
Abstract
The ability to generate DNA double-strand breaks (DSBs) at specified locations in a plant’s genome, thereby stimulating the cell’s DNA repair processes, represents a promising means of facilitating genetic modification for both basic studies of gene function as well as applied crop improvement. Zinc finger nucleases (ZFNs) are engineered restriction enzymes consisting of a nonspecific cleavage domain and sequence-specific DNA-binding domains designed to create site-specific DSBs. Since DSB repair in plants appears to occur primarily via error-prone nonhomologous end joining (NHEJ) processes, ZFNs designed to cleave endogenous genes is a path toward targeted mutagenesis. Similarly, ZFN-mediated induction of concurrent DSBs can give rise to targeted deletions of genomic segments between cleavage sites. In addition, homology-directed repair of targeted DSBs allows for site-specific transgene integration into transgenic and endogenous gene loci as well as the creation of specific sequence modifications. The combination of sequence-specific DNA cleavage by designed ZFNs and homology-directed DSB repair at investigator-specified break sites makes precision genome modification a reality. This capability, in combination with rapid advances in genome sequencing and bioinformatics, bodes well for the future of plant functional genomics and crop improvement.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
DNA Double-Strand Breaks (DSBs)
The creation and repair of DNA DSBs is of central importance to the recombination between DNA sequences (Xu and Price 2011). Pioneering studies in yeast have highlighted the importance of DSBs in both meiotic (Keeney 2001) and mitotic (Lisby and Rothstein 2007) DNA recombination. The induction of genomic DSBs and their repair via various homologous and nonhomologous processes is well established (Haber 2007). Many of the genes involved in DSB repair have been elucidated and found to be conserved across a broad range of life-forms (Li et al. 2011), although the contributions of each to the DNA repair process have dramatically changed during evolution (Sonoda et al. 2006). These studies have highlighted the dual role of DSB formation and resolution as a means of both promoting genetic diversity by facilitating DNA sequence exchange and conserving genomic integrity via DNA repair.
DSBs can be repaired using homologous sequences, i.e., from a sister chromatid or other related template DNA, via pathways involving a collection of proteins which facilitate strand resection, invasion, annealing, and synthesis reactions resulting in an intact DNA sequence (Rajesh et al. 2011). Alternative pathways of DSB repair involve nonhomologous end joining (NHEJ) of DNA sequences whereby cleaved ends are religated without regard for homology, often resulting in deletions or insertions at the cleavage site (Wu et al. 2012). These complexes of apparently competing processes effectively repair DSBs with varying degrees of fidelity (Shibata et al. 2011).
In higher plants, it appears as if DSBs are most typically repaired via NHEJ where sequence-independent repair often results in deletions, insertions, and/or rearrangements at the break site (Gorbunova and Levy 1999; Puchta 2005). Although not completely understood, it appears as if several NHEJ pathways in plants operate to repair DSBs (Charbonnel et al. 2011). If homologous sequences are in close proximity to the DSB, high-fidelity, homology-directed repair has been observed to occur in plant cells (Roth et al. 2012; Siebert and Puchta 2002).
The ability to generate DSBs, thereby stimulating the cell’s DNA repair processes, represents a means of facilitating genetic modification (Fig. 12.1). The error-prone nature of NHEJ repair makes induction of DSBs a method for inducing mutations (Carroll 2011). Intervening sequence elimination following the formation and repair of concurrent DSBs is a means of generating various sorts of gene deletions (Lee et al. 2010). Homology-directed repair of DSBs enables transgene integration (Lombardo et al. 2011) and genome editing (McMahon et al. 2012).

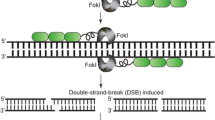
ZFNs facilitate targeted genome modifications. ZFNs can be designed to specific genomic sequences to enable targeted gene addition, gene editing, and targeted mutagenesis. Delivery of ZFNs into cells results in targeted double-strand DNA breaks that are repaired by cellular repair mechanisms such as NHEJ and HDR. Repair of the double-strand DNA break by NHEJ leads to introduction of indels (insertions/deletions) at the cut site and targeted mutagenesis in the genome. Repair in the presence of an exogenous DNA molecule carrying a gene of interest (donor) with homology to the break site leads to targeted gene insertion. Presence of specific mutations in the donor result in edits at desired locations in the genomic sequence. DSB double-strand break, NHEJ nonhomologous end joining, HDR homology-directed repair, indels insertions/deletions. Zinc finger DNA-binding domains are represented by green rectangles and the blue circle represents the Fok1 nuclease domain
Designed Zinc Finger Nucleases (ZFNs)
To take advantage of DSB repair for controlled genome modification, a method is required for targeted DNA cleavage (Puchta and Fauser 2013). Previously, targeted DSBs could only be made in plant genomes following pre-integration of restriction enzyme cleavage sites and expression of genes encoding the corresponding restriction enzyme (Salomon and Puchta 1998). Recently, ZFNs have been described that allow for DSB formation at endogenous plant loci (de Pater et al. 2013; Shukla et al. 2009; Townsend et al. 2009; Zhang et al. 2010). ZFNs are engineered restriction enzymes consisting of a nonspecific cleavage domain and sequence-specific DNA-binding domains designed to create site-specific DSBs (Porteus and Carroll 2005). In this way, DSBs can be targeted to investigator-specified sites by engineering and delivering novel sequence-specific restriction enzymes capable of binding and cleaving endogenous genomic DNA (Tzfira et al. 2012).
Zinc finger protein domains consist of ~ 30 amino acids which, upon chelating a zinc atom, fold into ββαstructures capable of binding specific DNA triplets (Pabo et al. 2001). Key amino acid residues in the α helix dictate sequence-specific binding, while the remaining amino acids maintain a consensus backbone structure with a modular architecture (Durai et al. 2005). Linking such modular structures together allows for the creation of DNA-binding domains capable of recognizing predetermined stretches of sequence (Fig. 12.2a). The development of designed ZFNs that cleave DNA at predetermined sites depends on the reliable creation of zinc finger protein domains that can specifically recognize the chosen target site within a genome. The design, assembly, and validation of such DNA-binding proteins based on modular zinc fingers are becoming more routine (Hurt et al. 2003; Isalan et al. 2001; Maeder et al. 2008; Mandell and Barbas 2006). ZFN design services are commercially available (e.g., ComposZr® from Sigma-Aldrich).

