
Abstract
Bioequivalence is applied in various facets of the pharmaceutical industry from drug development to regulations. Since the inception of FDA bioequivalence regulations in 1977, bioequivalence evaluation has evolved to include the two one-sided test procedure, the biopharmaceutical classification system, and new approaches for highly variable, narrow therapeutic index, and locally acting drugs. In this introductory chapter, we provide an overview of the history of bioequivalence from its emerging—1970s, to the current—2000s. We discuss the clinical problems that led to bioequivalence, the associated regulatory challenges, and methods to assure bioequivalence, over the course of bioequivalence development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Drug Product
- Reference Product
- Biopharmaceutics Classification System
- Bioequivalence Study
- Biopharmaceutics Drug Disposition Classification System
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1.1 Introduction
Bioequivalence (BE) is defined as the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study. Drug products are considered pharmaceutical equivalents if they contain the same active ingredient(s), are of the same dosage form, route of administration, are identical in strength or concentration, and meet the same or compendial or other applicable standards (i.e., strength, quality, purity, and identity). Drug products are considered pharmaceutical alternatives if they contain the same therapeutic moiety, but are different salt, esters, or complexes of that moiety, or are different dosage forms or strengths (21 CFR 320).
Bioequivalence studies are a major component in evaluating therapeutic equivalence by verifying that the active ingredient of the test drug product will be absorbed into the body to the same extent and at the same rate as the corresponding reference drug product. The significance of this study is that when two pharmaceutically equivalent products are shown to be bioequivalent, the two products are judged to be therapeutically equivalent. Therapeutically equivalent products are expected to have the same safety and efficacy profiles, when administered under the conditions listed in the product labeling. For generic drugs, bioequivalence studies confirm the clinical equivalence between the generic and reference products. For new drugs, these studies verify the clinical equivalence between different formulations and sometimes between different strengths. As such, bioequivalence is an integral part of development and regulations for both generic and new drugs.
This chapter discusses the evolution of bioequivalence by dividing the history of bioequivalence into three time periods: the 1970–1980s, the 1990s, and the 2000s. The 1970s and 1980s were when bioequivalence was first established with an important role in drug development and regulations. The 1990s marked an intense discussion of the individual bioequivalence concept as well as the development of the Biopharmaceutics Classification System (BCS) and its subsequent applications to regulatory guidances. This era also featured the development of the predictive compartmental absorption and transit (CAT) model. The turn of the millennia (2000s) saw the development of Biopharmaceutics Drug Disposition Classification System (BDDCS), evolution of BE standards for highly variable drugs, implementation of partial area under the curve (pAUC), creation of novel approaches for narrow therapeutic index (NTI) drugs, and the development of a number of BE approaches for locally acting drugs.
1.2 Bioequivalence Evolution in 1970s and 1980s
1.2.1 Bioequivalence Problems and Recognition
The early 1970s observed the start of serious investigation when patients that took digoxin had variable or poor responses to the medication. Lindenbaum et al. (1971) conducted a crossover study where 0.5 mg of digoxin was orally administrated to four normal volunteers. There were significant variations observed in peak serum levels from the same drug in different products made by various manufacturers. One product exhibited sevenfold higher peak serum levels than the other manufacturer’s formulation. Even within the same manufacturer, there was significant between-lot variation. Wagner et al. (1973), under the contract with the FDA (Skelly 1976), confirmed Lindenbaum’s findings of lack of equivalence in plasma levels of digoxin tablets made by different manufacturers.
A likely reason for the variation was a formulation defect where there was an insufficient or excessive amount of active ingredient in the dosage form. This was confirmed by the FDA through a systematic testing program initiated in April 1970 (Vitti et al. 1971). When digoxin tablet lots from Lindenbaum’s study were assayed, it was found that the tablets from B2 were out of potency specification (between 72 and 158.2 % of declared potency) whereas products A and B1 were within potency requirements (Vitti et al. 1971).
Other possible reasons for variation include particle size, disintegration time, dissolution rate, and the effects of various excipients. Wagner et al. (1973) found equivalence lacking in digoxin plasma levels even with tablets that met the acceptance criteria for both potency and disintegration. Similar observations were also made for other products such as tetracycline (Barnett et al. 1974; Barr et al. 1972), chloramphenicol (Glazko et al. 1968), phenylbutazone (Chiou 1972; Van Petten et al. 1971), and oxytetracycline (Barber et al. 1974). These drug products exhibited large variations in drug plasma levels exposing patients to potentially deadly hazards.
Recognizing the existence of bioequivalence problems in marketed products, the FDA Office of Technology Assessment (OTA) organized a drug bioequivalence study panel of ten senior clinicians and scientists in 1974. The panel examined the relationships between chemical and therapeutic equivalence of drug products on the market. They also assessed the capabilities of technology available at that time to determine whether drug products with the same physical and chemical composition produced comparable therapeutic effects (OTA 1974b). Among the panel’s eleven conclusions and recommendations, five are critical to the establishment of bioequivalence regulations (OTA 1974a):
-
1.
Current standards and regulatory practices do not assure bioequivalence.
-
2.
Variations in bioavailability are recognized as responsible for a few therapeutic failures. It is probable that other therapeutic failures (or toxicity) of a similar origin have escaped recognition.
-
3.
Most of the analytical methodology and experimental procedures for the conduct of bioavailability studies in man are available. Additional work may be required to develop means of applying them to certain drugs and to special situations of drug use.
-
4.
It is neither feasible nor desirable that studies or bioavailability be conducted for all drugs or drug products. Certain classes of drugs for which evidence of bioequivalence is critical should be identified. Selection of these classes should be based on clinical importance, ratio of therapeutic to toxic concentration in blood, and certain pharmaceutical characteristics.
-
5.
Additional research aimed at improving the assessment and prediction of bioequivalence is needed. This research should include efforts to develop in vitro tests or animal models that will be valid predictors of bioavailability in man.
