
Abstract
Increased blood serotonin in people with autism was first reported over 50 years ago, and this biogenic amine has remained a focus for the understanding, risk and treatment of Autism Spectrum Disorders (ASD). There is growing evidence that serotonergic transmission is altered by disparate genetic and environmental risk factors for ASD. This review will focus on recent developments regarding serotonin in ASD. Recent studies include epidemiology studies linking ASD with conditions that alter prenatal serotonin in the fetus, altered serotonin in diverse genetic and environmental animal models, and human pathology and molecular and functional brain imaging studies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Increased blood serotonin is the earliest biomarker reported for ASD (Schain and Freedman 1961). This biogenic amine has remained a focus for the understanding, risk and treatment of ASD. Current evidence now suggests that autism is likely to be caused by dysfunction of many genes (Zhao et al. 2007). Given that many different genes can cause autism, it is striking that 30–50 % of autistic individuals show elevated serotonin in the blood (Schain and Freedman 1961; Hoshino et al. 1984; Anderson et al. 1987; Cook et al. 1990). Furthermore, there is growing evidence that serotonergic transmission is altered by disparate genetic and environmental risk factors for ASD. This review will focus on recent developments on serotonin in autism. Recent studies include epidemiologic studies linking ASD with conditions that alter prenatal serotonin in the fetus, altered serotonin in diverse genetic and environmental animal models , and human pathology and molecular and functional brain imaging studies.
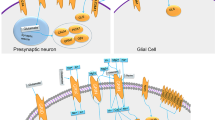
1 Overview of the Serotonin Pathway
Serotonin is synthesized from the precursor tryptophan, an essential amino acid constituting one percent of the total amino acid pool (Hamon et al. 1981). The majority of tryptophan in blood is bound to plasma protein, with only free plasma tryptophan being available for transport into the brain. Tryptophan is transported into the brain via the large neutral amino acid carrier (LAT1) where it competes for transport with the other large neutral amino acids (Pardridge et al. 1977; Smith et al. 1987). The first and rate-limiting step of serotonin synthesis is the formation of 5-hydroxytryptophan catalyzed by the enzyme tryptophan hydroxylase (TPH) . Tryptophan hydroxylase is only 50 % saturated with tryptophan, resulting in the dependence of brain serotonin levels on the plasma concentration of free tryptophan as well as the plasma levels of the other large neutral amino acids (Fernstrom and Wurtman 1971). This dependence of free plasma tryptophan on brain serotonin levels has been exploited to study the effects of tryptophan depletion on brain function in adults with ASD (Daly et al. 2012, see below). 5-hydroxytryptophan is further metabolized by aromatic amino acid decarboxylase (AADC) to form serotonin (Christenson et al. 1972). Serotonin is metabolized by the enzyme monoamine oxidase A (MAOA) to produce its major metabolite 5-hydorxyindoleacetic acid (5HIAA) (for review see Bortolato et al. 2008).
The serotonin transporter (5HTT or SERT) mediates uptake of serotonin following release at the synapse and at other sites such as the platelet. The gene for the serotonin transporter has 2 forms, the short and the long form, that are common in the general population. The short variant of the 5HTT gene has 30–40 % lower mRNA expression leading to 50 % lower serotonin uptake than transporters produced by the long form (Lesch et al. 1996). To date, fifteen receptors for serotonin have been identified, and these are expressed in the brain and throughout the body where they regulate numerous processes (for review see Berger et al. 2009). In addition to receptor-mediated mechanisms, serotonin has been recently reported to signal through a process called serotonylation (for review see, Walther et al. 2011). Serotonylation involves the covalent linkage of serotonin to proteins mediated by the enzyme transglutaminase. Linkage of serotonin to GTPases has been reported to increase alpha-granule exocytosis in platelets (Walther et al. 2003).
There are 2 genes encoding tryptophan hydroxylase , TPH1 and TPH2 (Sakowski et al. 2006). TPH2 is found in the brain and in the gastrointestinal nervous system (Zhang et al. 2004), while TPH1 is located in the pineal gland and throughout the rest of the body. Many current references suggest that the majority of the serotonin synthesized in the body is generated by the enterochromaffin cells in the gastrointestinal tract (Gershon and Tack 2007). However, tryptophan hydroxylase is located at many peripheral sites, and there have been no studies comparing the amount of serotonin produced at all of these sites to that synthesized in the gastrointestinal tract . There is much evidence that TPH1 is ubiquitously distributed and that serotonin is involved in many different functions throughout the body. Many of these functions might be related to different aspects of autism. In addition to the gastrointestinal tract and the central nervous system, serotonin is synthesized in bronchial epithelium, taste papillae, thyroid parafollicular cells, ovaries, thymus, pancreas, breast, skin and arteries (Eddahibi et al. 2006; Ortiz-Alvarado et al. 2006; Matsuda et al. 2004; Stull et al. 2007; Slominski et al. 2002, Ni et al. 2008). TPH and AADC mRNAs are expressed in the heart and serotonin is produced and released in cardiomyocytes (Ikeda et al. 2005). TPH1 mRNA and TPH protein are expressed in trigeminal neurons and regulated during the estrous cycle (Berman et al. 2006). Serotonin plays an important role in mammary gland function. TPH mRNA is elevated during pregnancy and lactation and serotonin is present in mammary epithelium and in milk (Matsuda et al. 2004). Elevated plasma serotonin in autism might also be affected by changes in storage and degradation. Plasma serotonin is taken up by platelets through serotonin transporters (Ni and Watts 2006), and is cleared by liver and lung endothelium (Nocito et al. 2007). Finally, serotonin and tryptophan are very tightly regulated at the fetal/maternal interface in the placenta. The serotonin transporter is highly expressed in the brush border membrane of the human placenta and may mediate transport of serotonin from the maternal circulation to the developing fetus (Balkovetz et al. 1989). Tryptophan is transported competitively at the placenta via the large neutral amino-acid carrier (LAT1) (Kudo and Boyd 2002). The placenta expresses TPH2 resulting in serotonin production (Correa et al. 2009). In addition, tryptophan in the placenta is catalyzed along the kynurenine pathway by tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase (IDO), both of which are expressed in the placenta and have a vital role in the prevention of allogeneic rejection of the fetus (Munn et al. 1998) and regulation of feto-placental blood flow in late gestation (Ligam et al. 2005). Finally, Bonnin et al. (2011) demonstrated that serotonin in the forebrain during the early fetal period in the rat has its origin in the placenta. Studies in mice deficient in peripheral serotonin synthesis have shown that maternal serotonin production is crucial for normal fetal neurogenesis and development (Cote et al. 2007).