Double-strand DNA binding by site-specific nucleases. Schematic representation of a ZFN (a) ZFN and a TALEN (b) bound to DNA. ZFNs recognize and bind DNA through the zinc finger domains (green, a) and each finger binds a nucleotide triplet. DNA binding by TALENs is mediated by TALE effector (red, b) domains with single nucleotide specificity. The nonspecific Fok-1 nuclease domain is shown in blue
DNA cleavage is facilitated by a sequence-independent nuclease domain from the bacterial type IIS restriction endonuclease FokI(Kim et al. 1996). To cut DNA and generate a DSB, the FokI Inuclease domain needs to dimerize at the cleavage site (Bitinaite et al. 1998). A ZFN is created by linking the FokI cleavage domain to the C-terminus of a tethered series of zinc finger protein domains designed to bind a specific DNA sequence. Upon binding of two adjacent ZFN pairs to sequences flanking the intended cleavage site in a precise orientation and spacing relative to each other, the FokI domains dimerize thereby facilitating DSB formation (Fig. 12.2a). ZFNs have been used to create targeted DSBs and enable genome modification in a broad spectrum of genomes, including human (Lombardo et al. 2007; Moehle et al. 2007; Perez et al. 2008; Porteus and Baltimore 2003; Provasi et al. 2012; Sebastiano et al. 2011; Urnov et al. 2005; Wilen et al. 2011), hamster (Santiago et al. 2008), mouse (Osiak et al. 2011), pig (Hauschild et al. 2011), frog (Young et al. 2011), zebra fish (Doyon et al. 2008), insect (Beumer et al. 2006; Bibikova et al. 2002), roundworm (Morton et al. 2006), and Plasmodium (Straimer et al. 2012). The present chapter reviews the use of designed ZFNs for inducing targeted DSBs and facilitating genome modification in plants (Table 12.1).
Targeted Mutagenesis
The ability to modify specific gene sequences is an indispensable tool for systematic analysis of plant gene function (Perry et al. 2003) . Since DSB repair in plants appears to be primarily via NHEJ (Gorbunova and Levy 1999; Puchta 2005) and, since NHEJ in plants tends to be rather error-prone (Britt 1999), targeted DSB formation is a path toward targeted mutagenesis (Lyznik et al. 2012). Designed ZFNs appear to be ideally suited for such an application.
The first proof-of-concept study demonstrating ZFN-mediated targeted mutagenesis in plants involved the mutation of an introduced construct comprising a ZFN cleavage site and a corresponding ZFN under the control of a heat shock promoter (Lloyd et al. 2005). The experimental system involved an EcoR1 restriction sequence within the ZFN cleavage site which could be lost upon mutation, due to NHEJ-induced deletion or insertion, thereby allowing mutations to be identified. TOPO-cloning of polymerase chain reaction (PCR) products amplified from genomic DNA from heat-treated T1 Arabidopsis seedlings with single copy integration of the construct revealed mutation frequencies across multiple independent transgenic events, measured by lost EcoR1 restriction sites, to be in the range of 1.7–19.6 % based on a random sampling of clones. Sequencing of the EcoR1-minus clones illustrated the types of mutations resulting from DSB repair. Most of the mutations (78 %) were simple deletions of 1–52 bp . Simple insertions (1–4 bp) and combinations of insertions and deletions were also observed at lower frequency. These frequencies should be considered to represent an underestimate of the actual mutation frequency. Based on the design of the targeting construct, larger deletions (> 62 bp) which removed the PCR primer binding sites would not have been observed in this study. In fact, NHEJ-mediated deletions of 0.2–2.0 kb have been commonly observed and 50 % of all such deletions were found to be > 100 bp (Gorbunova and Levy 1999). Nonetheless, this study clearly demonstrated that ZFN-mediated DSB formation can lead to targeted mutations.
In a similar study, Arabidopsis plants, stably transformed with a target construct comprising an EcoR1-containing ZFN cleavage site, were retransformed with corresponding ZFN-expressing constructs driven by various promoters (de Pater et al. 2009). Most EcoR1-resistant DNA fragments amplified from transgenic plants contained deletions ranging from 1 to 80 bp. Small insertions (1–14 bp) and larger deletions (up to 200 bp) were also observed. Mutation frequency was estimated to be about 2 % based on a random sampling of cloned PCR fragments. Reverse transcription polymerase chain reaction (RT-PCR) was used to estimate relative ZFN expression. Driving the ZFN gene with a stronger promoter appeared to be more effective at generating mutations .
Additional examples of the ability of ZFN expression to mediate targeted genome modification via NHEJ DSB repair involved the mutation of a disabled reporter gene (Cai et al. 2009; Tovkach et al. 2009). In one study (Tovkach et al. 2009), a GUS gene, engineered to carry a TGA stop codon within a ZFN cleavage site—and thereby rendered nonfunctional, was stably transformed into tobacco. As expected, transgenic tissue did not express the GUS reporter gene. Cocultivation of transgenic tissue with an Agrobacterium strain harboring a construct containing a corresponding ZFN expression cassette resulted in small sectors of positive GUS staining. Similarly, Arabidopsis plants stably transformed with the nonfunctional GUS gene and a ZFN under the control of a heat shock promoter expressed GUS following high-temperature induction. Sequence analysis of the target site following PCR amplification identified several single nucleotide deletions and substitutions resulting in an open reading frame expected to encode an active GUS gene. This mutation was also facilitated using viral delivery of a ZFN (Vainstein et al. 2011). In another study (Cai et al. 2009), a reporter construct carrying a GFP gene disabled by the insertion of a 2.8-kb stretch of heterologous DNA containing a ZFN-binding site was stably integrated into tobacco cell cultures. A tandem repeat of 540 bp in the two GFP gene fragments served as a substrate for intrachromosomal repair. Upon retransformation with a ZFN gene, fluorescent foci were visible and PCR analysis confirmed homology-directed repair of the targeted DSB .
Mutations at endogenous gene loci have also been demonstrated following expression of designed ZFNs (Maeder et al. 2008; Shukla et al. 2009; Townsend et al. 2009). Tobacco protoplasts were transformed with a ZFN designed to cleave a specific site within the SuRA gene. Among 66 transgenic plants regenerated, three displayed single base mutations in the SuRA gene (Maeder et al. 2008). Similarly, ZFNs designed to cleave SuRA and SuRB genes displayed varying degrees of specificity relative to creating site-specific mutations (Townsend et al. 2009). A ZFN designed to cleave within the maize IPK1 gene was transiently expressed in cultured maize cells after which multiple deletions and insertions were observed following deep sequencing of PCR amplified products (Shukla et al. 2009).