1.2.2 FDA 1977 Bioequivalence Regulation
Based on the recommendations provided by the drug bioequivalence study panel, the FDA issued regulations that set forth procedures to establish bioequivalence requirements. Effective February 7, 1977, these regulations define the terms of drug product, pharmaceutical equivalent, pharmaceutical alternative, bioequivalent drug product, and bioequivalence requirement (Federal Register 1977).
A bioequivalence requirement may be one or more of the following (Federal Register 1977):
-
An in vivo test in humans
-
An in vivo test in animals other than humans that has been correlated with human in vivo data
-
An in vivo test in animal other than humans that has not been correlated with human in vivo data
-
An in vitro bioequivalence standard, i.e., an in vitro test that has been correlated with human in vivo bioavailability data
-
A currently available in vitro test (usually a dissolution rate test) that has not been correlated with human in vivo bioavailability data
-
In vivo testing in humans shall ordinarily be required if there is well-documented evidence that pharmaceutical equivalents or pharmaceutical alternatives intended to be used interchangeably for the same therapeutic effect meet one of the following conditions:
-
They do not give comparable therapeutic effect
-
They are not bioequivalent drug product
-
They exhibit a narrow therapeutic ratio, e.g., there is less than a twofold difference in LD50 and ED50 values, or there is less than twofold difference in minimum toxic concentration and minimum effective concentration in the blood, and safe and effective use of the product requires careful dosage titration and patient monitoring
-
These regulations also required that all bioequivalence in vivo or in vitro testing records of any marketed batch of drug products must be maintained until 2 years after the batch expiration date and remain available to be submitted to the FDA on request.
1.2.3 Drug Price Competition and Patent Term Restoration Act
The FDA 1977 bioequivalence regulations played an important role in the establishment of the 1984 Drug Price Competition and Patent Term Restoration Act, informally known as the “Hatch-Waxman Act.” This act assumes that bioequivalence is an effective surrogate for safety and efficacy. It established the modern system of generic drugs where drug products must be therapeutically equivalent by meeting the following general criteria (FDA Orange Book 2013):
-
1.
Products are approved as safe and effective.
-
2.
Products are pharmaceutical equivalents in that they (a) contain identical amounts of the same active drug ingredient in the same dosage form and route of administration and (b) meet compendial or other applicable standards of strength, quality, purity, and identity.
-
3.
Products are bioequivalent in that (a) they do not present a known or potential bioequivalence problem, and they meet an acceptable in vitro standard, or (b) if they do present such a known or potential problem, they are shown to meet an appropriate bioequivalence standard.
-
4.
Products are adequately labeled.
-
5.
Products are manufactured in compliance with Current Good Manufacturing Practice regulations.
Upon meeting these requirements, generic products are expected to have the same clinical effect and safety profile when administered to patients under the conditions specified in the labeling (FDA 2013a).
Continual refinement of in vivo and in vitro science has led the FDA to revise methods to demonstrate bioequivalence. As of publication date, current methods used to meet the statutory bioequivalence requirement include (FDA 2003a):
-
1.
Pharmacokinetic (PK) studies
-
2.
Pharmacodynamic (PD) studies
-
3.
Comparative clinical trials
-
4.
In vitro studies
The selection of the type of bioequivalence studies to be conducted is based on the drug’s site of action and the study design’s ability to compare drug delivery.
1.2.4 Bioequivalence Decision Rules
There is extensive literature discussing the criteria for establishing bioequivalence. The FDA Orange Book mentions a common notion that “based on the opinions of FDA medical experts, a difference of greater than 20 % for each of the above tests (area under the curve (AUC) and C max) was determined to be significant, and therefore, undesirable for all drug products (FDA Orange Book 2013).” As such, the bioequivalence limits have generally been taken within 20 % of the standard (Hauck and Anderson 1984).
During the early development of bioequivalence, Skelly (2010) suggested the determination of AUC measurements by physically plotting serum concentration versus time on specially weighted paper, cutting out the respective plots, and weighing each plot separately for comparison. This method, known as the Canadian rule of ±20 %, requires that the mean AUC of the generic drug be within 20 % of the mean AUC of the approved product.
After the 1971 conference on bioavailability of drugs at the National Academy of Science (Brodie and Heller 1971), the FDA started using the power approach. This approach involved determining the AUC through integration instead of physical weights and required both AUC and C max to be within ±20 % of the innovator product at an estimated power of 80 %.
However, the power approach is limited in that it only considers differences in the calculated averages of AUC and C max. With this approach, two approved products can have equal AUC and C max mean values but differ in variability, which may be problematic for some drugs such as NTI drugs. Consideration of variability was deemed necessary for these drugs at the time. As a result, the FDA developed an additional 75/75 rule, under which bioequivalence would be met if:
-
(a)
There was no more than 20 % difference in mean AUC and C max between the test and reference products.
-
(b)
The relative bioavailability of the test product to the reference product exceeded 75 % in at least 75 % of the subjects studied.
The use of 75/75 rule would be responsible for ensuring that there is not a lack of efficacy in the event that there is variable plasma concentration (Patterson and James 2005; Cabana 1983). However, the opposite also applies in that it is possible that the 75/75 rule does not prevent side effects that result of potentially high concentrations. Haynes (1981) also demonstrated that the rule had undesirable performance characteristics and lacked statistical underpinning. As such, the 75/75 rule was later abandoned.
In 1983, Hauck and Anderson (1984) proposed the use of a bioequivalence analysis that incorporated two null hypotheses (H 0) t-tests as shown below:
For these equations, μ T is the logarithmic mean for the test (i.e., generic drug), μ R is the logarithmic mean for the reference (reference product), θ 1 is the lower limit (log 80 %), and θ 2 is the upper limit (log 125 %). By combining the two statistical one-sided tests, the null hypothesis (H 0) states that the means are not equivalent and the alternative hypothesis (H 1) states that the means are equivalent.
