
Abstract
Recent advances in genomic and transcriptomic technologies have revolutionized our knowledge of the genetic and molecular basis of pediatric brain tumors. These discoveries have pinpointed novel genes and pathways, identified distinct molecular subgroups, and have led to developments of new mouse models. This chapter details our current understanding of the basic science of pediatric brain tumors, providing an outline of disease mechanisms and potential targets for molecular therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Medulloblastoma
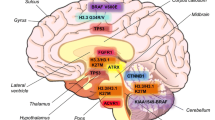
A small fraction of medulloblastomas (MBs) have inherited disorders with germline mutations, which have revealed the first pathways that underlie MB tumorigenesis. It is known that prominent tumor suppressor genes such as TP53 and APC, when germline mutated in patients with Li-Fraumeni syndrome and Turcot syndrome, respectively, predispose to MB. Furthermore, the identification of Gorlin’s syndrome, in patients harboring mutations in the patched-1 (PTCH1) gene, has led to the identification of aberrant sonic hedgehog signaling (SHH) in a subset of MBs (Fig. 7.1).

Summary of molecular subgroups and recurrent genomic alterations identified in pediatric brain tumors
Integrated genomic studies have revealed that MB comprises at least four molecular variants, which are genetically and clinically distinct. These four subgroups are termed WNT, Sonic Hedgehog (SHH), Group 3, and Group 4 [1] have significant prognostic value, and can be further subdivided into additional relevant molecular subtypes. The delineation of these four core subgroups underscores the heterogeneity that exists between MB patients. The WNT subgroup is characterized by activation of the WNT pathway, which commonly harbors mutations in β-catenin (CTNNB1). Patients with WNT activated tumors tend to have a favorable prognosis and occur primarily outside the infant age group. The SHH subgroup is characterized by activation of the Sonic Hedgehog (SHH) pathway and is more common in infants with desmoplastic tumors and in adults. Group 3 MBs have a poor prognosis and are commonly associated with metastatic disease. MYC amplification is common in Group 3 MBs, and survival in these patients is poor. Group 4 MBs have an intermediate prognosis and are commonly associated with isochromosome 17q and MYCN amplification.
There have been a number of chromosomal alterations reported in MB. The most commonly reported cytogenetic abnormality is isochromosome 17q (i17q), involving the loss of chromosome 17p and gain of 17q, which is found in 30–40 % of all primary MB tumors. This is also a genomic feature of the Group 4 subgroup, which is observed in more than 80 % of cases. Other less common aberrations include gains on chromosomes 1q, 3q, 7, and 17q, as well as loss on chromosome 5q, 9q, 10q, 11, 17p, and 22 [2].
Recent publications highlight the importance of examining the mutational landscape of MB according to subgroup affiliation [3, 4]. WNT subgroup MBs remain the most genomically balanced of the four subgroups without any focal recurrent somatic copy number alterations (SCNAs). The most common mutation observed in this subset is in CTNNB1, which highlights the important role of WNT signaling in this subgroup, and less frequently in DDX3X. Many groups have sought to identify the cell of origin for this subgroup. It has been suggested to be the progenitor cells of the lower rhombic lip. Further, a mouse model harboring activated Ctnnb1 in the Blbp expressing radial glial cells has been shown to generate MBs that are characteristic of WNT tumors [5].
SHH subgroup is the best characterized of the four subgroups, with the distinctive feature of activated SHH signaling. Somatic mutations targeting the SHH receptor PTCH1 and downstream genes, such as SUFU, are found exclusively in this subgroup. SCNAs targeting the PI3K signaling cascade have also been known to be aberrant in this subgroup. Most animal models of SHH MBs involve the inactivation of patched1 in either the cerebellar granule neuron precursors (CGNP; marked by Atoh1) or neural stem cells (NSC; marked by GFAP) [6].
Group 3 and 4 MBs are currently the least understood. Group 3 MB is associated with the worst prognosis and characterized by high-level amplification of the proto-oncogene MYC. Research examining somatic mutations have identified dysregulation of the epigenome in both Group 3 and 4 MBs. These events, although occurring across all MBs, are enriched in chromatin-associated genes such as MLL2, MLL3, SMARCA4, and KDM6A. These newly identified mutations point to the importance of the chromatin structure in MB pathogenesis. Efforts have also identified novel fusion proteins such as PVT1-MYC and novel mechanisms such as the tandem duplication of SNCAIP in Group 3 and 4 MBs, respectively. The biological and clinical relevance of these novel pathogenic mechanisms will need to be further studied. Although no transgenic models of Group 3 and 4 diseases exist, several orthotopic transplantation models are currently being studied. The activation of Myc with p53 inactivation generates medulloblastomas with characteristic Group 3 features [7].