2 Elevated Blood Serotonin in Autism
As mentioned above, elevated serotonin in ASD is a long-standing (Schain and Freedman 1961) and replicated finding (Hoshino et al. 1984; Anderson et al. 1987; Cook et al. 1990), and extended with the recognition that blood serotonin is also elevated in the first degree relatives of autistic individuals (Leventhal et al. 1990; Piven et al. 1991; Cook et al. 1994; Leboyer et al. 1999). The mechanism for the elevated platelet serotonin may be due to increased exposure to serotonin or altered handling of serotonin, as recently examined by Anderson et al. (2012). Based on the lack of difference in the serotonin concentration in platelet poor plasma in samples from an ASD group, an ASD subgroup with hyperserotonemia and a control group, these authors suggest that hyperserotonemia may be more related to platelet handling of serotonin than exposure to serotonin. Mechanisms they discussed for further study included study of the transporter, the granular transporter and dense granule storage mechanisms, serotonin release processes and platelet activation. In this regard, Veenstra-VanderWeele et al. (2012) developed a mouse model based upon the SERT Ala56 variant that is over-transmitted to ASD probands. The SERT Ala56 mouse model showed hyperserotonemia, consistent with a role of the transporter in elevated blood serotonin. However, this mechanism does not explain most cases of hyperserotonemia, as mutations in SERT are not found in the majority of cases of ASD.
3 Serotonin and Human Brain Pathology in Autism
Given the long standing recognition of altered serotonin in the blood in those with ASD, it is surprising that studies of serotonergic markers in postmortem brains from persons with ASD have not been examined until recently . Two reports from the same group have reported increased serotonin transporter immunoreactivity in human postmortem brains from individuals with autism aged 2–29 years (Azmitia et al. 2011a, b). Increased SERT immunoreactivity was demonstrated for the principle ascending fiber bundles of the medial and lateral forebrain bundles, Ansa lenticularis and stria terminalis, and in the innervation density of the globus pallidus, amygdala, and in the piriform, superior temporal and parahippocampal cortices. In addition, in cases over 8 years of age, the presence of thick, heavily stained dystrophic axons were observed. In contrast, Oblak et al. (2013) reported significant reduction in the density of SERT binding in the deep layers of the fusiform gyrus and normal binding levels in the superficial layers of the fusiform gyrus, as well as in both layers of the posterior cingulate cortex. The same investigators reported significant reductions in 5-HT1A receptor-binding density in superficial and deep layers of the posterior cingulate cortex and fusiform gyrus and reduced density of 5-HT2A receptors in superficial layers of the posterior cingulate cortex and fusiform gyrus. Differences in the results between the immunocytochemistry studies and the binding studies for SERT may be due to the use of different brain regions, differences in age or use of different investigational techniques. However, molecular imaging studies have reported SERT binding in both children and adults with ASD, including some of the same brain regions that showed an increase SERT immunoreactivity (see below) .
4 Molecular Imaging of Serotonin Precursor, Transporter and Receptor Studies
In the earliest molecular imaging studies of serotonin in ASD, Chugani et al. (1997) applied alpha[C-11]methyl-L-tryptophan (AMT) as a PET tracer in children with autism . AMT, which was developed as a tracer for serotonin synthesis with PET (Diksic et al. 1990), is an analogue of tryptophan, the precursor for serotonin synthesis. Two types of serotonergic abnormality were found in children with autism (Chugani et al. 1997, 1999; Chandana et al. 2005). The first was altered whole brain serotonin synthesis capacity in autistic children compared to age matched nonautistic children. Serotonin synthesis capacity was greater than 200 % of adult values until the age of 5 years followed by a decline to adult values in non-autistic children. In contrast, serotonin synthesis capacity in autistic children increased gradually between the ages of 2 years and 15 years to values 1.5 times the adult normal values (Chugani et al. 1999). These data suggested that developmental regulation of brain serotonin synthesis capacity postnally in humans, was disrupted in children with autism. Secondly, focal symmetries of AMT uptake in frontal cortex, thalamus and cerebellum were visualized in children with autism, suggesting a role of the dentato-thalamo-cortical pathway in autism (Chugani et al. 1997). Subsequently, the same group measured brain serotonin synthesis in a large group of autistic children (n = 117) with AMT PET and related these data to handedness and language function (Chandana et al. 2005) . Autistic children with left cortical AMT decreases showed a higher prevalence of severe language impairment, whereas those with right cortical decreases showed a higher prevalence of left and mixed handedness. These results suggest that global as well as focal abnormally asymmetric development in the serotonergic system could lead to miswiring of the neural circuits specifying hemispheric specialization .
Decreased serotonin transporter binding has been reported in both children and adults with autism. Makkonen et al. (2008) using the SPECT tracer [123I] nor-beta-CIT labeling both the dopamine and serotonin transporter, reported reduced serotonin transporter binding capacity in medial frontal cortex, midbrain and temporal lobes. Similarly, Nakamura et al. (2010) reported decreased serotonin transporter binding throughout the brain in adults with autism (20 men, 18–26 years) using [11C]McN-5652 imaged with PET . Furthermore, the reduction in binding in anterior and posterior cingulate cortices was correlated with impairment in social cognition, while the reduction in serotonin transporter binding in the thalamus was correlated with repetitive and/or obsessive behavior. In contrast, Girgis et al. (2011) reported no significant difference in brain serotonin transporter binding, measured with [11C]DASB and PET, in a group of 8 adults with Asperger’s Disorder (mean age 29.7 years) and 8 healthy control subjects matched for age, gender, and ethnicity. All subjects in this study had normal intelligence , while this was not the case for the other studies reporting changes in serotonin transporter binding .
Serotonergic neurotransmission was also studied using tracers for receptor binding. Murphy et al. (2006) measured 5HT2A receptors in eight men with Asperger’s Syndrome (mean age 26 years) using the SPECT tracer [123I]5-I-R91150, compared to 10 healthy age-matched men. The group with Asperger’s Syndrome has significantly reduced serotonin receptor binding in total, anterior and posterior cingulate cortex, bilaterally in frontal and superior temporal lobes and in the left parietal lobe. Interestingly, there were significant correlations with qualitative abnormalities in social interaction and binding reductions in anterior and posterior cortices, as well as right frontal cortex. More recently, 5-HT2 receptor distribution was measured with the PET tracer [18F] setoperone in six high functioning autistic adults compared to ten matched control subjects (Beversdorf et al. 2012) . In this study, reduced serotonin receptor binding was found in thalamus, and there was a negative relationship between thalamic binding and history of language impairment. Goldberg et al. (2009) compared the parents of children with autism (19 parents from 11 families, 8 females, 11 males) compared to adults who do not have children with autism (9 females, 8 males). The cortical 5HT2 binding potential, using [18F]setoperone, to measure cortical serotonin type-2 receptor (5-HT2) using PET, was significantly lower in the autism parent group compared to the control group. Furthermore, the 5HT-2 binding potential was inversely correlated with platelet serotonin levels in the parent group. These results are interesting in light of family members having what has been described as the broader phenotype of autism. Finally, Girgis et al. (2011) reported no difference in 5HT2A receptor binding in a PET study using the tracer [11C]MDL 100907 in a group of 17 adults with Asperger’s Disorder (mean age 34.3) compared to 17 healthy matched adults .