Genes encoding ZFNs designed to recognize Arabidopsis ADH1 and TT4 driven by an estrogen-inducible promoter resulted in somatic mutation frequencies of 7 and 16 %, respectively (Zhang et al. 2010) . The mutations were typically 1–142 bp insertions or deletions localized at the ZFN cleavage site and were often found to be biallelic, i.e., homozygous. A ZFN gene, designed to recognize the Arabidopsis ABI4 gene sequence, driven by a heat shock promoter, upon induction, resulted in up to 3 % mutagenesis of the binding site and the appearance of expected phenotypes, i.e., abscisic acid (ABA) and glucose insensitivity, in homozygous progeny (Osakabe et al. 2010). In a similar study, independent mutations in the paralogous DCLa and DCLb soybean genes involved in RNA silencing were generated using designed ZFNs (Curtin et al. 2011). Taken together, these results suggest the general utility for basic and applied studies of making site-specific mutations by expressing ZFNs designed to create targeted DSBs and induce NHEJ repair .
Mutation breeding in plants has resulted in numerous commercially relevant varieties in a broad spectrum of crop species (Maluszynski 2001). Conventional methods of mutagenesis used to generate genetically-modified crops typically involve random perturbations in the DNA sequence, using treatment with chemicals such as ethyl methanesulfonate (Watanabe et al. 2007), physical methods such as fast neutron radiation (Li et al. 2001) or naturally occurring genetic mechanisms such as transposable elements (Mathieu et al. 2009) combined most recently with sequence-specific screening (McCallum et al. 2000). Such approaches have serious limitations, such as the lack of observable phenotypes, in highly duplicated genomes such as those found in modern domesticated crop species (Pham et al. 2010). More targeted transgenic approaches, such as RNAi-based gene silencing, have been fraught with unanticipated phenotypic consequences presumably due to lack of specificity and potential off-target effects (Duxbury and Whang 2004). The ability to modify single or multiple gene copies in duplicated genomes of crop species would represent a powerful means of generating new genetic variants. Targeted mutagenesis via sequence-specific DSB formation and repair using designed ZFNs enables such a capability .
Gene Deletion
As complete plant genomic sequences become elucidated, the need to assign functions to unknown genes becomes increasingly important. This is most effectively approached via reverse genetics and the analysis of gene disruptions, including silencing (Baulcombe 1999), insertional mutants (Feldmann 1991), and deletions (Koornneeff et al. 1982). Conventional methods of creating plant gene deletions, such as exposure to fast neutron emission, combined with molecular analysis of pooled arrays of mutant DNA, have resulted in the assembly of large deletion libraries covering most known genes in Arabidopsis and rice (Li et al. 2001). The ability to generate investigator-specified deletions by creating targeted DSBs, followed by subsequent intervening sequence removal via DNA repair , represents an increasingly powerful refinement for genome modification. In human cell cultures, predetermined genomic DNA segments up to 15 mega-bp were deleted following expression of ZFNs designed to cleave at specific loci (Lee et al. 2010). Targeted deletions of promoter or exon sequences by generating DSBs in intergenic regions or introns could result in targeted gene knockouts, including multigene disruption. By virtue of the polyploid nature of most crop species, agronomically relevant genes exist as multiple copies such that single gene disruptions may not result in discernable phenotypes (Pham et al. 2010). The ability to knockout multiple homologous genes simultaneously with carefully designed ZFNs might be particularly useful for crop improvement.
Proof of concept for ZFN-mediated gene deletion was obtained in a recent study involving the removal of a ZFN cleavage site-flanked reporter gene from a stably transformed plant by crossing it with a second plant expressing a corresponding ZFN gene (Petolino et al. 2010). A target construct, containing a GUS reporter gene flanked by ZFN cleavage sites, was used to generate transgenic tobacco target events. A second construct, containing a ZFN gene driven by a strong constitutive promoter, was used to generate separate transgenic ZFN events. Homozygous T1 target plants, which expressed the GUS reporter gene, were crossed with homozygous T1 ZFN plants, which expressed the ZFN gene. Numerous GUS-negative hybrid plants were observed (up to 35 % in one cross). Evidence for complete deletion of a 4.3-kb sequence between the ZFN cleavage sites was obtained and sequence verified in hybrid plants and progenies. Since ZFNs can be designed to cleave a wide range of DNA sequences, the results from this study constitute a general strategy for creating targeted deletions.
Site-Specific Transgene Integration
The ability to introduce exogenous DNA into a predetermined location within the plant genome would greatly enhance the precision and predictability of transgenic technology. The potential mutagenic effects of random DNA integration and the unpredictable consequences of position effect on transgene behavior could be circumvented by targeting transgenes to specific genomic locations.
Early attempts at targeted transgene integration used a combination of integrated, nonfunctional selectable marker genes and exogenous DNA homologous and complementary to the integrated target (Offringa et al. 1990; Paszkowski et al. 1988). Transgene integration into the target site was achieved under selective conditions following correction of the nonfunctional selectable marker gene at very low frequency, i.e., estimated to be in the range of 10− 4–10− 5. In similar approaches, nonfunctional ALS gene fragments, carrying mutations that specified resistance to various herbicides, were used to target the endogenous gene loci in tobacco (Lee et al. 1990) and rice (Endo et al. 2007). Using herbicide selection, transgenic events were obtained that suggested that homologous recombination between the exogenous DNA and the endogenous gene had occurred at estimated frequencies in the range of 10− 4–10− 5. “Brute force” attempts at generating transgenic events via homologous recombination without direct selection corroborated the extremely low frequency of targeted transgene integration (Miao and Lam 1995) . Some success was reported using a combination of positive and negative selection to enrich for targeted events, whereby a targeting construct containing an antibiotic resistance gene within and a cytosine deaminase gene outside sequences homologous to an endogenous locus allow for selection against random integration in the presence of fluorocytocine (Xiaohui Wang et al. 2001). Subsequently, rice Waxy and adh2 genes were successfully targeted using a similar approach whereby a diphtheria toxin gene was used as a negative selectable marker (Terada et al. 2007; Terada et al. 2002). Attempts to enhance targeted transgene integration by modifying DNA repair pathways , such as co-expressing recombinase genes (Reiss et al. 2000; Shaked et al. 2005; Shalev et al. 1999), or knocking out genes associated with NHEJ (Jia et al. 2012), have met with limited success. Clearly, homology-directed repair does occur in plants and can facilitate targeted transgene integration; however, the frequency of targeted versus random integration appears to be too low for practical use with conventional transformation technology .