Pediatric High-Grade Glioma
Pediatric Glioblastoma
Whole-exome sequencing of pediatric glioblastoma (GBM) cases has revealed somatic recurrent mutations in a gene called H3F3A, which encodes a replication-independent histone variant H3.3 [2] (Fig. 7.1). Heterozygous mutations in H3F3A are present in 31 % of pediatric GBMs and result in amino acid substitutions within the N-terminal histone tail, specifically a lysine to methionine (K27M), a glycine to arginine (G34R), or a glycine to valine (G34V) substitution [2]. These mutations, which occur specifically in pediatric GBMs, and often with TP53 mutations (54 % of cases), are situated at sites which are important for post-translational modification of histone 3 (H3) and regulation of global chromatin structure. Recurrent mutations in ATRX and DAXX have also been reported in 31 % of pediatric GBMs and present always in tumors harboring a G34R/V mutation [2]. The ATRX and DAXX proteins are important for H3.3 incorporation at peri-centrometic heterochromatin and telomeres [2]. Together somatic mutations in the H3.3-ATRX-DAXX chromatin remodeling pathway have been identified in 44 % of pediatric GBMs [2]. Epigenomic differences between subsets of pediatric GBM patients were demonstrated in a study examining DNA methylation signatures in a series of 59 childhood and 77 adult GBM patients [8]. Here they identified robust, epigenetically distinct subgroups defined by GBM mutations and gene expression-defined classes. The subgroups were classified as: (1) IDH1 mutated (adults), (2) H3.3-K27 mutated, (3) H3.3-G34 mutated, (4) RTKI (PDGFRα amplified, proneural), (5) mesenchymal, and (6) RTKII (classic). Copy number events also defined subgroups of pediatric GBM, specifically PDGFRα amplifications in the RTKI subgroup, whole chromosome 7 gains, chromosome 10 loss, CDKN2A homozygous deletions, and EGFR amplifications in RTKII pediatric GBMs [8]. Additional copy number events have been observed in comprehensive copy number studies of high-grade gliomas, including chromosome 8p12 loss in ~16 % of cases, encompassing a potential tumor suppressor gene, ADAM3A, MYCN amplifications (5 %), and chromosome 1q gain [9].
Diffuse Intrinsic Pontine Glioma
The vast majority (up to 78 %) of diffuse intrinsic pontine gliomas (DIPGs) harbors heterozygous H3.3-K27M mutations [10] (Fig. 7.1). TP53 mutations have also been observed in up to 77 % of patients and are often concurrent with H3F3A mutations, PDGFRA gene amplifications, and MYC-PVT1 gene fusions. ATRX mutations have also been reported in older DIPG patients, albeit at lower frequency (9 %), further highlighting aberrant chromatin structure in DIPG patients. The copy number landscape of DIPGs pinpoints other pathways relevant to disease formation, namely the PI3K pathway which is affected in ~47 % of DIPGs, involving PDGFRa, MET, IGF1R, ERRB4, EGFR, KRAS, AKT1, AKT3, and PIK3CA focal gains [11]. Gross copy number events have also reported to be enriched in DIPGs, specifically gains of chromosomes 2q, 8q, and 9q and losses of 16q, 17p, and 20p [11].
Pediatric Low-Grade Glioma
Non-diffuse Low-Grade Glioma: Pilocytic Astrocytomas
Spontaneous pilocytic astrocytomas (PAs) occur typically in the cerebellum, and in the absence of NF1 mutations. The most common genetic alteration in spontaneous PAs results in increased activity of the MAPK pathway, through a tandem duplication event on chromosome 7q34, which forms an in-frame fusion of KIAA1549 with BRAF [12] (Fig. 7.1). Adding to this, genome sequencing studies of low-grade gliomas have shown that the MAPK pathway is affected in nearly all tumors and that PAs may represent a single pathway-driven disease [13]. Several other, albeit less frequent, genetic alterations convergent upon BRAF activation have been reported including FAM131B-BRAF, RNF130-BRAF, CLCN6-BRAF, MKRN1-BRAF, and GNAI1-BRAF fusions, all of which result in N-terminal loss of the BRAF regulatory region [13]. The significance of BRAF alterations is further highlighted by the presence of somatic mutations. These findings are supported functionally, in which PAs are generated by ectopic activation of BRAF in murine neural progenitor cells [14]. Recurrent, somatic, and activating mutations in PAs have also been identified at lower frequencies in other genes such as KRAS, FGFR1, PTPN11, and NTRK2 fusions, however all of which lead to downstream MAPK activation [13].