In summary, molecular imaging studies provide convincing evidence of altered serotonergic neurotransmission in both children and adults with autism, as well as in parents of autistic individuals. Decreased serotonin transporter and 5HT2A binding measured in vivo using molecular imaging techniques are consistent with postmortem binding studies (Oblak et al. 2013). These studies are discordant with postmortem immunocytochemistry studies reporting increased serotonin immunoreactivity (Azmitia et al. 2011a, b). The reason for the discrepancy between the binding and immunocytochemistry studies might offer clues regarding the nature of alterations in serotonergic fibers. For example, these results considered together, might indicate fewer serotonergic fibers with remaining serotonergic fibers showing higher density of transporters .
5 Serotonin and Functional Imaging
In addition to molecular imaging studies that use radiolabeled ligands and substrates to measure serotonergic metabolism, transport and receptor systems, functional MRI (fMRI) has been applied in adults with ASD following pharmacological manipulation of the serotonergic system using either tryptophan depletion or treatment with the selective serotonin reuptake inhibitor (SSRI) fluoxetine. Following treatment to decrease blood tryptophan, which has been long demonstrated to decrease brain serotonin (Moja et al. 1988, 1989), Daly et al. (2012) reported altered brain activation as measured by blood oxygenation labeled dependent (BOLD) response measured with fMRI to face emotion in adults with ASD compared to matched control subjects. Furthermore, the differences in brain activation varied by emotion, and brain regions showing altered brain activation in the ASD group include brain regions showing focal abnormalities in serotonin synthesis, and decreased numbers of receptors and transporter in the molecular imaging studies. The same group (Chantiluke et al. 2014) studied the effects of fluoxetine on medial prefrontal activation during a reward reversal learning task in boys with autism compared to a matched group of typically developing boys. This study showed that while placebo treatment was associated with decreased activation of the medial prefrontal cortex during the performance of the task in the ASD group, fluoxetine treatment lead to normalization of the activation in boys with ASD compared to controls. Despite normalization of brain activation in this study, the fluoxetine treatment did not improve behavioral performance on the task. Altered function related to serotonergic function in medial prefrontal cortex is consistent with decreased serotonin transporter binding in this region as reported by Makkonen et al. (2008).
The pharmacological treatments combined with fMRI measure of brain activation complement the postmortem and molecular imaging studies by assessing the functional significance of altered serotonergic neurotransmission in different brain regions. These results might be due to differences during brain development and ongoing changes in serotonergic mechanisms.
6 Genetic Evidence Linking Serotonin to Autism
Because of the long-standing link between serotonin and ASD, the serotonin transporter, its receptors and pathway enzymes have been studied as candidate genes for ASD. The serotonin transporter has 2 isoforms, the short and the long forms, that are common in the general population. The short variant of the 5HTT has 30–40 % lower mRNA expression and 50 % lower serotonin uptake than the long form (Lesch et al. 1996). There have been conflicting reports regarding the relative roles of the short and long forms in autism (reviewed in Devlin et al. 2005). More recently, the short allele of the 5HTT receptor was associated with higher gray matter volume in young boys with autism (Wassink et al. 2007). Altered serotonin synthesis, turnover and dynamic regulation in multiple brain regions have been reported in mice lacking the serotonin transporter (Kim et al. 2005). Veenstra-VanderWeele et al. (2012) reported that the SERT Ala56 variant was over-transmitted to ASD probands, but that it was also seen in some unaffected individuals, suggesting that associated ASD risk is influenced by the interaction of the SERT variant with other genetic factors. Subsequently, the same group showed that mice expressing the SERT Ala56 variant on a 129S6/S4 genetic background display multiple biochemical, physiological and behavioral changes, including hyperserotonemia, altered 5-HT receptor sensitivity, and abnormal social, communication, and repetitive behavior. The same group (Kerr et al. 2013) examined the effect of this same mutation on different mouse backgrounds. Presence of the SERT Ala56 variant on the B6 background caused a significant increase in mutant pup ultrasonic vocalizations, whereas vocalizations were decreased on the 129 background. Further, hyperserotonemia, 5-HT receptor hypersensivity, and repetitive behavior were not observed on the B6 background. These studies combined, demonstrated how epistatic interactions can modulate the effect of genetic mutations and how genes may modulate the risk of ASD.
Blood serotonin levels have been linked to an interaction of the serotonin transporter gene (SLC6A4) and integrin beta3 (ITGB3) (Weiss et al. 2006a, b). This finding was recently replicated and additional interactions between these 2 genes were found, including an additive effect of the HTR5A gene, an interaction of TPH1 and SLC6A4, and an interaction between 5HTR1D and SLC6A4 for blood serotonin level (Coutinho et al. 2007). Whyte et al. (2014) examined the effect of Itgb3 on SERT function using a mouse genetic approach. Using isolated synaptoneurosomes and citalopram binding, they reported significant Slc6a4-driven reductions in SERT expression in midbrain synapses, but no significant changes in hippocampal or cortical regions. They also measured 5-HT uptake activity in synaptoneurosomal preparations. Itgb3 heterozygous mice displayed significant reductions in 5HT Vmax, with no changes in Km, in midbrain preparations. These results support an interaction of SLC6A4 and ITGB3 in the regulation of serotonergic systems in midbrain but not cortical and hippocampal serotonin synapses.
Several studies report association of genes that may impact serotonin metabolism in autism. Single nucleotide polymorphisms in introns 1 and 4 of TPH2, the brain specific form of tryptophan hydroxylase , showed an association with autism (Coon et al. 2005). Brain serotonin synthesis in mouse strains differs depending on a single nucleotide polymorphism in the Tph2 gene (Siesser et al. 2010). Based upon this Tph2 polymorphism, the BALB/c strain shows lower brain serotonin synthesis, although serotonin levels were not significantly altered. Behavioral studies of the BALB/c strain have shown low sociability and other phenotypes relevant to ASD (Brodkin 2007). Kane et al. (2012) analyzed mice lacking brain serotonin via a null mutation in the Tph2 gene for behaviors that are relevant to ASD. Mice lacking brain TPH2 showed substantial deficits in validated tests of social interaction and communication, as well as repetitive and compulsive behaviors. Newborn TPH22/2 mutant mice showed diminished preference for maternal scents over the scent of an unrelated female. The authors conclude that lack of serotonin during development could lead to ASD behavioral traits.
There are several studies linking genetic variation in the MAOA gene to ASD. Cortical enlargement in autism was reported to be associated with the “low activity” allele of the MAOA gene (Davis et al. 2008). Jones et al. (2004) reported that maternal genotypes containing specific polymorphisms at the MAOA locus show significant negative effects on the intelligence quotient (IQ) in children with autism. These results are consistent with those of a study which found that a low activity MAOA allele, due to an upstream variable-number tandem repeat region, is associated with both lower IQ and more severe autistic behavior in children, as compared to the high-activity allele (Cohen et al. 2003). Tassone et al. (2011) also reported an association of ASD with repeats in the MAOA promoter. They found that boys carrying 4 tandem repeats in the promoter region of the MAOA gene showed a 2-fold higher risk of autism or other forms of ASD than those carrying allele 3. They also found that mothers homozygous for the 4 tandem repeat allele showed at least a 3-fold higher risk for having a child with ASD than mothers homozygous for allele 3. Tassone et al. concluded that the functional MAOA promoter allelles in male children and mothers play a role in the risk for ASD. Finally, Verma et al. (2013) analyzed 8 MAOA markers in 421 subjects including cases and ethnically-matched controls from West Bengal. MAOA marker, rs6323 and haplotypes formed between the markers showed a significant association with the ASD. In addition, gender stratification showed significant genetic effect of rs6323 with low activity T allele posing higher risk of ASD in males, but not in females. Taken together, genetic differences in genes for enzymes involved in the synthesis and degradation of serotonin play a role in the risk for ASD and associated phenotypes such as brain enlargement and intellectual disability. In addition, these studies suggest that serotonin alterations in mothers and gender may also play a role in the risk for ASD.