However, it is possible that the Hauck–Anderson t-test could conclude that two products are bioequivalent when they are not. Schuirmann (1987) proposed a solution called the “two one-sided tests procedure” that splits the alternative hypothesis into two parts:
This test eliminates the possibility of an infinitely large rejection region when certain criteria are met (typically when the observed means between the test and reference are similar). This two one-sided test procedure has been used to establish bioequivalence to this day.
To evaluate the performance of the two one-sided tests, Davit et al. (2009) collected a total over 2,000 single-dose bioequivalence studies of orally administered generic drug products approved by the Food and Drug Administration (FDA) from 1996 to 2007 for a period of 12 years. For each study, the measurements evaluated were drug plasma peak concentration (C max) and drug concentration in plasma over time (AUC). The average difference in C max and AUC between generic and innovator products was 4.35 % and 3.56 %, respectively. In addition, in nearly 98 % of the bioequivalence studies conducted during this period, the generic product AUC differed from that of the innovator product by less than 10 %. The resulting conclusion is that while the statistical test analyzes BE confidence from the limit of 80–125 %, the actual difference between test and reference drug is usually much smaller as noted by Fig. 1.1.

AUC distributions over 12 years of FDA BE data (Yu 2013)
1.3 Bioequivalence Evolution in 1990–2000
1.3.1 Individual Bioequivalence
One of the potential weaknesses of the two one-sided tests procedure lies in the fact that it cannot address the question of whether the bioequivalence outcome is sufficient to guarantee that an individual patient could be expected to respond similarly to two different products.
This is because the two one-sided tests procedure only assesses the difference between the test and reference means (average bioequivalence) while individual bioequivalence assesses the difference of the mean and variability. Anderson and Hauck proposed an individual bioequivalence test to provide reasonable assurance that an individual patient could be switched from a therapeutically successful product to another (Anderson and Hauck 1990; Hauck and Anderson 1994).
In the 1990s, the FDA published guidance documents on the proposed criterion and statistical methodology for an individual bioequivalence approach (Chen and Lesko 2001; FDA 1999b). These guidances would allow comparison of intra-subject variances, scaling of bioequivalence criterion to the reference variability, and detection of possible subject-by-formulation interactions. The new criterion would also promote inclusion of heterogeneous population of volunteers in bioequivalence studies. Based on these considerations, the FDA had intended for use of individual bioequivalence to replace average bioequivalence (Chen et al. 2000; Hauck et al. 2000).
Despite the advantages and benefits, there were challenges for using the individual bioequivalence approach. Most questions were focused on the following three general areas (Chen and Lesko 2001):
-
1.
Justification and need for an individual bioequivalence criterion
-
2.
Financial and human resource burden of conducting replicate study designs
-
3.
Appropriateness of the statistical methodology
To address these questions, there were many AAPS public workshops and conferences (AAPS 1997, 1998, 1999) as well as the FDA Advisory Committee for Pharmaceutical Science meetings (FDA 1996, 1997, 1998, 1999a, 2000a). The FDA Individual Bioequivalence Expert Panel chaired by Leslie Benet reported at the 1999 FDA Advisory Committee for Pharmaceutical Science meeting that (Benet 1999):
-
Individual bioequivalence is a promising, clinically relevant method that should theoretically provide further confidence to clinicians and patients that generic drug products are indeed equivalent in an individual patient.
-
Even today, considering the studies summarized and analyzed by the FDA, the data is inadequate to validate the theoretical approach and provide confidence to the scientific community that the methodology required and the expense entailed are justified.
-
At this time, individual bioequivalence still remains a theoretical solution to solve a theoretical clinical problem. We have no evidence that we have a clinical problem, either a safety or an efficacy issue, and we have no evidence that if we have the problem that individual bioequivalence will solve the problem.
As a result, the average bioequivalence approach remains the key method for evaluation of bioequivalence today.
1.3.2 Biopharmaceutics Classification System
Amidon et al. (1995) developed a BCS for correlating in vitro drug product dissolution and in vivo bioavailability. This classification was derived from the physical properties of solubility and permeability on drug absorption. According to the devised BCS system, drug substances are classified into four classes, as shown in Table 1.1.
The BCS solubility classification is derived from an in vitro experiment that tests the highest strength of a drug product. If the highest strength drug of a specified dosage form is soluble in 250 mL or less of aqueous media over the pH range of 1.0–7.5, the drug is considered highly soluble. The 250 mL volume estimate is derived from typical bioequivalence study protocols that prescribe administration of drug product with a glass of water (~8 oz) to fasted human volunteers.
The BCS permeability classification is based directly on the extent of intestinal absorption of a drug substance in humans or indirectly based on measurements of the mass transfer rate across the human intestinal membrane. Animal or in vitro models capable of predicting the extent of intestinal absorptions in humans may also be used as alternatives, e.g., in situ rat perfusion models and in vitro epithelial cell culture models (FDA 2000b). A drug substance is considered highly permeable when the extent of intestinal absorption is determined to be 90 % or higher.
1.3.3 Biowaiver Based on BCS
In 2000, the FDA issued a guidance describing the waiver of in vivo bioavailability and bioequivalence studies for immediate-release (IR) solid oral dosage forms based on the BCS. This guidance allows applicants to request biowaivers for highly soluble and highly permeable drug substances (Class I) in immediate-release solid oral dosage forms provided the following conditions are met (FDA 2000b):
-
(a)
The drug must be stable in the gastrointestinal tract
-
(b)
Excipients used in the IR solid oral dosage forms have no significant effect on the rate and extent of oral drug absorption
-
(c)
The drug must not have an NTI
-
(d)
The product is designed not to be absorbed in the oral cavity
-
(e)
The drug dissolves rapidly in vitro
An IR drug product is considered to have a rapid dissolution when not less than 85 % of the labeled amount of the drug substance dissolves within 30 min using USP Apparatus I at 100 rpm or USP Apparatus II at 50 rpm in a volume of 900 mL or less of each of the following media (FDA 2000b):
-
(a)
Acidic media, such as 0.1 N HCl or USP simulated gastric fluid without enzymes (SGF)
-
(b)
A pH 4.5 buffer
-
(c)
A pH 6.8 buffer or USP simulated intestinal fluid without enzymes (SIF)
If the drug product does not meet these requirements, it is not considered to be a rapidly dissolving product.