The yeast mitochondrial endonuclease, I-sceI, which has an 18-bp recognition sequence, has been used to demonstrate the importance of homology-directed repair of DSBs for targeted transgene integration (Puchta et al. 1996). A target construct containing an I-sceI restriction site flanking a partially deleted antibiotic resistance gene was transformed stably into tobacco. Retransformation with a repair construct containing sequences homologous to the target construct and complementary to the deleted antibiotic resistance gene together with an I-sceI expression construct resulted in targeted transgene integration at the I-sceI cleavage site. Using different ratios of Agrobacterium strains harboring the repair versus the I-secI construct, it appeared as if the induction of DSBs by the I-sceI was rate limiting, i.e., the best targeting frequency (18.8 × 10− 3) was achieved using a 1:9 ratio of repair: I-sceI strain. Thus, the induction of DSB formation and its repair via homology-directed processes are a key to targeted transgene integration .
Using analogous strategies, targeted transgene integration into transgenic reporter loci via homology-directed repair has also been demonstrated after ZFN-mediated DSB formation in tobacco (Cai et al. 2009; Wright et al. 2005) . Following stable integration of a defective GUS/NPTII reporter gene containing a 600-bp deletion and a ZFN cleavage site, transgenic protoplasts were electroporated with DNA encoding the corresponding ZFN and donor DNA homologous to the target and capable of correcting the deletion. Homology-directed repair of the reporter gene occurred in more than 10 % of the protoplasts across multiple transgenic events, i.e., target chromosomal positions (Wright et al. 2005). In a similar study, a pre-integrated reporter construct containing a 3′ partial herbicide resistance gene fragment flanked by ZFN binding sites allowed for in vitro selection following targeted integration of a complementary 5′ sequence from an incoming donor DNA co-transformed with a ZFN-expressing construct (Cai et al. 2009). Approximately 6 kb of target sequence between two ZFN cleavage sites was excised and replaced by 1.9 kb of donor DNA sequence using 1.2 and 1.7 kb of homology directly flanking each of two induced DSBs. These studies clearly illustrate the efficacy of ZFN-mediated DSB induction and the ability to effectively target exogenous DNA using homology-directed repair. NHEJ-mediated repair of DSBs has also been used to integrate DNA sequences in a targeted manner (Weinthal et al. 2013). ZFN-mediated cassette exchange was facilitated between an incoming promoter-less hpt gene and a pre-integrated GFP reporter gene both flanked with the same ZFN cleavage sites.
The ability to design ZFNs to cleave virtually any DNA sequence and thereby create investigator-modified, site-specific DSBs has allowed for targeted transgene integration into endogenous gene loci. Using a yeast-based system for screening ZFN efficacy (Doyon et al. 2008), ZFNs were designed against native gene sequences including, tobacco endochitinase (Cai et al. 2009) and maize IPK1 (Shukla et al. 2009) . An herbicide resistance gene driven by a constitutive promoter flanked on each side by 750 bp of endochitinase, CHN50, gene sequence was co-delivered with a ZFN expression cassette via Agrobacterium (Cai et al. 2009). Although the majority of the resulting transgenic events were the result of random integration, 5–10 % of the events appeared to be targeted to the CHN50 locus. Four different ZFN pairs targeting exon 2 of the maize IPK1 gene were independently co-delivered with donor constructs containing a herbicide resistance gene cassette flanked by 815 bp of sequence homologous to IPK1 (Shukla et al. 2009). Two different donor constructs were used for targeted integration into the maize IPK1 gene locus. One carried an autonomous herbicide resistance gene with its own promoter, whereas a second comprised a nonautonomous, i.e., promoter-less, gene that relied on precise trapping of the endogenous IPK1 promoter for expression and herbicide resistance. All four ZFN pairs drove targeted gene addition into their respective target sites, albeit with different efficiencies. In addition, site-specific transgene integration was successful using either donor construct with frequencies ranging from 3.4–22.3 % and 16.7–100 % for autonomous and nonautonomous constructs, respectively. Moreover, both monoallelic and biallelic insertions into the IPK1 locus were observed. These exciting results with designed ZFNs not only extend transgenic technology to targeted transgene integration into endogenous genomic loci but also to include important crop species .
Genome Editing
The ability to make specific modifications to plant genome sequences in order to truly edit genes in a precise and predicable fashion would not only enhance basic understanding of plant biology but also ultimately result in genetically enhanced crops with new traits and improved performance. A recent study suggests that this capability might not be too far from reality (Townsend et al. 2009). Specific mutations in SuR genes in tobacco result in resistance to different imidazolinone herbicides. ZFNs were designed to cleave a specific sequences within the tobacco SuRA and SuRB genes. Electroporation of protoplasts with DNA encoding these engineered ZFNs along with donor DNA templates containing specific mutations resulted in herbicide resistance resulting from homology-directed processes. A surprising outcome was that mutation frequencies in the range of 2 % were observed with up to 1.3 kb removed from the DSB. Although this study relied on herbicide resistance for identifying edited events, the frequencies observed were high enough for screening via high-throughput DNA analysis. A ZFN designed to recognize the Arabidopsis PPO gene was co-delivered with a truncated PPO gene containing two mutations resulting in tolerance to the herbicide butafenacil using Agrobacterium floral dip transformation (de Pater et al. 2013). Targeted PPO modification was observed at a frequency of 3.1 × 10− 3. The combination of sequence-specific DNA cleavage by designed ZFNs and homology-directed DSB repair at investigator-specified break sites makes precise genome modification a reality. This capability, in combination with rapid advances in genome sequencing and bioinformatics, bodes well for the future of plant functional genomics and crop improvement.
Alternative Nuclease Technologies
Although ZFNs have become the most well-established tools for precise genome engineering, alternative nucleases are also available, such as those based on DNA binding domains from transcription activator-like effector (TALE) proteins (Boch and Bonas 2010) or “meganucleases” encoded by mobile introns (Arnould et al. 2011). TALEs are a family of proteins, first discovered in the plant pathogen Xanthomonas sp., that contain variable N- and C-terminal domains and a conserved central domain for DNA binding (Boch et al. 2009). The DNA-binding domain consists of a variable number of tandem 34 amino acid repeats (Fig. 12.2b), whereby binding specificity is determined by the repeat-variant di-residues (RVDs) at positions 12 and 13, which specifically recognize a single nucleotide (Bogdanove and Voytas 2011; Deng et al. 2012; Moscou and Bogdanove 2009). A one-to-one correspondence of the RVDs to a single nucleotide enables TALE designs for any target DNA sequence of interest with a high degree of specificity, though the RVD binding is not completely independent of its neighbor in TALE derivatives (Streubel et al. 2012). TALE-Fok1 nuclease (TALENs) fusions have been shown to facilitate genome modifications in several species, including human (Hockemeyer et al. 2011), rat (Tesson et al. 2011), zebra fish (Sander et al. 2011), worms (Wood et al. 2011), and plants (Cermak et al. 2011).