Metabolism of tryptophan by other metabolic pathways may also affect serotonin synthesis, due to the dependence of brain serotonin synthesis on plasma tryptophan levels (Fernstrom and Wurtman 1971). One such study reports the presence of a susceptibility mutation in a promoter variant of the tryptophan 2,3 dioxygenase gene (Nabi et al. 2004). Tryptophan 2,3-dioxygenase is a rate-limiting enzyme in the metabolism of tryptophan by the kynurenine pathway. A mutation that results in decreased activity of this enzyme could decrease the metabolism of tryptophan by this pathway and increase the level of whole body serotonin content.
7 Altered Serotonin in Autism Caused by Genes not Related to Serotonin
In addition to genetic variation in genes directly involved with serotonergic function, there is evidence that serotonergic function is altered in humans and in animal models having genetic changes associated with ASD. These genetic changes include mutations involved in causation of Rett syndrome, 15q duplication syndrome and Fragile X syndrome . There are several studies showing alterations in serotonergic neurotransmission in Rett syndrome, as well as in the MECP2-null mouse model of Rett syndrome. Decreased levels of the serotonin metabolite 5HIAA were reported in the CSF from 32 patients with Rett syndrome compared to age matched control subjects (Zoghbi et al. 1989). Serotonin concentrations in whole brains from mecp2-null mice showed no differences at birth, but were significantly lower at 42 days of age compared to wild-type mice (Ide et al. 2005). However, these changes were not specific to serotonin and changes in other transmitters were also reported. Further evidence that the serotonergic system is involved in MeCP2, is the report that the selective serotonin uptake inhibitor fluoxetine induced up regulation of Mecp2_e1 and Mecp2_e2 transcripts in adult rat brain (Cassel et al. 2006). Samaco et al. (2009) confirmed lower serotonin metabolite 5HIAA in the cerebral spinal fluid of 64 individuals with Rett syndrome and decreased serotonin in Mecp2-null male mice. In addition, this study deleted Mecp2 in serotonergic neurons positive for the marker PET-1 and showed decreased levels of serotonin and decreased expression of TPH2. These data suggest that Mecp2 is involved in the regulation of serotonin synthesis by promoting the transcription of TPH2.
The duplication of human chromosome 15q11-13 is a chromosome rearrangement associated with ASD (Cook et al. 1997). Tamada et al. (2010) studied serotonin signaling in a mouse model in which chromosome 7C (orthologous to human chromosome 15q11-13) was duplicated. In this study, compared to wild type mice, there were decreased serotonin levels measured at 1, 2 and 3 weeks of age in cerebellum, cerebral cortex, hippocampus, hypothalamus, midbrain and pons. In adult animals, serotonin levels remained decreased in midbrain.
Fragile X syndrome is caused by a CGG expansion greater than 200 repeats in the 5’ untranslated region in the fragile X mental retardation 1 gene, encoding the fragile X mental retardation 1 protein (FMRP) , and is a leading single gene cause of autism (Hagerman et al. 2010). Zhang et al. (2005) used a Drosophila model and a proteomic approach to find targets of dFMRP in the brain. They found two upregulated enzymes, phenylalanine hydroxylase (Henna) and GTP cyclohydrolase (Punch), which mediate the dopamine and serotonin synthetic pathways. They confirmed a nearly 2-fold elevation of Punch activity in dfmr1 null mutants and significantly increased dopamine and serotonin in dfmr1 null mutants.
Thus, serotonin mechanisms may be impacted by disparate genetic risks for ASD. These examples illustrate that in the different disorders serotonin levels can be decreased or increased. Furthermore, changes in serotonergic function may differ depending on the developmental stage. These results have implications for pharmacological treatments aimed at serotonergic function.
8 Environmental Exposures and ASD
There is evidence for increased risk of autism with several environmental exposures. Three types of exposure will be discussed here as they relate to serotonin: prenatal antidepressants acting at the serotonin transporter (SSRIs) , prenatal sodium valproate and maternal infection/inflammation . A recent epidemiology study reported a modest increased risk for ASD in offspring of mothers taking antidepressants, which inhibit serotonin uptake by the serotonin transporter (Croen et al. 2011). However, Hviid et al. (2013) did not replicate a significant association between maternal use of SSRIs during pregnancy and autism spectrum disorder in the offspring, although their study could not rule out a relative risk of up to 1.61. Animal studies of SSRI treatment during pregnancy have demonstrated changes in brain development and behavior. For example, Smit-Rigter et al. (2012) reported changes in cortical cytoarchitecture and anxiety in mice exposed to the SSRI fluoxetine in utero. Further study is required to determine the risk for the development of ASD with antidepressant treatment in pregnant women.
Epidemiological studies have shown increased risk of developing ASD due to in utero exposure to the anticonvulsant sodium valproate (Christensen et al. 2013; Bromley et al. 2013). Among other changes, the rodent valproate model of ASD shows changes in serotonergic function (Wang et al. 2013). In this model, there was increase tryptophan hydroxylase immunoreactivity in the dorsal and medial raphe nuclei and an increase number of TPH immunoreactive neurons in the medial raphe but not in the dorsal raphe of the valproate-exposed group. Using the SERT ligand [123I]ADAM and small animal SPECT, valproate exposed animals showed an increase in the uptake of [123I]ADAM in the amygala. Furthermore, long term potentiation (LTP) was shown to be enhanced in the lateral amygdala at thalamic-amygdala synapses (Lin et al. 2013). Treatment of amygdala slices with the 5HT1A agonist 5-OH-DPAT reversed the excitatory/inhibitory imbalance in tissue from VPA-exposed animals, as measured by the miniature excitatory postsynaptic currents and paired pulse facilitation. Furthermore, chronic treatment with 5-OH-DPAT improved social deficits and fear extinction memory in the valproate exposed animals.