Based on these BCS scientific principles, the cause of two pharmaceutically equivalent solid oral products exhibiting in vivo differences in the rate and extent of drug absorption may be due to in vivo differences in drug dissolution. If the in vivo dissolution of an IR oral dosage form is rapid relative to gastric emptying, then the rate and extent of drug absorption is likely to be independent of drug dissolution. In terms of in vivo behavior, a highly soluble and rapidly dissolving drug product is similar to an oral solution. Demonstration of in vivo bioequivalence may not be necessary as long as the inactive ingredients used in the dosage form do not significantly affect absorption of the active ingredient. For BCS Class I (both high solubility and high permeability) drug products, demonstration of rapid in vitro dissolution using required test conditions is sufficient for assurance of similarly rapid in vivo dissolution. This avoids unnecessary costs and risks involved in conducting clinical trials to demonstrate bioequivalence.
1.3.4 CAT Model
Although it was well known that small intestine transit time plays an important role in absorption, there was little development in this area before 1990s. In 1996, Yu et al. developed a CAT model constructed from the understandings of small intestinal transit flow and its characterization (Yu et al. 1996a, b; Yu and Amidon 1998a). This model is able to predict both the rate and extent of absorption (Yu et al. 1996a; Yu and Amidon 1998b).
When compared to the dispersion and single compartment model, it was found that the CAT model was superior to the single-compartment model and less complex than the dispersion model. The single compartment model characterizes the drug as being distributed into the body as a single volume while the dispersion model characterizes the drug distribution through convection and dispersion.
To extend the original CAT model’s capabilities in determining the rate, extent, and approximate gastrointestinal location of drug liberation (for controlled release formulations), an advanced compartmental absorption and transit (ACAT) model was developed later (Agoram et al. 2001). The ACAT model is essentially the same as the integrated absorption model which estimates fraction of dose absorbed and provides a framework to determine when the absorption is limited by permeability, dissolution, and solubility (Yu 1999).
The subsequent development of computer software transformed the ACAT models into commercially available software for research and evaluation. Continued development has led to more accurate prediction models of in vitro–in vivo correlations for oral absorption in comparison to previous models (Grbic et al. 2011). Combined with biorelevant solubility, the modern computer programs are also able to predict the magnitude of food effects and oral pharmacokinetics of different drugs in both fasted and fed conditions. In addition to its use for predicting oral drug absorption in the GI tract, whole body Physiologically Based Pharmacokinetic Modeling (PBPK) and combined Pharmacokinetic/Pharmacodynamic models have been constructed for predicting whole body PK/PD consequences in humans (Huang et al. 2009). Recently, the computer models have been also used to conduct virtual bioequivalence simulations (Zhang et al. 2011).
1.4 Bioequivalence Evolution from 2000s to Present
1.4.1 Biopharmaceutics Drug Disposition Classification System
In 2005, Wu and Benet (2005) proposed the BDDCS. This work expanded on the BCS’ foundations of solubility and permeability by incorporating transporter effects and elimination mechanisms. In particular, the BDDCS was developed to predict drug disposition and drug–drug interactions in both the intestine and liver. According to the BDDCS, a drug can be classified into one of the four classes as shown in Table 1.2.
An example of how inclusion of metabolic and transporter analysis can allow for predictions of drug delivery behavior when high-fat meals are taken into account is given by Fleisher et al. (1999). Similar thought processes can also be used to predict scenarios such as in vivo drug–drug interaction (i.e., competing transporters; Benet 2013). This approach leads to a significant distinction between BDDCS and BCS as the former focuses on metabolism and the latter on absorption.
1.4.2 Bioequivalence Approach for Highly Variable Drugs
Highly variable drugs are defined as those for which within-subject variability (%CV) of bioequivalence (BE) measures is 30 % or greater (Haidar et al. 2008). The sources of within-subject variability include:
-
Physiological factors affecting bioavailability such as regional pH in the gastrointestinal tract, bile and pancreatic secretions, luminal and mucosal enzymes, gastrointestinal motility, gastric emptying, small intestinal transit time, and colonic residence time.
-
Inherent properties of the drug such as distribution, first-pass metabolism, systemic metabolism, and elimination.
-
Physicochemical properties of drug substance such as solubility.
-
Formulation factors such as drug release.
-
Other factors such as food intake.
Because of the nature of the average bioequivalence approach, bioequivalence studies for highly variable drugs may need to enroll a large number of subjects even when the generic and reference products have very little difference in mean bioavailability. This is a consequence of high within-subject variability as shown in Fig. 1.2. It is even possible that a highly variable reference product will fail to demonstrate bioequivalence when compared with itself in a bioequivalence study using the average bioequivalence approach and usual sample size (Midha et al. 2005).

Effect of variability on BE studies. With the same number of subjects, high variability will lead to the wide confident intervals, making the study more difficult to pass the BE limit of 80–125 %
The belief is that highly intra-subject variable drugs generally have a wide therapeutic window where products have been demonstrated to be both safe and effective despite the high variability (Benet 2006). With this in mind, applying the same average bioequivalence criteria to highly variable drugs/products may unnecessarily expose large number of healthy subjects to the drug (Benet 2006). To minimize unnecessary human testing, various approaches with alternate study designs, statistical methods, and other considerations have been proposed and investigated to demonstrate bioequivalence of highly variable drugs. These approaches include bioequivalence studies with multiple doses at steady state, a limited sampling method, individual bioequivalence, direct expansion of bioequivalence limits to prefixed values, and widening of bioequivalence limits by scaling approaches (Zhang et al. 2013). Because each method has its advantages and disadvantages, there is no universally accepted approach to demonstrating bioequivalence for highly variable drugs.