In contrast, designing ZFNs is more complex as each finger can only recognize a nucleotide triplet and there are multiple zinc finger designs for a given triplet of base pairs, with complex contextual interactions. Detailed knowledge of DNA binding of individual zinc fingers as well as the influence of various combinations of zinc fingers on binding specificity and affinity is required. Ease of design, high degree of specificity, minimal documented off-target effect, and low cost make TALENs an attractive alternative to ZFNs. Indeed, several recent reports of successful targeted mutagenesis following expression of designed TALENs suggest that this type of nuclease may represent a powerful addition to the arsenal of tools for plant genome modification (Li et al. 2012; Zhang et al. 2013). However, the larger size of TALENs (~ 3 ×) might limit their activity in plant cells primarily by effecting their expression negatively. Also, due to their pathogenic origins, TALENs might have a higher regulatory hurdle to cross for product development. Well controlled, comparative studies of ZFNs, and TALENs in plants will be critical for understanding their relative merits for precision genome engineering.
“Meganucleases” are naturally occurring gene-targeting proteins that function as homodimers comprising two identical subunits each 160–200 amino acid residues in size, but also active as a single peptide of two tandem repeat monomers joined together by a linker sequence (Stoddard 2011). Meganucleases typically bind to 20–30 bp DNA target sites which provide remarkable specificity, a primary reason for pursuing these proteins as for genome modification. In contrast to ZFNs and TALENs, the cleavage and DNA-binding domains of meganucleases are not clearly separated. Attempts to reengineer DNA contact points of the endonuclease can be challenging and often compromise nuclease activity (Taylor et al. 2012). Because of these engineering challenges, only a handful of academic groups and companies routinely engineer meganucleases that target novel DNA sites.
Most recently, RNA-guided nucleases from bacteria and archaea, referred to as “clustered regulatory interspaced short palindromic repeats” or CRISPRs have been adapted for genome modification whereby short segments of DNA are transcribed into RNAs which direct sequence-specific cleavage by Cas proteins (Wiedenheft et al. 2012). Using this system, targeted mutations were made in Arabidopsis BRI1, JAZ1 and GAI, and in rice ROC5 (Feng et al. 2013).
One of the main challenges associated with the routine deployment of designed nuclease technology for crop improvement is the relative inefficiencies of transgenic event production in all but a few plant species. Recently, in planta gene targeting was demonstrated using the meganuclease I-SceI (Fauser et al. 2012). In this study, three constructs were transformed independently into Arabidopsis: (i) a target with a broken reporter gene and nuclease cleavage sites, (ii) a donor with sequences complementary to the broken reporter, nuclease cleavage sites, and sequences homologous to the target, and (iii) the meganuclease which cuts in both the target and donor. Single copy, homozygous plants for each construct were generated and intermated in the following manner, [(target × donor) × nuclease]. The target contained a 3′ partial GUS reporter gene sequence and two I-SceI nuclease cleavage sites. The donor contained a 5′ partial GUS reporter gene, two I-SceI nuclease cleavage sites, sequences homologous to the target and two flanking identical sequences for single strand annealing repair following excision. Nuclease cleavage at the donor locus released the 5′ GUS gene fragment and the homologous sequences which provided a template for repair of the target. Observed targeting frequencies were as high as ~ 1 % on a progeny seed basis. This approach was corroborated in maize whereby inducible expression of I-SceI, combined with in vitro selection on kanamycin, allowed for the detection of the somatic repair of an NPTII gene (Ayar et al. 2013).
Future Prospects
The availability of custom targeting reagents such as designed ZFNs, together with the development of high-resolution molecular methods and bioinformatics for trait characterization, is likely to rapidly advance precision genome engineering in plants to enable product development in the near future. It is anticipated that targeted mutagenesis , gene excision, and genome editing will be routinely deployed for functional genomics and trait discovery. Some of these applications of precision genome engineering are likely to be regulated differently, i.e., as non-transgenic (Waltz 2011) and, as such, resulting changes in regulatory policies may have positive economic and social consequences. Similarly, current transgenic product development methods involve the random integration of transgenes into the plant genome, such that generating events and screening them for a trait of interest is time and cost intensive. The ability to target transgene integration into a predetermined genomic site should result in events whereby undesired side effects would be minimized and cycle times associated with product development reduced as event-specific analysis and characterization is simplified. Moreover, additional routes to product development are also likely through retargeting of transgenic loci leading to transgene stacking (Ainley et al. 2013; D’Halluin et al. 2013). In addition, from a trait discovery standpoint, targeting experimental constructs to specific genomic loci effectively removes variability associated with position effect thereby providing a uniform background against which genes and gene constructs can be screened to find lead candidates for new traits. Clearly, the enhanced precision relative to DNA manipulation, made possible by designed ZFNs, opens up some intriguing possibilities for both basic and applied research.
References
Ainley WM, Sastry-Dent L, Welter ME, Murray MG, Zeitler B, Amora R, Corbin DR, Miles RR, Arnold NL, Strange TL, Simpson MA, Cao Z, Carroll C, Pawelczak KS, Blue R, West K, Rowland LM, Perkins D, Samuel JP, Dewes CM, Shen L, Sriram S, Evans SL, Rebar EJ, Zhang L, Gregory PD, Urnov FD, Webb SR, Petolino JF (2013) Trait stacking via targeted genome editing. Plant Biotechnol J 11:1126–1134
Arnould S, Delenda C, Grizot S, Desseaux C, Paques F, Silva GH, Smith J (2011) The I-CreI meganuclease and its engineered derivatives: applications from cell modification to gene therapy. Protein Eng Des Sel PEDS 24:27–31