Case reports and epidemiological studies have linked autism risk to prenatal viral infections, such as cytomegalovirus, herpes simplex virus, rubella, measles, and mumps (Chess 1971; Deykin and MacMahon 1979; Ghaziuddin et al. 1992; Mason-Brothers et al. 1990; Yamashita et al. 2003). In a more recent epidemiologic study, Zerbo et al. (2013) showed no overall association between diagnoses of any maternal infection during pregnancy and ASD (Zerbo et al. 2013). However, this study did find an increased risk for ASD when maternal infection was diagnosed during a hospital admission. The risk was higher for bacterial infections. Maternal intrauterine inflammation resulting in immune dysfunction during development has been implicated in the development of neurodevelopmental disorders such as autism, schizophrenia, and cerebral palsy (Fatemi et al. 2008). A common link among these disorders appears to be the presence of activated microglia and evidence for immune dysregulation in the developing brain (Patterson 2009). Brain tissues of autistic patients were found to have increased number of activated microglia and astrocytes along with an increase in the levels of proinflammatory cytokines (Vargas et al. 2005). One link between maternal infection and intrauterine inflammation and fetal brain development may be the transport and metabolism of serotonin and its precursor tryptophan in the placenta. As kynurenine pathway enzyme IDO is induced by the cytokine interferon-γ, it is upregulated in the placenta with maternal infection (Mackler et al. 2003). Intrauterine infections in women are associated with upregulation of kynurenine pathway enzyme mRNA expression in the placenta (Manuelpillai et al. 2005). Increased metabolism of tryptophan by the kynurenine pathway at the placenta may result in abnormalities in fetal brain development because of decreased tryptophan available for serotonin synthesis and the neurotoxic effects of kynurenine metabolites (Stone 2001). ASD animal models of maternal inflammation have shown alterations in brain serotonin in the postnatal brains. Viral infections in the prenatal period have been associated with alterations in serotonin in the cerebellum of P14 mice (Winter et al. 2008). Maternal intrauterine endotoxin administration in a rabbit model results in microglial activation in the brain of the newborn rabbit (Kannan et al. 2007) and decreased serotonin synthesis in the cortex of the neonatal rabbit (Kannan et al. 2011). Kannan et al. (2011) demonstrated that maternal intrauterine endotoxin administration decreased multiple serotonergic markers in the offspring. Tryptophan metabolism to serotonin as measured by AMT PET in vivo, and serotonin levels in the frontal and parietal cortices measured in vitro, were significantly decreased following intrauterine endotoxin exposure. In addition to the decreased serotonin content, there was a loss of serotonin-immunoreactive terminals and decreased expression of 5HTT measured in the parietal sensory cortex of endotoxin-exposed. In contrast, serotonergic raphe nuclei cell bodies and TPH2-positive cortical fibers were intact in the endotoxin-treated kits. The authors concluded that the loss of serotonin-immunoreactive fibers in the cortex was due to loss of thalamic neurons and thalamocortical afferents that transiently express 5HTT to uptake and store serotonin during development. Diminished serotonin signaling during development due to intrauterine inflammatory mechanisms may result in defects in thalamocortical circuit formation. These results are significant because they demonstrate that maternal intrauterine inflammation can disrupt the serotonergic developmental regulation of thalamocortical innervation in somatosensory cortex of newborn rabbit kits.
9 Conclusions
Although a role for serotonin in ASD has been known for over 50 years, mechanisms by which serotonin may be altered in ASD are only now being elucidated. This new understanding of the role of serotonin in autism has been aided by technological advances, including the discovery of autism risk genes and the ability to model these genetic variants in animal models. Molecular and functional imaging techniques now offer insight into altered serotonin function in vivo in human brain. Understanding of how prenatal factors such as maternal inflammation, maternal antidepressant use, and maternal genetic factors such as MAOA variants may lead to new strategies for the prevention of ASD. Understanding how different genetic factors affect serotonin may lead to individualized pharmacotherapy targeting different aspects of serotonergic function. Finally, understanding how serotonergic function during development differs in ASD may guide treatments aimed at particular points during development.
References
Anderson GM, Freedman DX, Cohen DJ, Volkmar FR, Hoder EL, McPhedran P, Minderaa RB, Hansen CR, Young JG (1987) Whole blood serotonin in autistic and normal subjects. J Child Psychol Psychiatry 28:885–900
Anderson GM, Hertzig ME, McBride PA (2012) Brief report: platelet-poor plasma serotonin in autism. J Autism Dev Disord 42:1510–1514
Azmitia EC, Singh JS, Whitaker-Azmitia PM (2011a) Increased serotonin axons (immunoreactive to 5-HT transporter) in postmortem brains from young autism donors. Neuropharmacology 60:1347–1354
Azmitia EC, Singh JS, Hou XP, Wegiel J (2011b) Dystrophic serotonin axons in postmortem brains from young autism patients. Anat Rec, 294:1653–1662
Balkovetz DF, Tiruppathi C, Leibach FH, Mahesh VB, Ganapathy V (1989) Evidence for an imipramine-sensitive serotonin transporter in human placental brush-border membranes. J Biol Chem 264:2195–2198
Berger M, Gray JA, Roth BL (2009) The expanded biology of serotonin. Annu Rev Med 60:355–366.
Berman NEJ, Puri V, Chandrala S, Puri S, Macgregor R, Liverman CS, Klein RM (2006) Serotonin in trigeminal ganglia of female rodents: relevance to menstrual migraine. Headache 46:1230–1245
Beversdorf DQ, Nordgren RE, Bonab AA, Fischman AJ, Weise SB, Dougherty DD, Felopulos GJ, Zhou FC, Bauman ML (2012) 5-HT2 receptor distribution shown by [18F]setoperone PET in high-functioning autistic adults. J Neuropsychiatry Clin Neurosci 24:191–197