The FDA has chosen to evaluate the following approaches for demonstration of bioequivalence for highly variable drugs and products: direct expansion of BE limits, expansion of BE limits based on fixed sample size, widening of BE limits based on reference variability, and expansion of BE limits based on sample size and scaling (FDA 2004). Based on these evaluations, the FDA developed a reference-scaled average bioequivalence approach with a point-estimate constraint, where the bioequivalence acceptance limits are scaled to the variability of the reference product.
This approach adjusts the bioequivalence limits of highly variable drugs by scaling to the within-subject variability of the reference product in the study. The use of reference-scaling is based on the general concept that reference variability should be used as an index for setting the public standard of the bioequivalence limit. This effectively decreases the sample size needed to demonstrate bioequivalence of highly variable drugs.
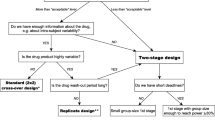
The FDA’s final approach includes the additional requirement of a point-estimate constraint that imposes a limit on the difference between the test and reference means. This eliminates the potential that a test product could enter the market with a large mean difference from the reference product. The use of the reference-scaling approach necessitates a study design that would allow for determination of reference variability, i.e., multiple administrations of the reference treatment to each subject. The FDA recommended partial replicate design as the most efficient way to obtain this information. The reference-scaled average bioequivalence approach has been used successfully at the FDA. To date, this new approach has supported many approvals of high variable generic drug products.
1.4.3 Bioequivalence for NTI Drugs
Although the use of 80–125 % bioequivalence limits has been historically proven to be a rigorous criterion after approval of thousands of generic drugs and post-marketing drug product changes, this criterion may not be conservative enough for NTI drugs as small changes in blood concentration of these drugs can potentially have serious therapeutic consequences and/or adverse drug reactions in patient use. Because of the risks that can arise from the NTI drugs, there have been debates, among health care professionals, pharmaceutical scientists, regulatory agencies, and consumer advocates, about how much assurance is needed for a generic NTI drug product to be considered bioequivalent to its reference product. In 2010 and 2011, the FDA held two advisory committee meetings to discuss the definition of NTI drugs and the BE approaches to establishing therapeutic equivalence of these drug products (FDA 2011d).
Historically, a variety of terms have been used to describe the drugs in which comparatively small differences in dose or concentration may lead to serious therapeutic failures and/or serious adverse drug reactions in patients. These may include NTI, narrow therapeutic range, narrow therapeutic ratio, narrow therapeutic window, and critical-dose drugs. The FDA advisory committee recommended the use of the term “narrow therapeutic index (NTI)” and defined NTI drugs as drugs where small differences in dose or blood concentration may lead to serious therapeutic failures and/or adverse drug reactions that are life-threatening or result in persistent or significant disability or incapacity (Yu 2011):
-
(a)
There is little separation between therapeutic and toxic doses or associated blood/plasma concentrations.
-
(b)
Subtherapeutic concentrations may lead to serious therapeutic failure and/or above-therapeutic concentrations may lead to serious adverse drug reactions in patients.
-
(c)
Subject to therapeutic monitoring based on pharmacokinetic or pharmacodynamics measures.
-
(d)
Possess low-to-moderate (i.e., no more than 30 %) within-subject variability.
-
(e)
In clinical practice, doses are often adjusted in very small increments (less than 20 %).
Based on the input from the advisory committee, FDA conducted simulations to investigate the application of different BE approaches for NTI drugs, including the use of (1) direct tightening of average BE limits and (2) tightening BE limits based on the variability of the reference product (the reference-scaled average BE approach) (FDA 2011d). Variables evaluated in the simulations included within-subject variability, sample size, and point-estimate limit. The powers of a given study design were compared using the reference-scaled average BE approach versus the average BE approach. Simulation results indicated that an approach that tightens BE limits based on reference variability is the preferred approach for evaluating the BE of NTI drugs. A four-way, crossover, fully replicated study design is preferred because such a study design will permit variability comparison in addition to the mean comparison. Both comparisons have to be considered when declaring bioequivalent.
The baseline BE limits for NTI drugs is 90–111 %, which would be scaled based on the within-subject variability of the reference product. When the reference variability is ≤10 %, the BE limits will be narrower than 90–111 %. Conversely, when the reference variability is >10 %, the BE limits will be wider than 90–111 %, but are capped at 80–125 %. To ensure that the BE limits for NTI drugs are never wider than those for conventional drugs, it is critical that every study pass the scaled average BE and the unscaled average BE limits of 80–125 %. Because most NTI drugs have low within-subject variability, the BE limits for these drug products would almost always be tightened to less than 80–125 % accordingly.
The four-way, crossover, fully replicated study design will also permit the comparison of within-subject variability in the test and reference products to confirm that their variances do not differ significantly. FDA’s simulation studies demonstrated that test and reference products with unacceptably large differences in within-subject variability may still pass the reference-scaled BE limits, suggesting that the reference-scaled average bioequivalence approach alone is not adequate to ensure the similarity of test and reference products for NTI drugs. FDA proposed an F-test to evaluate whether the within-subject variability of test and reference products are comparable by calculating the 90 % confidence interval of the ratio of the within-subject standard deviation of the test to reference product (FDA 2011d). To determine the appropriate upper limit of the confidence interval of the variability test, FDA evaluated the limit value of 2, 2.5, and 3 and concluded that the appropriate upper limit of the 90 % confidence interval should be ≤2.5.
