended commercial facility that demonstrate manufacturing process consistency with regard to meeting pre-specified process parameter ranges, process performance indicators, and CQAs. When considering a scale-down model, additional experimental work is typically required to demonstrate that the data generated using the small-scale model is adequately representative of the commercial manufacturing scale.
Recently, several case study examples for both Drug Substance and Drug Product manufacturing process have been published and provided useful information for define CPP and process design space during QbD process. Harms et al. have presented a case study, involving P. pastoris fermentation process to demonstrate a stepwise approach for defining process design space (Harms et al. 2008). Similar work has also been conducted to define process design space for Drug Product manufacturing process. Martin Moe et al. have recently published a paper that describes the use of QbD concept for Drug Product process development for an antibody.
3.5 Control Strategy and Control System
3.5.1 Definition
The control strategy is a key element of the QbD process. The control strategy refers to a set of planned controls, derived from current product and process understanding that ensures process performance and product quality . One of the important parts of control strategy is to establish a control system . A control system is a set of defined controls and their established acceptance criteria (or limit) based on product understanding that assures product quality. The control strategy comprises several elements including
-
Raw material controls
-
Process control via procedural and process parameter control
-
In-process, lot release, and stability testing
-
Testing to demonstrate comparability
-
Testing done as part of process monitoring.
Raw material controls are controls relating to raw materials, excipients, buffer components, etc. used in the formulation and manufacturing processes, including supplier quality management, raw material qualification, and raw material specifications. Procedural controls are a comprehensive set of facility, equipment, and quality system controls that result in robust and reproducible operations and product quality. Process parameter controls are linked to CPPs that must be controlled within the limits of the design space to ensure product quality. In-process testing is conducted using analytical test methods or functionality test to ensure that selected manufacturing operations are performing satisfactorily to achieve the intended product quality. Lot release testing is related to the testing at final lot release on a set of quality attributes to confirm quality of the Drug Substance or Drug Product. Some of the attributes will also be tested as part of the stability testing. Characterization and comparability testing are often used to test certain attributes beyond lot release testing for the purpose of intermittent process monitoring or demonstration of comparability when a change is being implemented (e.g., licensing a new production facility or modified manufacturing process). Process monitoring is the testing or evaluation of selected attributes or parameters to trend product quality or process performance within the design space and/or enhance confidence in an attribute’s normal distribution. The frequency of monitoring is periodically reviewed and adjusted based on trends. The process monitoring program may include limits for evaluating data trends.
3.5.2 Interpretation/Considerations for Biopharmaceuticals
The focus of the control strategy for a biopharmaceutical is typically the testing strategy for each attribute. It should be determined using a risk-based assessment related to the understanding of the potential impact of the quality attribute on the safety and efficacy of the product and the ability to control the level of the attribute through the manufacturing process and during storage (see Fig. 3.3). The testing strategy for each attribute is typically developed using a risk assessment tool and is also often confirmed using a separate risk assessment to determine the robustness of the resulting testing strategy.

Interrelationship between control strategy and design space
One approach for determining the testing strategy for each identified attribute is to use a risk assessment tool that incorporates that quality attribute criticality and the risk that an attribute will exceed the acceptable range for the CQA when the process is operated within its design space or during with Drug Substance and Drug Product in the recommended conditions. The assessment would be performed for each quality attribute during Drug Substance manufacturing, Drug Product manufacturing, Drug Substance stability, and Drug Product stability (see Fig. 3.4).

Establishing the control strategy
From this evaluation, one of three possible outcomes is identified for each quality attribute
-
1.
Control system testing is required (in-process, lot release, and/or stability testing)
-
2.
Testing is required as part of process monitoring or to support comparability
-
3.
No testing is required.
Once a testing strategy has been defined for each attribute, an overall robustness assessment should be performed using a risk assessment to determine the risk to the overall program that a more critical quality attribute is not controlled adequately by the proposed control strategy . In this evaluation, the type of measurement (i.e., direct versus indirect measurement), as well as its sensitivity and robustness, are considered in the overall risk assessment for each attribute.