Diffuse Low-Grade Glioma: Diffuse Grade II Astrocytomas, Ganglioglioma, Angiocentric Glioma, and Pleomorphic Xanthoastrocytoma
Diffuse low-grade gliomas are also affected by BRAF alterations; however, these occur mostly in the setting of BRAF-V600E mutations [15]. In the case of diffuse Grade II gliomas, recurrent amplifications of MYC and intragenic duplications of FGFR1 have been reported, and shown to be largely mutually exclusive [16]. Copy number profiling in diffuse Grade II gliomas has identified other candidates, such as a partial duplication of the MYBL1 transcription factor in 28 % of cases, which results in loss of its C-terminus negative regulatory domain [17]. In the same pathway, MYB amplifications have also been observed at lower frequency, in addition to deletion-truncation breakpoints in the regulatory terminus of MYB, seen preferentially in angiocentric gliomas [17]. Loss of chromosome 9, encompassing the CDKN2A/p14ARF/CDKN2B locus, has been reported in ~50 % of pleomorphic xanthoastrocytomas, along with less frequent loss of chromosome 17 [18].
Desmoplastic Infantile Astrocytomas/Ganglioglioma
Desmoplastic infantile astrocytomas (DIA)/gangliogliomas (DIG) displayed only a few nonrecurrent genomic imbalances or normal karyotypes. Only loss of chromosome 9p and 22q was recurrently observed in a limited number of studies to date [19]. Characteristic genomic imbalances were not observed when DIA were compared with DIG [19]. BRAF V600E mutation, EGFR and MYCN amplification have been described in single cases.
Central Nervous System Germ Cell Tumors
Several studies have investigated cytogenetic alterations in central nervous system germ cell tumors (CNS-GCTs). A study of 15 malignant CNS-GCTs revealed recurrent gains of 12p12 which is also commonly amplified in adult testicular germ cell tumors [20]. Recurrent gains of 1q and 8q and recurrent losses on chromosome 11, 18, and 13 were also detected. The genomic alterations identified in this series were almost identical to those found in gonadal and extragonadal germ cell tumors. Moreover, there were no differences in the cytogenetic profiles of germinomas compared to non-germinomatous CNS-GCT. This suggests strongly that the pathogenesis of CNS-GCTs is similar to systemic GCTs. At a transcriptional level, there are several differences between germinomas and non-germinomatous germ cell tumors. Genes responsible for self-renewal (OCT4, NANOG, and KLF4) and immune response are more highly expressed in germinomas whereas genes involved in neuronal differentiation, Wnt/β-catenin pathway, invasiveness, and epithelial-mesenchymal transition are more commonly observed in malignant non-germinomatous germ cell tumors. The transcriptional profiles of non-germinomatous germ cell tumors closely resemble the profiles observed in embryonic stem cells consistent with their more undifferentiated nature.
Craniopharyngioma
Craniopharyngiomas are thought to arise from squamous-cell rests along the path of the primitive craniopharyngeal duct and adenohypophysis. The adenohypophysis arises from Rathke’s pouch. As such it is believed that these squamous-cell rests represent the cell of origin for craniopharyngioma, and it is generally felt that adamantinomatous craniopharyngioma represents a developmental anomaly. The rare papillary histological variant more common in adults appears to arise from the adenohypophysis; however, this remains to be confirmed. Recent studies have shown activating mutations in exon 3 of the β-catenin gene (CTNNB1) to be common in adamantinomatous craniopharyngioma suggesting a likely role for Wnt signaling in the pathogenesis of craniopharyngioma [21].