Bonnin A, Goeden N, Chen K, Wilson ML, King J, Shih JC, Blakely RD, Deneris ES, Levitt P (2011) A transient placental source of serotonin for the fetal forebrain. Nature 472:347–350
Bortolato M, Chen K, Shih JC (2008) Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev 60:1527–1533
Brodkin ES (2007) BALB/c mice: low sociability and other phenotypes that may be relevant to autism. Behav Brain Res 176:53–65
Bromley RL, Mawer GE, Briggs M, Cheyne F, Clayton-Smith J, García-Fiñana M, Kneen R, Lucas SB, Shallcross R, Baker GA; Liverpool and Manchester Neurodevelopment Group (2013) The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J Neurol Neurosurg Psychiatry 84:637–643
Cassel S, Carouge D, Gensburger C, Anglard P, Burgun C, Dietrichm JB, Aunis D, Zwiller J (2006) Fluoxetine and cocaine induce the epigenetic factors MeCP2 and MBD1 in adult rat brain. Mol Pharmacol 70:487–492
Chandana SR, Behen ME, Juhasz C, Muzik O, Rothermel RD, Mangner TJ, Chakraborty PK, Chugani HT, Chugani DC (2005) Significance of abnormalities in developmental trajectory and asymmetry of cortical serotonin synthesis in autism. Int J Dev Neurosci 23:171–182
Chantiluke K, Barrett N, Giampietro V, Brammer M, Simmons A, Murphy DG, Rubia K (2014) Inverse effect of fluoxetine on medial prefrontal cortex activation during reward reversal in ADHD and autism. Cereb Cortex [Epub ahead of print]
Chess S (1971) Autism in children with congenital rubella. Journal of Autism and Childhood Schizophrenia 1:33–47
Christenson JG, Dairman W, Udenfriend S (1972) On the identity of DOPA decarboxylase and 5-hydroxytryptophan decarboxylase (immunological titration-aromatic L-amino acid decarboxylase-serotonin-dopamine-norepinephrine). Proc Nat Acad Sci U S A 69:343–347
Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, Vestergaard M (2013) Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 309:1696–1703
Chugani DC, Muzik O, Rothermel R, Behen M, Chakraborty P, Mangner T, da Silva EA, Chugani HT (1977) Altered serotonin synthesis in the dentatothalamo-cortical pathway in autistic boys. Ann Neurol 14:666–669
Chugani DC, Muzik O, Behen M, Rothermel R, Janisse JJ, Lee J, Chugani HT (1999) Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann Neurol 45:287–295
Cohen IL, Liu X, Schutz C, White BN, Jenkins EC, Brown WT, Holden JJ (2003) Association of autism severity with a monoamine oxidase a functional polymorphism. Clin Genet 64:190–197
Cook EH Jr Charak DA Arida J Spohn JA Roizen NJ Leventhal BL (1994) Depressive and obsessive–compulsive symptoms in hyperserotonemic parents of children with autistic disorder. Psychiatry Res 52:25–33
Cook EH Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E (1997) Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet 60:928–934
Cook EH Jr Leventhal BL Heller W Metz J Wainwright M Freedman DX (1990) Autistic children and their first-degree relatives: relationships between serotonin and norepinephrine levels and intelligence. J Neuropsychiatry Clin Neurosci 2:268–274
Coon H, Dunn D, Lainhart J, Miller J, Hamil C, Battaglia A, Tancredi R, Leppert MF, Weiss R, McMahon W (2005) Possible association between autism and variants in the brain-expressed tryptophan hydroxylase gene (TPH2). Am J Med Genet B Neuropsychiatr Genet 135B:42–46
Correa RR, Barrilari SE, Guimaraes CS, Rossi e Silva RC, Olegario JG, Cavellani CL, Oliveira FA, Salge AK, Teixeira VP, Castro EC (2009) Expression of the melatonin receptor and tryptophan hydroxylase in placentas of the fetus with intra-uterine stress. Eur J Obstet Gynecol Reprod Biol 147:234–236
Cote F, Fligny C, Bayard E, Launay JM, Gershon MD, Mallet J, Vodjdani G (2007) Maternal serotonin is crucial for murine embryonic development. Proc Natl Acad Sci U S A 104:329–334
Coutinho AM, Sousa I, Martins M, Correia C, Morgadinho T, Bento C, Marques C, Ataíde A, Miguel TS, Moore, JH, Oliveira G, Vicente AM (2007) Evidence for epistasis between SLC6A4 and ITGB3 in autism etiology and in the determination of platelet serotonin levels. Hum Genet 121:243–256
Croen LA, Grether JK, Yoshida CK, Odouli R, Hendrick V (2011) Antidepressant use during pregnancy and childhood autism spectrum disorders. Arch Gen Psychiatry 68:1104–1112
Daly EM, Deeley Q, Ecker C, Craig M, Hallahan B, Murphy C, Johnston P, Spain D, Gillan N, Brammer M, Giampietro V, Lamar M, Page L, Toal F, Cleare A, Surguladze S, Murphy DG (2012) Serotonin and the neural processing of facial emotions in adults with autism: an fMRI study using acute tryptophan depletion. Arch Gen Psychiatry 69:1003–1013
Davis LK, Hazlett HC, Librant AL, Nopoulos P, Sheffield VC, Piven J, Wassink TH (2008) Cortical enlargement in autism is associated with a functional VNTR in the monoamine oxidase a gene. Am J Med Genet B Neuropsychiatr Genet 147B:1145–1151
Devlin B, Cook EH Jr, Coon H, Dawson G, Grigorenko EL, McMahon W, Minshew N, Pauls D, Smith, M, Spence MA, Rodier PM, Stodgell C, Schellenberg GD (2005) CPEA Genetics Network Autism and the serotonin transporter, the long and short of it. Mol Psychiatry 10:1110–1116
Deykin EY, MacMahon B (1979) Viral exposure and autism. Am J Epidemiol 109:628–638
Diksic M, Nagahiro S, Sourkes TL, Yamamoto YL (1990) A new method to measure brain serotonin synthesis in vivo. I. Theory and basic data for a biological model. J Cereb Blood Flow Metab 9:1–12
Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, Dartevelle P, Humbaert M, Simonneau G, Hanoun N, Saurini F, Hamon M, Adnot S (2006) Cross talk between endothelial and smooth muscle cells in pulmonary hypertension. Circulation 113:1857–1864
Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, Smee DF, Pearce DA, Winter C, Sohr R, Juckel G (2008) Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res 99:56–70
Fernstrom JD, Wurtman RJ (1971) Brain serotonin content: physiological dependence on plasma tryptophan levels. Science 173:149–151
Gershon MD, Tack J (2007) The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology 132:397–414
Ghaziuddin M, Tsai LY, Eilers L, Ghaziuddin N (1992) Brief report: Autism and herpes simplex encephalitis. J Autism Dev Disord 22:107–113