1.4.4 Partial Area Under the Curve
Since the inception of bioequivalence, the peak exposure (C max) and total exposure (AUC) have been used to measure the rate and extent of absorption. These two metrics along with the time to peak concentration (T max) generally work well for immediate release and even for many modified release dosage forms. However, for some modified products that exhibit multiphasic pharmacokinetic behavior which is clinically important and meaningful, the traditional metrics of AUC and C max may not be sufficient to ensure BE. In these cases, AUC and C max may be equivalent for two products, but the rate or extent of exposure during a clinically relevant time interval may not be equivalent (Heald 2010). Consequently, an additional PK metric, such as a pAUC to assess partial exposure, may be necessary to demonstrate bioequivalence.
Chen proposed to use a pAUC approach for the evaluation of equivalence in the rate of absorption for immediate-release formulations (Chen 1992; Chen et al. 2011). For orally administered immediate-release drug products, bioequivalence can generally be demonstrated by measurements of peak and total exposure. An early exposure measure may be informative on the basis of appropriate clinical efficacy/safety trials and/or pharmacokinetic/pharmacodynamic studies that call for better control of drug absorption into the systemic circulation (e.g., to ensure rapid onset of an analgesic effect or to avoid an excessive hypotensive action of an antihypertensive). Although the FDA general BA/BE guidance recommended the use of partial AUC as an early exposure measure, it was rarely used (FDA 2003a).
In 2011 and 2012, the FDA implemented the use of pAUC for the determination of bioequivalence of zolpidem extended-release tablets and methylphenidate hydrochloride extended-release capsules and tablets (FDA 2011c, 2012c). Pharmacokinetic/pharmacodynamic relationship is the foundation for recommending use of pAUC for these products. Modeling and simulation studies were performed to aid in understanding the need for pAUC measures and also the proper pAUC truncation times (Lionberger et al. 2012; Stier et al. 2012; Fourie Zirkelbach et al. 2013).
The choice of truncation of the area under the curve is most appropriately based on PK/PD relationship or efficacy/safety data for the drug under examination. When PK/PD relationship is lacking, the selection of the truncation point for pAUCs is challenging. When pAUC is highly variable, the reference-scaling approach can be employed for bioequivalence evaluation.
1.4.5 Bioequivalence for Locally Acting Gastrointestinal Drugs
The function of locally acting gastrointestinal (GI) drug products is to deliver active ingredients directly to the site of action in the GI tract, which allows the intended therapeutic effect to occur without entering the systemic circulation as shown in Fig. 1.3.

Absorption process and action site of locally acting and orally administered gastrointestinal drugs
While local delivery is excellent from a therapeutic effect standpoint, it presents challenges when attempting to evaluate bioequivalence using standard techniques. Some locally acting GI drugs such as mesalamine are permeable to the intestinal membrane and can enter the systemic circulation while others such as vancomycin hydrochloride are not as permeable and have very low systemic availability (Zhang et al. 2013). There is a strong possibility that systemic exposure may not be directly correlated to the local concentration of the drug in the GI tract. In order to confirm bioequivalence, a selection of BE methods are often used depending on considerations of various factors, such as mechanism of drug delivery, mechanism of drug release, systemic absorption of the drug, drug physiochemical properties, and study feasibility.
It is currently recommended that bioequivalence methods for mesalamine include in vitro dissolution studies as well as in vivo BE studies with PK endpoints (Zhang et al. 2013). Because mesalamine is well absorbed from the GI tract, it is likely that the PK profiles obtained may reflect the local availability of the drug. In vitro dissolution in different solutions will confirm that the release profile is similar throughout the GI tract. On the other hand, vancomycin HCl is highly soluble and expected to be solubilized before reaching the site of action in the lower GI tract. As such, the FDA recommends that in vitro dissolution studies be conducted for the vancomycin HCl formulations that are quantitatively and qualitatively the same or an in vivo BE study be conducted with clinical endpoints if formulations are not quantitatively and qualitatively the same. The same quantitative and qualitative requirements ensure that there is no excipient interaction on the transport of vancomycin in vivo. Table 1.3 shows an example of BE methods for some locally acting gastrointestinal drug products.
It should be noted that if there is a safety concern related to systemic exposure or there are contributions of systemic exposure to efficacy, then the FDA Office of Generic Drugs (OGD) may recommend a PK study intended to demonstrate equivalent systemic exposure, in addition to any other study requested to demonstrate equivalent local delivery (FDA 2008b).
Detail discussions of locally acting GI drug bioequivalence will be presented in a subsequent chapter.
1.4.6 Bioequivalence for Nasal and Inhalation Products
1.4.6.1 Bioequivalence for Nasal Sprays for Local Action
Nasal spray products deliver drug to the nasal cavity by spraying a metered dose of the active ingredient that is dissolved or suspended in solutions or mixtures of excipients in nonpressurized or pressurized dispensers. Because of the delivery form and the target site of activity, the bioequivalence of locally acting nasal drug products is currently not believed to be evaluable via traditional bioequivalence methods used for systemically targeted drug products (i.e., blood plasma). For solution formulations of locally acting nasal drug products, the bioequivalence standard is based on the premise that in vitro studies would be more sensitive indicators of drug delivery to nasal sites of action than clinical studies (FDA 2003b) and there is no local in vivo drug dissolution step that might lead to differences in local bioavailability. The following in vitro tests can demonstrate equivalent product performance if there is formulation sameness and device comparability between test and reference products (Li et al. 2013):
-
1.
Single actuation content through container life
-
2.
Droplet size distribution (by laser diffraction)
-
3.
Drug in small particles/droplet size distribution
-
4.
Spray pattern
-
5.
Plume geometry
-
6.
Priming and repriming
In the case of formulation sameness, the inactive ingredient of the test and reference formulations must be qualitatively and quantitatively the same. Device comparability defines that the dimensions of all critical components that are involved in the dispensing of the formulation is comparable.