In some cases, starting with a “minimum” control system and then adding additional tests based on the outcome of the risk assessments may be required by the health authorities as those tests are considered useful in monitoring product consistency and for further mitigating risk to patients due to unanticipated sources of variation.
3.5.2.1 Control System Testing
Control system testing includes in-process testing (e.g., bioburden, endotoxin), product release testing (e.g., product attributes, adventitious agents, impurities) and stability testing (e.g., stability indicating product attributes) .
3.5.2.2 Process Monitoring
Process monitoring programs should be designed to provide ongoing assurance and verification that product quality is appropriately controlled during routine commercial manufacturing. The process-monitoring program is designed to meet the following criteria:
-
Provide assurance that the process is operating in a validated state
-
Provide knowledge to enhance process understanding
-
Identify adverse trends and opportunities for process improvements
Continuous process monitoring is key element in a lifecycle approach for process validation. A process monitoring system collects data on CPPs , key performance indicators (KPIs), and CQAs. The attributes monitored have been selected based on knowledge gained during development and execution of the process validation lots.
3.5.2.3 Comparability Assessments
Comparability assessments of both product and, if appropriate, process, are performed to ensure that there is no adverse impact on the quality, safety, or efficacy of the product as a result of a change made to the manufacturing process or licensing a new manufacturing site. The comparability assessment considers product quality (physicochemical characterization of the product), stability (degradation), and process performance (key performance indicators and removal of process-related impurities) .
3.6 Lifecycle and Knowledge Management
3.6.1 Definitions
A post-approval lifecycle management (PALM) plan is a formal document that explains how a product is managed within the QbD framework post regulatory licensure. Health authorities expect that a product developed using QbD has a formalized lifecycle management plan. The health authorities also expect that there is a formal knowledge management program that archives and updates documents associated with product and process knowledge, as well as the documents summarizing the outputs of the QbD strategy.
3.6.2 Interpretation/Considerations for Biopharmaceuticals
The elements of the PALM for a biologic product and the interrelationships between them are illustrated in Fig. 3.5. The LMP for a biologic should include a description of how process and product attributes will be monitored to ensure that both remain in a state of control post regulatory licensure. The frequency that a product attribute is measured is attribute-specific and dependent on the risk associated with that attribute. Some attributes may be monitored in every production batch (more critical) or intermittently on some subset of batches (e.g., every fifth batch). Additionally, product and process monitoring results, including adverse trends, serve as the scientific basis for continuous verification and improvement of the initial control system and manufacturing process. The lifecycle management plan also explains how changes to the critical process parameter (CPP) operating targets are managed within and outside of design space. For a change to a CPP target within the design space, the level of pre- and post-implementation testing is determined by the level of risk and the potential for the change to impact a CQA. The risk is assessed based on considerations such as CPP criticality , the product quality attributes affected, and the classification of those product quality attributes. There is pre-implementation and post-implementation/verification testing. The post-change assessment testing is meant to verify that the change had the desired result and that the design space continues to be valid for the manufacturing process. The PALM also explains how changes to a non-CPP (operating target and/or ranges) are managed as well. Since non-CPPs do not impact CQAs, the assessment of these changes typically focus on key performance indicators. If a non-CPP is associated with a step that has an influence on product quality, changes to the acceptable range for non-CPPs for these steps usually require additional justification, based on scientific literature, historical data, or new studies performed similar to the ones that established the acceptability of the original range.

Components of the lifecycle management plan (PALM)
The strategy for updating CQAs, the overall control strategy, and CPPs as further process or product knowledge gained post approval is also described in the LMP.
It is also an expectation of the health authorities that the elements of the PALM are integrated in the company’s pharmaceutical quality system (PQS) and that changes to CQAs, CPPs, etc. are documented and justified appropriately. In the majority of the cases, the same risk assessment tools are used to support those changes.
The lifecycle management plan may also be included in the product registration documentation and, if so, then the PALM becomes a regulatory agreement between the health authority and the company. In that circumstance, the company may be able to get agreement on some level of regulatory flexibility following the plan. Any change that does not meet the predefined requirements specified in the PALM would be reported to the health authorities following the standard regulatory reporting approach.