Girgis RR, Slifstein M, Xu X, Frankle WG, Anagnostou E, Wasserman S, Pepa L, Kolevzon A, Abi-Dargham A, Laruelle M, Hollander E (2011) The 5-HT(2A) receptor and serotonin transporter in Asperger’s disorder: A PET study with [¹¹C]MDL 100907 and [¹¹C]DASB. Psychiatry Res 194:230–234
Goldberg J, Anderson GM, Zwaigenbaum L, Hall GB, Nahmias C, Thompson A, Szatmari P (2009) Cortical serotonin type-2 receptor density in parents of children with autism spectrum disorders. J Autism Dev Disord 2009:97–104
Hagerman R, Hoem G, Hagerman P (2010) Fragile X and autism: Intertwined at the molecular level leading to targeted treatments. Mol Autism 1:12
Hamon M, Bourgoin S, Artaud FEl, Mestikawy S (1981) The respective roles of tryptophan uptake and tryptophan hydroxylase in the regulation of serotonin synthesis in the central nervous system. J Physiol 77:269–279
Hoshino Y, Yamamoto T, Kaneko M, Tachibana R, Watanabe M, Ono Y, Kumashiro H (1984) Blood serotonin and free tryptophan concentration in autistic children. Neuropsychobiol 11:22–27
Hviid A, Melbye M, Pasternak B (2013) Use of selective serotonin reuptake inhibitors during pregnancy and risk of autism. N Engl J Med 369:2406–2415
Ide S, Itoh M, Goto Y (2005) Defect in normal developmental increase of the brain biogenic amine concentration in the mecp2-null mouse. Neurosci Lett 386:14–17
Ikeda K, Tojo K, Otsubo C, Udagawa T, Kumazawa K, Ishikawa M, Tokudome G, Hosoya T, Tajima N, Claycomb WC, Nakao K, Kawamura M (2005) 5-hydroxytryptamine synthesis in HL-1 cells and neonatal rat cardiocytes. Biochem Biophys Res Commun 328:522–525
Jones MB, Palmour RM, Zwaigenbaum L, Szatmari P (2004) Modifier effects in autism at the MAO-A and DBH loci. Am J Med Genet B Neuropsychiatr Genet 126:58–65
Kane MJ, Angoa-Peréz M, Briggs DI, Sykes CE, Francescutti DM, Rosenberg DR, Kuhn DM (2012) Mice genetically depleted of brain serotonin display social impairments, communication deficits and repetitive behaviors: possible relevance to autism. PLoS One 7:e48975
Kannan S, Saadani-Makki F, Muzik O, Chakraborty P, Mangner TJ, Janisse J, Romero R, Chugani DC (2007) Microglial activation in perinatal rabbit brain induced by intrauterine inflammation: detection with 11C-(R)-PK11195 and small-animal PET. J Nucl Med 48:946–954
Kannan S, Saadani-Makki F, Balakrishnan B, Dai H, Chakraborty PK, Janisse J, Muzik O, Romero R, Chugani DC (2011) Decreased cortical serotonin in neonatal rabbits exposed to endotoxin in utero. J Cereb Blood Flow Metab 31:738–749
Kerr TM, Muller CL, Miah M, Jetter CS, Pfeiffer R, Shah C, Baganz N, Anderson GM, Crawley JN, Sutcliffe JS, Blakely RD, Veenstra-Vanderweele J (2013) Genetic background modulates phenotypes of serotonin transporter Ala56 knock-in mice. Mol Autism 4:35
Kim DK, Tolliver TJ, Huang S J, Martin BJ, Andrews AM, Wichems C, Holmes A, Lesch KP, Murphy DL (2005) Altered serotonin synthesis, turnover and dynamic regulation in multiple brain regions of mice lacking the serotonin transporter. Neuropharmacol 49:798–810
Kudo Y, Boyd CA (2002) Human placental amino acid transporter genes: expression and function. Reproduction 124:593–600
Leboyer M, Philippe A, Bouvard M, Guilloud-Bataille M, Bondoux D, Tabuteau F, Feingold J, Mouren-Simeoni MC, Launay JM (1999) Whole blood serotonin and plasma beta-endorphin in autistic probands and their first-degree relatives. Biol Psychiatry 45:158–163
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, Benjamin J Müller, CR Hamer DH, Murphy DL (1996) Association of anxiety-related with a polymorphism in the serotonin transporter gene regulatory region. Science 274:1527–1531
Leventhal BL, Cook Jr, EH, Morford M, Ravitz A, Freedman DX (1990) Relationships of whole blood serotonin and plasma norepinephrine within families. J Autism Dev Disord, 20:499–511
Ligam P, Manuelpillai U, Wallace EM, Walker D (2005) Localisation of indoleamine 2,3-dioxygenase and kynurenine hydroxylase in the human placenta and decidua: implications for role of the kynurenine pathway in pregnancy. Placenta 26:498–504
Lin HC, Gean PW, Wang CC, Chan YH, Chen PS (2013) The amygdala excitatory/inhibitory balance in a valproate-induced rat autism model. PLoS One 8:e55248
Mackler AM, Barber EM, Takikawa O, Pollard JW (2003) Indoleamine 2,3-dioxygenase is regulated by IFN-gamma in the mouse placenta during Listeria monocytogenes infection. J Immunol 170:823–830
Makkonen I, Riikonen R, Kokki H, Airaksinen MM, Kuikka JT (2008) Serotonin and dopamine transporter binding in children with autism determined by SPECT. Dev Med Child Neurol 50:593–597
Manuelpillai U, Ligam P, Smythe G, Wallace EM, Hirst J, Walker DW (2005) Identification of kynurenine pathway enzyme mRNAs and metabolites in human placenta: up-regulation by inflammatory stimuli and with clinical infection. Am J Obstet Gynecol 192:280–288
Mason-Brothers A, Ritvo ER, Pingree C, Petersen PB, Jenson WR, McMahon WM, Freeman BJ, Jorde LB, Spencer MJ, Mo A (1990) The UCLA-University of Utah epidemiologic survey of autism: prenatal, perinatal, and postnatal factors. Pediatrics 86:514–519
Matsuda M, Imaoka T, Vomachka AJ, Gudelsky GA, Hou Z, Mistry M, Bailey JP, Nieport KM, Walther DJ, Bader M, & Horseman ND (2004) Serotonin regulates mammary gland development via an autocrine–paracrine loop. Development Cell 6:193–203
Moja EA, Stoff DM, Gessa GL, Castoldi D, Assereto R, Tofanetti O (1988) Decrease in plasma tryptophan after tryptophan-free amino acid mixtures in man. Life Sci 42:1551–1556
Moja EA, Cipolla P, Castoldi D, Tofanetti O (1989) Dose-response decrease in plasma tryptophan and in brain tryptophan and serotonin after tryptophan-free amino acid mixtures in rats. Life Sci 44:971–976
Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, Brown C, Mellor AL (1998) Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 281:1191–1193
Murphy DG, Daly E, Schmitz N, Toal F, Murphy K, Curran S, Erlandsson K, Eersels J, Kerwin R, Ell P, Travis M (2006) Cortical serotonin 5-HT2A receptor binding and social communication in adults with Asperger’s syndrome: an in vivo SPECT study. Am J Psychiatry 163:934–936.