For suspension formulations, due to the presence of in vivo local dissolution of solid drug particles, the FDA’s bioequivalence requirements are based on weight-of-evidence which includes the six in vitro tests as well as the following two in vivo studies (Li et al. 2013):
-
1.
A clinical endpoint (PD) study to ensure equivalent delivery of drug substance to nasal sites of action.
-
2.
A PK endpoint study to establish equivalence of systemic exposure and potential systemic toxicity of the drug.
The addition of in vivo bioequivalence testing for suspension formulations stems from the current inability of particle sizing technologies to adequately distinguish between the active ingredient and suspending agent. The result is a potential difference of the active ingredient’s particle size distribution (PSD) between two formulations. Different particle size of drugs in different products could result in distinctive rate and extent of local in vivo dissolution, leading to different bioavailability/clinical results. Because of this concern, in vivo BE testing is needed for suspension formulations.
1.4.6.2 Bioequivalence for Locally Acting Orally Inhaled Drugs Products
Similar to locally acting nasal spray suspensions, locally acting orally inhaled drug products do not depend on systemic circulation for drug delivery and intended action. As such, bioequivalence for products such as dry powder inhalers (DPI) is established based on an aggregate weight of evidence approach that includes in vitro studies to demonstrate comparative in vitro performance, pharmacokinetic or pharmacodynamic studies to establish equivalence of systemic exposure, and pharmacodynamics or clinical endpoint studies to demonstrate equivalence in local action (Lee et al. 2009), as shown in Fig. 1.4.

The aggregate-weight-of-evidence approach for establishing bioequivalence of dry powder inhalers
Evaluation of formulation and device are considered to ensure bioequivalence. Because excipients can influence performance, such as the addition of magnesium stearate to drug-lactose mixture to improve particle deagglomeration, it is generally recommended that the qualitative and quantitative formulation aspects between test and reference products remain the same (within ±5 %). Pharmaceutical development data, involving in vitro testing of multiple drug-to-excipient ratios that encompass combinations below and above the ratios used in the test and reference products are needed to justify a test product formulation that is quantitatively different from the reference product. Likewise, although there are several types of DPI dosing systems (premetered single-dose units, drug reservoir (device metered), and premetered multiple dose units), it is recommended that the generic product device’s mechanism of function remain the same as that of the reference product. Furthermore, the generic product device itself should maintain a similar shape to ensure equivalence and decrease patient confusion when a generic product is substituted (Chrystyn 2007; Molimard et al. 2003).
Because in vitro testing is less variable and more sensitive to differences in bioequivalence, the following tests can be conducted to detect differences in test and reference products (FDA 2013b):
-
1.
Single inhalation content at different flow rates
-
2.
PSD at different flow rates
Although similar to the locally acting gastrointestinal drug products in that systemic circulation occurs after delivery to the local site, the drug moieties detected from systemic circulation for locally acting orally inhaled products include drugs from potentially multiple sites including the lung, buccal, and GI tract areas. Therefore, a systemic BE study is recommended to ensure equivalent systemic exposure of generic and reference drugs (Adams et al. 2010).
An additional part of the bioequivalence approach for demonstrating equivalence for locally inhalation products is the pharmacodynamics or clinical endpoint study. An example of a typical measurement is the Forced Expiratory Volume in 1 s (FEV1) which is the maximal amount of air an individual can exhale in 1 s.
1.4.7 Bioequivalence for Liposomal Products
A liposome is an artificially prepared vesicle comprises a lipid bilayer shell and an inner core of aqueous compartment. The drug substance may be encapsulated in the lipid bilayer or inner core. Liposome drug products may be designed to release drug to a particular target tissue, or to act as a parenteral dosage form for sustained release in systemic circulation. Due to the engineered properties, these nanoparticle drug products have altered pharmacokinetic and pharmacodynamics profiles. The success of liposome use as a drug carrier has been reflected in a number of liposome-based products which are commercially available or currently undergoing clinical trials. The first liposome drug product Doxil, a PEGylated liposome formulation of doxorubicin HCl shown in Fig. 1.5, was approved by the FDA in 1995.

Representation of a PEGylated liposomal doxorubicin (Jiang et al. 2011)
Liposomes such as Doxil can be biocompatible, biodegradable, and locally targeting. They can also avoid in vivo clearance by various mechanisms such as reticuloendothelial systems, renal clearance, and chemical or enzymatic inactivation (Scott 2008). Although the expected clinical behavior can be ideal, these liposomes are designed to exploit the enhanced permeability properties at the tissue site, and thus traditional bioequivalence methods such as pharmacokinetic measurements of systemic exposure alone may not be indicative of equivalent drug concentrations in the targeted tumor tissues. No direct correlations between plasma and target tissue concentrations have been established so far. As such, bioequivalence to Doxil can be demonstrated based on the following in vivo and in vitro tests recommended by FDA (FDA 2010a):
-
Same drug product composition
-
Same active loading process with an ammonium sulfate gradient
-
Equivalent in vitro liposome characteristics including liposome composition, state of encapsulated drug, internal environment of liposome, liposome size distribution, number of lamellar, grafted PEG at the liposome surface, electrical surface potential or charge, and in vitro leakage
-
Equivalent in vivo plasma pharmacokinetics of free and encapsulated drug
Requiring the same drug product composition, qualitatively and quantitatively, ensures that test and reference products use the same amounts of the same excipients. Requiring the same active loading manufacturing process with an ammonium sulfate gradient ensures equivalent contents within the liposome. Equivalent liposome size distribution and pharmacokinetics ensures equivalence in the mononuclear phagocyte system avoidance, long half-life, and liposome tumor distribution. Table 1.4 lists some of the proposed methods for evaluating in vitro leakage as well as the corresponding justification.