Ayar A, Wehrkamp-Richter S, Laffaire JB, Le Goff S, Levy J, Chaignon S, Salmi H, Lepicard A, Sallaud C, Gallego ME, White CI, Paul W (2013) Gene targeting in maize by somatic ectopic recombination. Plant Biotechnol J 11:305–314
Baulcombe DC (1999) Fast forward genetics based on virus-induced gene silencing. Curr Opin Plant Biol 2:109–113
Beumer K, Bhattacharyya G, Bibikova M, Trautman JK, Carroll D (2006) Efficient gene targeting in Drosophila with zinc-finger nucleases. Genetics 172:2391–2403
Bibikova M, Golic M, Golic KG, Carroll D (2002) Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161:1169–1175
Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I (1998) FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci U S A 95:10570–10575
Boch J, Bonas U (2010) Xanthomonas AvrBs3 family-type III effectors: discovery and function. Annu Rev Phytopathol 48:419–436
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Bogdanove AJ, Voytas DF (2011) TAL effectors: customizable proteins for DNA targeting. Science 333:1843–1846
Britt AB (1999) Molecular genetics of DNA repair in higher plants. Trends Plant Sci 4:20–25
Cai CQ, Doyon Y, Ainley WM, Miller JC, Dekelver RC, Moehle EA, Rock JM, Lee YL, Garrison R, Schulenberg L, Blue R, Worden A, Baker L, Faraji F, Zhang L, Holmes MC, Rebar EJ, Collingwood TN, Rubin-Wilson B, Gregory PD, Urnov FD, Petolino JF (2009) Targeted transgene integration in plant cells using designed zinc finger nucleases. Plant Mol Biol 69:699–709
Carroll D (2011) Genome engineering with zinc-finger nucleases. Genetics 188:773–782
Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, Baller JA, Somia NV, Bogdanove AJ, Voytas DF (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 39:e82
Charbonnel C, Allain E, Gallego ME, White CI (2011) Kinetic analysis of DNA double-strand break repair pathways in Arabidopsis. DNA repair 10:611–619
Curtin SJ, Zhang F, Sander JD, Haun WJ, Starker C, Baltes NJ, Reyon D, Dahlborg EJ, Goodwin MJ, Coffman AP, Dobbs D, Joung JK, Voytas DF, Stupar RM (2011) Targeted mutagenesis of duplicated genes in soybean with zinc-finger nucleases. Plant Physiol 156:466–473
D’Halluin K, Vanderstraeten C, Van Hulle J, Rosolowska J, Van Den Brande I, Pennewaert A, D’Hont K, Bossut M, Jantz D, Ruiter R, Broadhvest J (2013) Targeted molecular trait stacking in cotton through targeted double-strand break induction. Plant Biotechnol J 11(8):933–41
de Pater S Neuteboom LW Pinas JE Hooykaas PJ van der Zaal BJ (2009) ZFN-induced mutagenesis and gene-targeting in Arabidopsis through Agrobacterium-mediated floral dip transformation. Plant Biotechnol J 7:821–835
de Pater S Pinas JE Hooykaas PJ van der Zaal BJ (2013) ZFN-mediated gene targeting of the Arabidopsis protoporphyrinogen oxidase gene through Agrobacterium-mediated floral dip transformation. Plant Biotechnol J 11:510–515
Deng D, Yan C, Pan X, Mahfouz M, Wang J, Zhu J-K, Shi Y, Yan N (2012) Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 335:720–723
Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Amacher SL (2008) Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26:702–708
Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S (2005) Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res 33:5978–5990
Duxbury MS, Whang EE (2004) RNA interference: a practical approach. J Surg Res 117:339–344
Endo M, Osakabe K, Ono K, Handa H, Shimizu T, Toki S (2007) Molecular breeding of a novel herbicide-tolerant rice by gene targeting. Plant J 52:157–166
Fauser F, Roth N, Pacher M, Ilg G, Sanchez-Fernandez R, Biesgen C, Puchta H (2012) In planta gene targeting. Proc Natl Acad Sci U S A 109:7535–7540
Feldmann KA (1991) T-DNA insertion mutagenesis in Arabidopsis: mutational spectrum. Plant J 1:71–82
Feng Z, Zhang B, Ding W, Liu X, Yang DL, Wei P, Cao F, Zhu S, Zhang F, Mao Y, Zhu JK (2013) Efficient genome editing in plants using a CRISPR/Cas system. Cell Res 23:1229–1232
Gorbunova V, Levy AA (1999) How plants make ends meet: DNA double-strand break repair. Trends Plant Sci 4:263–269
Haber J (2007) Multiple mechanisms of repairing meganuclease-induced double-sdtrand DNA breaks in budding yeast. In: Aguilera A, Rothstein R (eds) Molecular genetics of recombination. Springer, Berlin/Heidelberg, p 285–316
Hauschild J, Petersen B, Santiago Y, Queisser A-L, Carnwath JW, Lucas-Hahn A, Zhang L, Meng X, Gregory PD, Schwinzer R, Cost GJ, Niemann H (2011) Efficient generation of a biallelic knockout in pigs using zinc-finger nucleases. Proc Natl Acad Sci U S A 108:12013–12017
Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, Zeitler B, Cherone JM, Meng X, Hinkley SJ, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R (2011) Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol 29:731–734
Hurt JA, Thibodeau SA, Hirsh AS, Pabo CO, Joung JK (2003) Highly specific zinc finger proteins obtained by directed domain shuffling and cell-based selection. Proc Natl Acad Sci U S A 100:12271–12276
Isalan M, Klug A, Choo Y (2001) A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat Biotechnol 19:656–660
Jia Q, Bundock P, Hooykaas PJJ, de Pater S (2012) Agrobacterium tumefaciens T-DNA integration and gene targeting in Arabidopsis thaliana non-homologous end-joining mutants. J Bot 2012:1–13
Keeney S (2001) Mechanism and control of meiotic recombination initiation. Curr Top Dev Biol 52:1–53
Kim YG, Cha J, Chandrasegaran S (1996) Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 93:1156–1160
Koornneeff M, Dellaert LWM, van der Veen JH (1982) EMS- and relation-induced mutation frequencies at individual loci in Arabidopsis thaliana (L.) Heynh. Mutation Res 93:109–123
Lee KY, Lund P, Lowe K, Dunsmuir P (1990) Homologous recombination in plant cells after Agrobacterium-mediated transformation. Plant Cell 2:415–425
Lee HJ, Kim E, Kim JS (2010) Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res 20:81–89
Li X, Song Y, Century K, Straight S, Ronald P, Dong X, Lassner M, Zhang Y (2001) A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J 27:235–242
Li R, Yang Y, An Y, Zhou Y, Liu Y, Yu Q, Lu D, Wang H, Jin L, Zhou W, Qian J, Shugart YY (2011) Genetic polymorphisms in DNA double-strand break repair genes XRCC5, XRCC6 and susceptibility to hepatocellular carcinoma. Carcinogenesis 32:530–536
Li T, Liu B, Spalding MH, Weeks DP, Yang B (2012) High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat Biotechnol 30:390–392