Nabi R, Serajee FJ, Chugani DC, Zhong H, Huq AH (2004) Association of tryptophan 2,3 dioxygenase gene polymorphism with autism. Am J Med Genet B Neuropsychiatr Genet 125:63–68
Nakamura K, Sekine Y, Ouchi Y, Tsujii M, Yoshikawa E, Futatsubashi M, Tsuchiya KJ, Sugihara G, Iwata Y, Suzuki K, Matsuzaki H, Suda S, Sugiyama T, Takei N, Mori N (2010) Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch Gen Psychiatry 67:59–68
Ni W, Watts SW (2006) 5-hydroxytryptamine in the cardiovascular systems: focus on the serotonin transporter (SERT). Clin Exp Pharmacol Physiol 33:575–583
Ni W, Geddes TJ, Priestley JR, Szasz T, Kuhn DM, Watts SW (2008) The existence of a local 5-hydroxytryptaminergic system in peripheral arteries. Br J Pharmacol 154:663–674
Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M, Renner EL, Clavien PA (2007) Serotonin mediates oxidative stress and mitochondrial toxicity in a murine model of nonalcoholic steatohepatitis. Gastroenterology 133:608–618
Oblak A, Gibbs TT, Blatt GJ (2013) Reduced serotonin receptor subtypes in a limbic and a neocortical region in autism. Autism Res 6:571–583
Oritz-Alvarado R, Guzmán-Quevedo O, Meracado-Camargo R, Haertle T, Vignes C, Bolaños-Jiménez F (2006) Expression of tryptophan hydroxylase in developing mouse taste papillae. FEBS Letters 580:5371–5376
Pardridge, WM (1977) Kinetics of competitive inhibition of neutral amino acid transport across the blood-brain barrier. J Neurochem 28:103–108
Patterson PH (2009) Immune involvement in schizophrenia and autism: etiology, pathology and animal models. Behav Brain Res 204:313–321
Piven J, Tsai GC, Nehme E, Coyle JT, Chase GA, Folstein SE (1991) Platelet serotonin, a possible marker for familial autism. J Autism Dev Disord 21:51–59
Sakowski SA, Geddes TJ, Thomas DM, Levi E, Hatfield JS, Kuhn DM (2006) Differential tissue distribution of tryptophan hydroxylase isoforms 1 and 2 as revealed with monospecific antibodies. Brain Res 1085:11–18
Samaco RC, Mandel-Brehm C, Chao HT, Ward CS, Fyffe-Maricich SL, Ren J, Hyland K, Thaller C, Maricich SM, Humphreys P, Greer JJ, Percy A, Glaze DG, Zoghbi HY, Neul JL (2009) Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci U S A 106:21966–21971
Schain RJ, Freedman DX (1961) Studies on 5-hydroxyindole metabolism in autism and other mentally retarded children. J Pediatr 59:315–320
Siesser WB, Zhang X, Jacobsen JP, Sotnikova TD, Gainetdinov RR, Caron MG (2010) Tryptophan hydroxylase 2 genotype determines brain serotonin synthesis but not tissue content in C57Bl/6 and BALB/c congenic mice. Neurosci Lett 481:6–11
Slominski A, Pisarchik A, Semak I, Sweatman T, Worstman J, Szczesniewski A, Slugocki G, McNulty J, Kauser S, Tobin DJ (2002) Serotoninergic and melatoninergic systems are fully expressed in human skin. FASEB J 16:896–898
Smith QR, Monna S, Aoyagi M, & Rapoport SI (1987) Kinetics of neutral amino acid transport across the blood-brain barrier. J Neurochem 49:1651–1658
Smit-Rigter LA, Noorlander CW, von Oerthel L, Chameau P, Smidt MP, van Hooft JA (2012) Prenatal fluoxetine exposure induces life-long serotonin 5-HT3 receptor-dependent cortical abnormalities and anxiety-like behaviour. Neuropharmacology 62:865–870
Stone TW (2001) Endogenous neurotoxins from tryptophan. Toxicon 39:61–73
Stull MA, Pai V, Vomachka AJ, Marshall AM, Joacb GA, Horseman ND (2007) Mammary gland homeostasis employs serotonergic regulation of epithelial tight junctions. Proc Natl Acad Sci 104:16708–16713
Tamada K, Tomonaga S, Hatanaka F, Nakai N, Takao K, Miyakawa T, Nakatani J, Takumi T (2010) Decreased exploratory activity in a mouse model of 15q duplication syndrome; implications for disturbance of serotonin signaling. PLoS One 5e:15126
Tassone F, Qi L, Zhang W, Hansen RL, Pessah IN, Hertz-Picciotto I (2011) MAOA, DBH and SLC6A4 variants in CHARGE: A case control study of autism spectrum disorders. Autism Res 4:250–261
Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA (2005) Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol 57:67–81
Veenstra-VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR, Cohen J, Mannangatti P, Jessen T, Thompson BJ, Ye R, Kerr TM, Carneiro AM, Crawley JN, Sanders-Bush E, McMahon DG, Ramamoorthy S, Daws LC, Sutcliffe JS, Blakely RD (2012) Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci U S A 109:5469–5474
Verma D, Chakraborti B, Karmakar A, Bandyopadhyay T, Singh AS, Sinha S, Chatterjee A, Ghosh S, Mohanakumar KP, Mukhopadhyay K, Rajamma U (2013) Sexual dimorphic effect in the genetic association of monoamine oxidase A (MAOA) markers with autism spectrum disorder. Prog Neuropsychopharmacol Biol Psychiatry 50C:11–20
Walther DJ, Peter JU, Winter S, Höltje M, Paulmann N, Grohmann M, Vowinckel J, Alamo-Bethencourt V, Wilhelm CS, Ahnert-Hilger G, Bader M (2003) Serotonylation of small GTPases is a signal transduction pathway that triggers platelet alpha-granule release. Cell 115:851–862
Walther DJ, Stahlberg S, Vowinckel J (2011) Novel roles for biogenic monoamines: from monoamines in transglutaminase-mediated post-translational protein modification to monoaminylation deregulation diseases. FEBS J 278:4740–4755
Wang CC, Lin HC, Chan YH, Gean PW, Yang YK, Chen PS (2013) 5-HT1A-receptor agonist modified amygdala activity and amygdala-associated social behavior in a valproate-induced rat autism model. Int J Neuropsychopharmacol 16:2027–2039
Wassink TH, Hazlett HC, Epping EA, Arndt S, Dager SR, Schellenberg GD, Dawson, G, Piven J (2007) Cerebral cortical gray matter overgrowth and functional variation of the serotonin transporter gene in autism. Arch Gen Psychiatry 64:709–717
Weiss LA, Ober C, Cook EH Jr (2006a) ITGB3 shows genetic and expression interaction with SLC6A4. Hum Genet 120:93–100
Weiss LA, Kosova G, Delahanty RJ, Jiang L, Cook EH, Ober C, Sutcliffe JS (2006b) Variation in ITGB3 is associated with whole-blood serotonin level and autism susceptibility. Eur J Hum Genet 14:923–931
Whyte A, Jessen T, Varney S, Carneiro AM (2014) Serotonin transporter and integrin beta 3 genes interact to modulate serotonin uptake in mouse brain. Neurochem Int 73:122–126
Winter C, Reutiman TJ, Folsom TD, Sohr R, Wolf RJ, Juckel G, Fatemi SH (2008) Dopamine and serotonin levels following prenatal viral infection in mouse—implications for psychiatric disorders such as schizophrenia and autism. Eur Neuropsychopharmacol 18:712–716
Yamashita, Y, Fujimoto, C, Nakajima E, Isagai, T, Matsuishi, T (2003) Possible association between congenital cytomegalovirus infection and autistic disorder. J Autism Dev Disord 33:455–459
Zerbo O, Qian Y, Yoshida C, Grether JK, Van de Water J, Croen LA (2013) Maternal infection during pregnancy and autism spectrum disorders. J Autism Dev Disord [Epub ahead of print]
Zhang X, Beaulieu, JM, Sotnikova TD, Gainetdinov RR, Caron MG (2004) Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science 305:217
Zhang YQ, Friedman DB, Wang Z, Woodruff E 3rd, Pan L, O’Donnell J, Broadie K (2005) Protein expression profiling of the drosophila fragile X mutant brain reveals up-regulation of monoamine synthesis. Mol Cell Proteomics 4:278–290
Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, Law P, Qiu S, Lord C, Sebat, J, Ye K, Wigler M (2007) A unified genetic theory for sporadic and inherited autism. Proc Nat Acad Sci U S A 104:12831–12836
Zoghbi HY, Milstien S, Butler IJ, Smith EO, Kaufman S, Glaze DG, Percy AK (1989) Cerebrospinal fluid biogenic amines and biopterin in Rett syndrome. Ann Neurol 25:56–60
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Benza, N., Chugani, D. (2015). Serotonin in Autism Spectrum Disorder: Insights from Human Studies and Animal Models. In: Fatemi, S. (eds) The Molecular Basis of Autism. Contemporary Clinical Neuroscience. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2190-4_13
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2190-4_13
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2189-8
Online ISBN: 978-1-4939-2190-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)