Central Nervous System Primitive Neuroectodermal Tumors
Primitive neuroectodermal tumors of the central nervous system (CNS-PNET) are a heterogeneous group of pediatric neoplasms composed of poorly differentiated neuroepithelial cells with varying degrees of divergent neural, astrocytic, and ependymal differentiation. Using global profiling, Picard et al. (2012) identified that CNS-PNETs comprise three distinct molecular groups: primitive-neural (Group 1), oligoneural (Group 2), and mesenchymal (Group 3) [22] (Fig. 7.1). Group 1 tumors are enriched in primitive-neural genes (CD133, CRABP1, LIN28, and ASCL1) and display activation of SHH and WNT signaling. Group 2 tumors are composed of genes with roles in oligoneural differentiation (OLIG1/2, SOX8/10, and BCAN) and exhibit down-regulation of SHH components. Lastly, Group 3 tumors comprise genes involved in epithelial and mesenchymal differentiation (COL1A2, COL5A, FOXJ1, and MSX1) and display up-regulation of genes involved in TGF-β and PTEN signaling. Copy number analyses reveal that Group 2 tumors have frequent gains of chromosome 8p, 13, and 20, whereas Group 3 have frequent loss of chromosome 14. Also Group 2 and 3 tumors have frequent chromosome 9p loss centered on the CDKN2A/2B locus. In 2000, Eberhart et al. [23] described a new CNS-PNET variant (termed “embryonal tumor with abundant neuropil and true rosettes” or ETANTR), which, based on gene expression profiling, are subgroups with Group 1 CNS-PNETs [22, 24]. Hallmark cytogenetic features of ETANTRs include frequent gains of chromosome 2 and 3, and focal amplification of an miRNA amplicon on chr19q13.41, which encompasses the oncogenic C19MC miRNA cluster [24]. Li et al. (2009) identified that chr19q13.41 amplification characterizes CNS-PNET variants labeled as ETANTRs, medulloepithelioma, supratentorial PNET with ependymal differentiation, and ependymoblastoma, suggesting that these tumors represent closely related molecular entities [24]. Although the mechanisms by which C19MC miRNAs mediate oncogenesis remain unclear, these miRNAs are implicated in cell survival, transformation, activation of noncanonical WNT-JNK2 signaling, and inhibition of differentiation of human neural stem cells [25]. Furthermore, ETANTRs are also distinguished by the presence of distinct primitive markers, including the RNA binding protein, Lin28 [22].
Ependymoma
Using gene expression profiling, ependymomas have been divided into three principal molecular subgroups, which are separated largely according to anatomical location: (1) supratentorial (ST), (2) posterior fossa (PF), and (3) spinal cord [26]. These three subgroups have been further divided into molecularly and biologically distinct subtypes of ST and PF ependymoma as defined by diverse clinical and genomic features [27–29] (Fig. 7.1). In the case of PF ependymoma, three studies have independently reported the existence of two principle subgroups of disease [27–29]. These PF subgroups termed Group A and B are transcriptionally, clinically, and biologically distinct entities. Group A PF ependymomas occur in young patients, invade laterally along the cerebellar-pontine angle, and are associated with increased tumor recurrence and decreased survival. Conversely, Group B PF ependymomas occur in older patients, grow along the midline, and are associated with a favorable prognosis. In addition, to the clinical differences, Group A and B are delineated by distinct genomic alterations. Group A PF ependymomas harbor chromosome 1q gain, which has been reported as a poor prognosis marker of ependymoma, but are largely characterized by balanced genomes [29]. In contrast, Group B ependymomas are defined by increased genomic instability as evidenced by increased numerical chromosome gains and losses. Group A ependymomas are characterized largely by pathways involved in angiogenesis (HIF-1α signaling, VEGF pathway), PDGF signaling, MAPK signaling, EGFR signaling, TGF-β signaling, tyrosine-receptor kinase signaling, RAS signaling, and integrin/ECM signaling, while Group B ependymomas are overrepresented by pathways involving ciliogenesis and microtubule assembly.
While these subgroup gene signatures may represent unique tumorigenic pathways, Taylor et al. (2005) proposed that these were potential marks of anatomically distinct cells of origin giving rise to different subgroups of ependymoma [26]. They suggested that ependymoma might originate from radial glia, a primitive neural and multipotent precursor important for neurogenesis and neuronal migration. Further evidence implicating radial glia as cells of origin of ependymoma was demonstrated by Johnson et al. (2010), in which over-expression of EPHB2 in p16/INK4A-deficient RGCs led to the formation of the first mouse model of supratentorial ependymoma [27].