Lisby M, Rothstein R (2007) The cell biology of mitotic recombination in Saccharomyces cerevisiae. In: Aguilera A, Rothstein R (eds) Molecular genetics of recombination. Springer, Berlin/Heidelberg, p 317–333
Lloyd A, Plaisier CL, Carroll D, Drews GN (2005) Targeted mutagenesis using zinc-finger nucleases in Arabidopsis. Proc Natl Acad Sci U S A 102:2232–2237
Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, Holmes MC, Naldini L (2007) Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25:1298–1306
Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF, Neri M, Magnani Z, Cantore A, Lo Riso P, Damo M, Pello OM, Holmes MC, Gregory PD, Gritti A, Broccoli V, Bonini C, Naldini L (2011) Site-specific integration and tailoring of cassette design for sustainable gene transfer. Nat Methods 8:861–869
Lyznik LA, Djukanovic V, Yang M, Jones S (2012) Double-strand break-induced targeted mutagenesis in plants. In: Dunwell JM, Wetten AC (eds) Transgenic plants. Humana Press, New York, p 399–416
Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, Unger-Wallace E, Sander JD, Muller-Lerch F, Fu F, Pearlberg J, Gobel C, Dassie JP, Pruett-Miller SM, Porteus MH, Sgroi DC, Iafrate AJ, Dobbs D, McCray PB Jr, Cathomen T, Voytas DF, Joung JK (2008) Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31:294–301
Maluszynski M (2001) Officially releasvarieties—the FAO/IAEA database. Plant Cell Tissue Organ Cult 65:175–177
Mandell JG, Barbas CF 3rd (2006) Zinc finger tools: custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res 34:W516–523
Mathieu M, Winters EK, Kong F, Wan J, Wang S, Eckert H, Luth D, Paz M, Donovan C, Zhang Z, Somers D, Wang K, Nguyen H, Shoemaker RC, Stacey G, Clemente T (2009) Establishment of a soybean (Glycine max Merr. L) transposon-based mutagenesis repository. Planta 229:279–289
McCallum CM, Comai L, Greene EA, Henikoff S (2000) Targeting Induced Local Lesions IN Genomes (TILLING) for plant functional genomics. Plant Physiology 123:439–442
McMahon MA, Rahdar M, Porteus M (2012) Gene editing: not just for translation anymore. Nat Methods 9:28–31
Miao ZH, Lam E (1995) Targeted disruption of the TGA3 locus in Arabidopsis thaliana. Plant J 7:359–365
Moehle EA, Rock JM, Lee YL, Jouvenot Y, DeKelver RC, Gregory PD, Urnov FD, Holmes MC (2007) Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc Natl Acad Sci U S A 104:3055–3060
Morton J, Davis MW, Jorgensen EM, Carroll D (2006) Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc Natl Acad Sci U S A 103:16370–16375
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501
Offringa R, de Groot MJ, Haagsman HJ, Does MP, van den Elzen PJ, Hooykaas PJ (1990) Extrachromosomal homologous recombination and gene targeting in plant cells after Agrobacterium mediated transformation. EMBO J 9:3077–3084
Osakabe K, Osakabe Y, Toki S (2010) Site-directed mutagenesis in Arabidopsis using custom-designed zinc finger nucleases. Proc Natl Acad Sci U S A 107:12034–12039
Osiak A, Radecke F, Guhl E, Radecke S, Dannemann N, Lütge F, Glage S, Rudolph C, Cantz T, Schwarz K, Heilbronn R, Cathomen T (2011) Selection-independent generation of gene knockout mouse embryonic stem cells using zinc-finger nucleases. PloS ONE 6:e28911
Pabo CO, Peisach E, Grant RA (2001) Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem 70:313–340
Paszkowski J, Baur M, Bogucki A, Potrykus I (1988) Gene targeting in plants. EMBO J 7:4021–4026
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, Guschin DY, Rupniewski I, Waite AJ, Carpenito C, Carroll RG, Orange JS, Urnov FD, Rebar EJ, Ando D, Gregory PD, Riley JL, Holmes MC, June CH (2008) Establishment of HIV-1 resistance in CD4 + T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26:808–816
Perry JA, Wang TL, Welham TJ, Gardner S, Pike JM, Yoshida S, Parniske M (2003) A TILLING reverse genetics tool and a web-accessible collection of mutants of the legume Lotus japonicus. Plant Physiol 131:866–871
Petolino JF, Worden A, Curlee K, Connell J, Strange Moynahan TL, Larsen C, Russell S (2010) Zinc finger nuclease-mediated transgene deletion. Plant Mol Biol 73:617–628
Pham AT, Lee JD, Shannon JG, Bilyeu KD (2010) Mutant alleles of FAD2-1A and FAD2-1B combine to produce soybeans with the high oleic acid seed oil trait. BMC Plant Biol 10:195
Porteus MH, Baltimore D (2003) Chimeric nucleases stimulate gene targeting in human cells. Science 300:763
Porteus MH, Carroll D (2005) Gene targeting using zinc finger nucleases. Nat Biotechnol 23:967–973
Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, Chu V, Paschon DE, Zhang L, Kuball J, Camisa B, Bondanza A, Casorati G, Ponzoni M, Ciceri F, Bordignon C, Greenberg PD, Holmes MC, Gregory PD, Naldini L, Bonini C (2012) Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med 18:807–815
Puchta H (2005) The repair of double-strand breaks in plants: mechanisms and consequences for genome evolution. J Exp Bot 56:1–14
Puchta H, Fauser F (2013) Gene targeting in plants: 25 years later. Int J Dev Biol 57:629–637
Puchta H, Dujon B, Hohn B (1996) Two different but related mechanisms are used in plants for the repair of genomic double-strand breaks by homologous recombination. Proc Natl Acad Sci U S A 93:5055–5060
Rajesh C, Baker DK, Pierce AJ, Pittman DL (2011) The splicing-factor related protein SFPQ/PSF interacts with RAD51D and is necessary for homology-directed repair and sister chromatid cohesion. Nucleic Acids Res 39:132–145
Reiss B, Schubert I, Kopchen K, Wendeler E, Schell J, Puchta H (2000) RecA stimulates sister chromatid exchange and the fidelity of double-strand break repair, but not gene targeting, in plants transformed by Agrobacterium. Proc Natl Acad Sci U S A 97:3358–3363
Roth N, Klimesch J, Dukowic-Schulze S, Pacher M, Mannuss A, Puchta H (2012) The requirement for recombination factors differs considerably between different pathways of homologous double-strand break repair in somatic plant cells. Plant J 72:781–790
Salomon S, Puchta H (1998) Capture of genomic and T-DNA sequences during double-strand break repair in somatic plant cells. EMBO J 17:6086–6095
Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, Yeh JR (2011) Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol 29:697–698
Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, Urnov FD, Holmes MC, Guschin D, Waite A, Miller JC, Rebar EJ, Gregory PD, Klug A, Collingwood TN (2008) Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci U S A 105:5809–5814
Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, Goodwin MJ, Hawkins JS, Ramirez CL, Batista LFZ, Artandi SE, Wernig M, Joung JK (2011) In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 29:1717–1726