The PALM is a key facilitator of knowledge management as it requires the outputs of the QbD strategy to be re-evaluated as new process and product knowledge is gained and requires that information and any changes to CQAs, CPPs, design space, and control strategy to be documented and justified.
3.7 Summary
QbD has provided opportunities for biopharmaceutical companies to develop better understanding of their products and their associated manufacturing processes. This approach can provide a higher level of assurance for product quality, and offer the potential of improved efficiency for industry and regulatory approval. Over the past several years, significant progress has been made to establish the industry approach and framework for applying QbD concepts to biopharmaceutical product development . The key tools and strategies are developed and implemented to assess the key elements of QbD, including CQAs, CPPs, and the control strategy. Some of these key QbD elements have been successfully included in the recent regulatory filling for complex biopharmaceutical products and likely will be a requirement for biopharmaceuticals in future. Close attention will be paid both by industry and regulators on how this novel approach will help to realize these potential benefits.
3.8 Acknowledgments
The authors would like to thank Sherry Martin-Moe, Paul Motchnik, Reed Harris, Mary Cromwell, Brian Kelley, Niklas Engler, Lynne Krummen, and the Roche Large Molecule Quality by design team members for their valuable input, discussion, and contributions that helped to implement the basic concepts for QbD at Genentech/Roche.
References
CMC Working Group (2009) A-Mab case study. From http://www.casss.org/displaycommon.cfm?an=1&subarticlenbr=286
Elliott P, Billingham S et al (2013) Quality by design for biopharmaceuticals: a historical review and guide for implementation. Pharm Bioprocess 1(1):105–122
FDA (2004) Guidance for industry. PAT-A framework for innovative pharmaceutical development, manufacturing, and quality assurance.
Harms J, Wang X et al (2008) Defining process design space for biotech products: case study of Pichia pastoris fermentation. Biotechnol Prog 24(3):655–662
Haut LL, Alfrey AC et al (1980) Renal toxicity of phosphate in rats. Kidney Int 17(6):722–731
ICH (2009) Pharmaceutical Development, accessed at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf on Jan. 29, 2015
ICH (2005) Quality Risk Management, accessed at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf on Jan. 29, 2015
ICH (2008) Pharmaceutical Quality System, accessed at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf on Jan. 29, 2015
ICH (2012) Development and manufacturing of drug substance, Q11, accessed at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Q11_Step_4.pdf on Jan. 29, 2015
Jameel F, Khan MA (2009) Quality-by-Design as applied to the development and manufacturing of a lyophilized protein product. Am Pharm Rev
Li F, Hashimura Y et al (2006) A systematic approach for scale-down model development and characterization of commercial cell culture processes. Biotechnol Prog 22(3):696–703
Lionberger RA, Lee SL et al (2008) Quality by design: concepts for ANDAs. AAPS J 10(2):268–276
Martin-Moe S, Lim FJ et al (2011) A new roadmap for biopharmaceutical drug product development: Integrating development, validation, and quality by design. J Pharm Sci 100(8):3031–3043
Smith CR, Lipsky JJ et al (1980) Double-blind comparison of the nephrotoxicity and auditory toxicity of gentamicin and tobramycin. N Engl J Med 302(20):1106–1109
Stevenson D, Cochrane T (2011a) Implementation of QbD part 1- Setting product specifications. Regul Rapporteur 8(2):3
Stevenson D, Cochrane T (2011b) Implementation of QbD part 2- organizational implications. Regul Rapporteur 8(2):3–16
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Taticek, R., Liu, J. (2015). Definitions and Scope of Key Elements of QbD. In: Jameel, F., Hershenson, S., Khan, M., Martin-Moe, S. (eds) Quality by Design for Biopharmaceutical Drug Product Development. AAPS Advances in the Pharmaceutical Sciences Series, vol 18. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2316-8_3
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2316-8_3
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2315-1
Online ISBN: 978-1-4939-2316-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)