Chromosome 22 loss has been shown to be the most frequent genomic alteration in ependymoma with a frequency ranging from 26 to 71 % [30]. Further, chromosome 22q loss has been observed preferentially in spinal versus intracranial ependymoma, and in adult versus pediatric cases [30]. NF2 is thought to be the candidate tumor suppressor gene of this region, as patients with neurofibromatosis type II develop a variety of central nervous system tumors including ependymoma, schwannoma, and meningioma [30]. However, NF2 has been shown to be mutated exclusively in spinal ependymomas, thus suggesting alternate mechanisms of down-regulation, or another putative 22q tumor suppressor gene in the case of intracranial ependymoma. Other recurrent gross chromosomal abnormalities in ependymoma involve losses of chromosome 1p, 3, 6q, 9p, 10q, 13q, 16p, 17, 21, and 22q and gains of 1q, 4q, 5, 7, 8, 9, 12q, and 20 [30].
The telomerase pathway and its role in ependymoma pathogenesis have also been studied by several groups. Specifically, the expression of hTERT, the enzyme responsible for telomere extension, and its cofactor nucleolin have both been shown to be independent predictors of ependymoma patient survival [31]. Castelo-Branco et al. (2013) suggest that a possible mechanism leading to hTERT over-expression could be due to epigenetic silencing of repressor regions in the promoter of hTERT by DNA hypermethylation [32].
As novel ependymoma targets are discovered, evaluation, validation, and prioritization of candidates will require accurate preclinical models of ependymoma. Atkinson et al. (2011) demonstrate the utility and promise of this approach in ST-Group D ependymomas, generated by EPHB2 over-expression and CDKN2A/Ink4a deletion in forebrain radial glia [33]. They performed a compound library screen of 7890 compounds, in both tumor and matched normal neural stem cells, and identified inhibitors of thymidylate synthase (TYMS) and dihydrogolate reductase, namely 5-FU as highly and specifically active in ST ependymoma cells.
Choroid Plexus Tumors
Choroid plexus tumors (CPTs) are classified as three distinct entities according to pathological examination: choroid plexus papillomas (CPPs, Grade I), atypical choroid plexus papillomas (aCPPs, Grade II), and choroid plexus carcinomas (CPCs, Grade III). CPCs are considered a hallmark tumor of the Li-Fraumeni syndrome (LFS). Germline and somatic mutations in TP53, a classical tumor suppressor gene involved in DNA repair, cellular differentiation, and apoptosis, are commonly found in CPCs [34]. The frequency of TP53 mutations is much higher in the carcinomas (~50 %) than papillomas (~5 %), which may contribute to the aggressiveness and poor outcome of these malignant tumors [35] (Fig. 7.1). No CPPs, or aCPPs, harbored germline mutations in TP53, while at least 36 % of CPCs harbored a germline TP53 mutation [35].
CPTs are characterized by a high degree of chromosomal imbalances. Generally, CPPs are characterized by extensive chromosomal gains, and CPCs by both gains and losses, yet CPCs demonstrate greater imbalances per tumor than CPPs [36]. CPPs exhibit recurrent gains in chromosomes 7 and 12, while CPCs exhibit gains and losses throughout the genome with a higher frequency of chromosome 1 gains. Association of chromosomal aberrations with survival revealed a significant association between gain of 9p and loss of 10q with improved survival [36]. Cytogenetics studies in aCPPs, although limited, have revealed variable aneuploidy phenotypes characterized by polyploidy.
Atypical Teratoid Rhabdoid Tumors
Atypical teratoid rhabdoid tumors (ATRT) are of mesenchymal origin and are characterized by biallelic loss of SMARCB1/INI1, which encodes the SNF5 protein, a core subunit of the SWI/SNF chromatin remodeling complex [37] (Fig. 7.1). SNF5 is a tumor suppressor gene located on chromosome 22q11.23 and is inactivated by a variety of genetic lesions including homozygous deletions and nonsense, missense, and frameshift mutations. The SWI/SNF complex is an ATP-dependent regulator of gene expression which acts by remodeling chromatin and repositioning nucleosomes through catalyzing the insertion and removal of histone proteins. Despite being extremely aggressive, SNF5-deficient cancers are diploid and genomically stable, highlighting the importance of this gene as a central driver of ATRT tumorigenesis [38]. Mechanistically, inactivation of SNF5 has been linked to broad repression of polycomb repressor complex 2 (PRC2) regulated genes [39]. Specifically loss of SNF5 leads to up-regulation of EZH2, the core enzymatic component of the PRC2 complex, and subsequent loss of the tumor suppressors such as p16INK4a. ATRT development is also affected by the Sonic Hedgehog (SHH) signaling pathway. SNF5 localizes to Gli1-regulated promoters of the SHH pathway, thus repressing Gli1targets [40]. Hence, loss of Snf5 leads to up-regulation of Gli1 targets and activation of the Hh-Gli pathway driving oncogenesis [40].