Shaked H, Melamed-Bessudo C, Levy AA (2005) High-frequency gene targeting in Arabidopsis plants expressing the yeast RAD54 gene. Proc Natl Acad Sci U S A 102:12265–12269
Shalev G, Sitrit Y, Avivi-Ragolski N, Lichtenstein C, Levy AA (1999) Stimulation of homologous recombination in plants by expression of the bacterial resolvase RuvC. Proc Natl Acad Sci U S A 96:7398–7402
Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, Jeggo PA (2011) Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 30:1079–1092
Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, Mitchell JC, Arnold NL, Gopalan S, Meng X, Choi VM, Rock JM, Wu YY, Katibah GE, Zhifang G, McCaskill D, Simpson MA, Blakeslee B, Greenwalt SA, Butler HJ, Hinkley SJ, Zhang L, Rebar EJ, Gregory PD, Urnov FD (2009) Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature 459:437–441
Siebert R, Puchta H (2002) Efficient repair of genomic double-strand breaks by homologous recombination between directly repeated sequences in the plant genome. Plant Cell 14:1121–1131
Sonoda E, Hochegger H, Saberi A, Taniguchi Y, Takeda S (2006) Differential usage of non-homologous end-joining and homologous recombination in double strand break repair. DNA Repair 5:1021–1029
Stoddard BL (2011) Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19:7–15
Straimer J, Lee MCS, Lee AH, Zeitler B, Williams AE, Pearl JR, Zhang L, Rebar EJ, Gregory PD, Llinas M, Urnov FD, Fidock DA (2012) Site-specific genome editing in Plasmodium falciparum using engineered zinc-finger nucleases. Nat Methods 9:993–998
Streubel J, Blucher C, Landgraf A, Boch J (2012) TAL effector RVD specificities and efficiencies. Nat Biotechnol 30:593–595
Taylor GK, Petrucci LH, Lambert AR, Baxter SK, Jarjour J, Stoddard BL (2012) LAHEDES: the LAGLIDADG homing endonuclease database and engineering server. Nucleic Acids Res 40:W110–116
Terada R, Urawa H, Inagaki Y, Tsugane K, Iida S (2002) Efficient gene targeting by homologous recombination in rice. Nat Biotechnol 20:1030–1034
Terada R, Johzuka-Hisatomi Y, Saitoh M, Asao H, Iida S (2007) Gene targeting by homologous recombination as a biotechnological tool for rice functional genomics. Plant Physiol 144:846–856
Tesson L, Usal C, Menoret S, Leung E, Niles BJ, Remy S, Santiago Y, Vincent AI, Meng X, Zhang L, Gregory PD, Anegon I, Cost GJ (2011) Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol 29:695–696
Tovkach A, Zeevi V, Tzfira T (2009) A toolbox and procedural notes for characterizing novel zinc finger nucleases for genome editing in plant cells. Plant J 57:747–757
Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF (2009) High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 459:442–445
Tzfira T, Weinthal D, Marton I, Zeevi V, Zuker A, Vainstein A (2012) Genome modifications in plant cells by custom-made restriction enzymes. Plant Biotechnol J 10:373–389
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651
Vainstein A, Marton I, Zuker A, Danziger M, Tzfira T (2011) Permanent genome modifications in plant cells by transient viral vectors. Trends Biotechnol 29:363–369
Waltz E (2011) GM grass eludes outmoded USDA oversight. Nat Biotech 29:772–773
Watanabe S, Mizoguchi T, Aoki K, Kubo Y, Mori H, Imanishi S, Yamazaki Y, Shibata D, Ezura H (2007) Ethylmethanesulfonate (EMS) mutagenesis of Solanum lycopersicumcv. Micro-Tom for large-scale mutant screens. Plant Biotechnol 24:33–38
Weinthal DM, Taylor RA, Tzfira T (2013) Nonhomologous end joining-mediated gene replacement in plant cells. Plant Physiol 162:390–400
Wiedenheft B, Sternberg SH, Doudna JA (2012) RNA-guided genetic silencing systems in bacteria and archaea. Nature 482:331–338
Wilen CB, Wang J, Tilton JC, Miller JC, Kim KA, Rebar EJ, Sherrill-Mix SA, Patro SC, Secreto AJ, Jordan APO, Lee G, Kahn J, Aye PP, Bunnell BA, Lackner AA, Hoxie JA, Danet-Desnoyers GA, Bushman FD, Riley JL, Gregory PD, June CH, Holmes MC, Doms RW (2011) Engineering HIV-resistant human CD4 + T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathol 7:e1002020
Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, Amora R, Miller JC, Leung E, Meng X, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Meyer BJ (2011) Targeted genome editing across species using ZFNs and TALENs. Science 333:307
Wright DA, Townsend JA, Winfrey RJ Jr, Irwin PA, Rajagopal J, Lonosky PM, Hall BD, Jondle MD, Voytas DF (2005) High-frequency homologous recombination in plants mediatedted by zinc-finger nucleases. Plant J 44:693–705
Wu Q, Sibanda L, Ochi T, Bolanos-Garcia V, Blundell T, Chirgadze D (2012) Spatial and temporal organisation of multiprotein systems of cell regulation and signalling: what can we learn from NHEJ system of double-strand break repair? In: Carrondo MA, Spadon P (eds) Macromolecular crystallography. Springer, Netherlands, pp 1–31
Xiaohui Wang H Viret J-F Eldridge A Perera R Signer ER Chiurazzi M (2001) Positive–negative selection for homologous recombination in Arabidopsis. Gene 272:249–255
Xu Y, Price BD (2011) Chromatin dynamics and the repair of DNA double strand breaks. Cell Cycle 10:261–267
Young JJ, Cherone JM, Doyon Y, Ankoudinova I, Faraji FM, Lee AH, Ngo C, Guschin DY, Paschon DE, Miller JC, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Harland RM, Zeitler B (2011) Efficient targeted gene disruption in the soma and germ line of the frog Xenopus tropicalis using engineered zinc-finger nucleases. Proc Natl Acad Sci U S A 108:7052–7057
Zhang F, Maeder ML, Unger-Wallace E, Hoshaw JP, Reyon D, Christian M, Li X, Pierick CJ, Dobbs D, Peterson T, Joung JK, Voytas DF (2010) High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc Natl Acad Sci U S A 107:12028–12033
Zhang Y, Zhang F, Li X, Baller JA, Qi Y, Starker CG, Bogdanove AJ, Voytas DF (2013) Transcription activator-like effector nucleases enable efficient plant genome engineering. Plant Physiol 161:20–27
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Petolino, J., Sastry-Dent, L., Samuel, J. (2015). Zinc Finger Nuclease-Mediated Gene Targeting in Plants. In: Azhakanandam, K., Silverstone, A., Daniell, H., Davey, M. (eds) Recent Advancements in Gene Expression and Enabling Technologies in Crop Plants. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2202-4_12
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2202-4_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2201-7
Online ISBN: 978-1-4939-2202-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)