References
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012;123(4):465–72.
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–31.
Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488(7409):49–56.
Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488(7409):100–5.
Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–9.
Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14(2):135–45.
Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, et al. An animal model of MYC-driven medulloblastoma. Cancer Cell. 2012;21(2):155–67.
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22(4):425–37.
Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28(18):3061–8.
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44(3):251–3.
Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, et al. Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol. 2011;29(30):3999–4006.
Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–7.
Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–32.
Gronych J, Korshunov A, Bageritz J, Milde T, Jugold M, Hambardzumyan D, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011;121(4):1344–8.
Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011;121(3):397–405.
Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45(6):602–12.
Ramkissoon LA, Horowitz PM, Craig JM, Ramkissoon SH, Rich BE, Schumacher SE, et al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci USA. 2013;110(20):8188–93.
Weber RG, Hoischen A, Ehrler M, Zipper P, Kaulich K, Blaschke B, et al. Frequent loss of chromosome 9, homozygous CDKN2A/p14(ARF)/CDKN2B deletion and low TSC1 mRNA expression in pleomorphic xanthoastrocytomas. Oncogene. 2007;26(7):1088–97.
Kros JM, Delwel EJ, de Jong TH, Tanghe HL, van Run PR, Vissers K, et al. Desmoplastic infantile astrocytoma and ganglioglioma: a search for genomic characteristics. Acta Neuropathol. 2002;104(2):144–8.
Schneider DT, Zahn S, Sievers S, Alemazkour K, Reifenberger G, Wiestler OD, et al. Molecular genetic analysis of central nervous system germ cell tumors with comparative genomic hybridization. Mod Pathol. 2006;19(6):864–73.
Sekine S, Shibata T, Kokubu A, Morishita Y, Noguchi M, Nakanishi Y, et al. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol. 2002;161(6):1997–2001.
Picard D, Miller S, Hawkins CE, Bouffet E, Rogers HA, Chan TS, et al. Markers of survival and metastatic potential in childhood CNS primitive neuro-ectodermal brain tumours: an integrative genomic analysis. Lancet Oncol. 2012;13(8):838–48.
Eberhart CG, Brat DJ, Cohen KJ, Burger PC. Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol. 2000;3(4):346–52.
Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C, et al. Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell. 2009;16(6):533–46.
Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet. 2009;41(7):843–8.
Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8(4):323–35.
Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;466(7306):632–6.
Wani K, Armstrong TS, Vera-Bolanos E, Raghunathan A, Ellison D, Gilbertson R, et al. A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 2012;123(5):727–38.
Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–57.
Mack SC, Taylor MD. The genetic and epigenetic basis of ependymoma. Childs Nerv Syst. 2009;25(10):1195–201.
Tabori U, Ma J, Carter M, Zielenska M, Rutka J, Bouffet E, et al. Human telomere reverse transcriptase expression predicts progression and survival in pediatric intracranial ependymoma. J Clin Oncol. 2006;24(10):1522–8.
Castelo-Branco P, Choufani S, Mack S, Gallagher D, Zhang C, Lipman T, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. 2013;14(6):534–42.
Atkinson JM, Shelat AA, Carcaboso AM, Kranenburg TA, Arnold LA, Boulos N, et al. An integrated in vitro and in vivo high-throughput screen identifies treatment leads for ependymoma. Cancer Cell. 2011;20(3):384–99.
Kamaly-Asl ID, Shams N, Taylor MD. Genetics of choroid plexus tumors. Neurosurg Focus. 2006;20(1):E10.
Tabori U, Shlien A, Baskin B, Levitt S, Ray P, Alon N, et al. TP53 alterations determine clinical subgroups and survival of patients with choroid plexus tumors. J Clin Oncol. 2010;28(12):1995–2001.
Rickert CH, Wiestler OD, Paulus W. Chromosomal imbalances in choroid plexus tumors. Am J Pathol. 2002;160(3):1105–13.
Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–92.
Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012;122(8):2983–8.
Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316–28.
Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16(12):1429–33.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Mack, S.C. et al. (2015). Basic Science of Pediatric Brain Tumors. In: Scheinemann, K., Bouffet, E. (eds) Pediatric Neuro-oncology. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1541-5_7
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1541-5_7
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1540-8
Online ISBN: 978-1-4939-1541-5
eBook Packages: MedicineMedicine (R0)