Once equivalent liposome distribution in target tissues is reached, equivalent in vivo pharmacokinetics and in vitro liposome characteristics will ensure equivalent drug delivery into cells. For example, the characterization of liposome surface chemistry can be used to assess liposome–cell interactions involved in liposome fusion or uptake mechanisms by tumor cells. In addition, equivalence in liposome internal environment, size distribution, state of encapsulated doxorubicin, and drug leakage can ensure equivalent drug leakage around tumor tissues or inside tumor cell endosomes or lysosomes. Plasma pharmacokinetics of free drug also accounts for drug release from liposomes.
Detail discussions of liposome product bioequivalence will be presented in a subsequent chapter.
1.5 Future Development
While the development of bioequivalence concept and standards, as well as its subsequent rise to become the regulatory requirement has made a monumental impact since the 1970s, there are still many unanswered questions in the field. For example, the current bioequivalence methods for many locally acting products use clinical endpoints for determination. The overall qualification and assessment of these endpoints may be lacking in sensitivity and need to be reevaluated. Similarly, questions have been raised regarding the sensitivity of in vitro testing for BE assessments, and how to develop better in vitro methods for implementation to improve BE standards. The benefits of in vitro testing, modeling, and simulation are enormous, but further investigations are needed to study the sensitivity, reliability, and correlation to clinical significance of these methods. For controlled release dosage forms such as monthly doses, the question is how the bioequivalence should be assessed for these dosage forms since it takes a long time to complete an in vivo study. These are only some of the questions that need to be answered to stimulate future improvements in bioequivalence methodology.
References
21 CFR 320. 21 CFR 320 Bioavailability and bioequivalence requirements. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=320.1. Accessed 9 July 2013
AAPS (1997) Science and regulations: individual and population bioequivalence—regulatory approaches and issues. America Association of Pharmaceutical Scientists (AAPS) Annual Meeting, Nov 2–6 1997, Boston
AAPS (1998) Scientific and regulatory issues in product quality: narrow therapeutic index drugs and individual bioequivalence. American Association of Pharmaceutical Scientists (AAPS) Workshop, Mar 16–18 1998, Arlington
AAPS (1999) Individual bioequivalence: realities and implementation. American Association of Pharmaceutical Scientists (AAPS) International Workshop, Aug 30–Sep 1 1999, Montreal
Adams WP, Ahrens RC, Chen ML, Christopher D, Chowdhury BA, Conner DP, Dalby R, Fitzgerald K, Hendeles L, Hickey AJ, Hochhaus G, Laube BL, Lucas P, Lee SL, Lyapustina S, Li B, O’Connor D, Parikh N, Parkins DA, Peri P, Pitcairn GR, Riebe M, Roy P, Shah T, Singh GJ, Sharp SS, Suman JD, Weda M, Woodcock J, Yu L (2010) Demonstrating bioequivalence of locally acting orally inhaled drug products (OIPs): workshop summary report. J Aerosol Med Pulm Drug Deliv 23:1–29
Agoram B, Woltosz WS, Bolger MB (2001) Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv Drug Deliv Rev 50(Suppl 1):S41–S67
Amidon GL, Lennernas H, Shah VP, Crison JR (1995) A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 12:413–420
Anderson S, Hauck WW (1990) Consideration of individual bioequivalence. J Pharmacokinet Biopharm 18:259–273
Barber HE, Calvey TN, Muir K, Hart A (1974) Biological availability and in vitro dissolution of oxytetracycline dihydrate tablets. Br J Clin Pharmacol 1:405–408
Barnett DB, Smith RN, Greenwood ND, Hetherington C (1974) Bioavailability of commercial tetracycline products. Br J Clin Pharmacol 1:319–323
Barr WH, Gerbracht LM, Letcher K, Plaut M, Strahl N (1972) Assessment of the biologic availability of tetracycline products in man. Clin Pharmacol Ther 13:97–108
Benet LZ (1999) Individual bioequivalence: have the opinions of the scientific community changed? http://www.fda.gov/ohrms/dockets/ac/01/slides/3804s2_09_benet.ppt. Accessed 8 Aug 2013
Benet LZ (2006) Therapeutic considerations of highly variable drugs. http://www.fda.gov/ohrms/dockets/ac/06/slides/2006-4241s2_2_files/frame.htm. Accessed 18 July 2013
Benet LZ (2013) The role of BCS (biopharmaceutics classification system) and BDDCS (biopharmaceutics drug disposition classification system) in drug development. J Pharm Sci 102:34–42
Brodie BB, Heller WM (1971) Bioavailability of drugs: proceedings. S. Karger, Basel
Cabana BE (1983) Assessment of 75/75 rule: FDA viewpoint. J Pharm Sci 72:98–100
Chen ML (1992) An alternative approach for assessment of rate of absorption in bioequivalence studies. Pharm Res 9:1380–1385
Chen ML, Davit B, Lionberger R, Wahba Z, Ahn HY, Yu LX (2011) Using partial area for evaluation of bioavailability and bioequivalence. Pharm Res 28:1939–1947
Chen ML, Lesko LJ (2001) Individual bioequivalence revisited. Clin Pharmacokinet 40:701–706
Chen ML, Patnaik R, Hauck WW, Schuirmann DJ, Hyslop T, Williams R (2000) An individual bioequivalence criterion: regulatory considerations. Stat Med 19:2821–2842
Chiou WL (1972) Determination of physiologic availability of commercial phenylbutazone preparations. J Clin Pharmacol New Drugs 12:296–300
Chrystyn H (2007) The Diskus: a review of its position among dry powder inhaler devices. Int J Clin Pract 61:1022–1036
Davit BM, Nwakama PE, Buehler GJ, Conner DP, Haidar SH, Patel DT, Yang Y, Yu LX, Woodcock J (2009) Comparing generic and innovator drugs: a review of 12 years of bioequivalence data from the United States Food and Drug Administration. Ann Pharmacother 43:1583–1597
FDA Orange Book Preface (2013) http://www.fda.gov/Drugs/DevelopmentApprovalProcess/ucm079068.htm. Accessed 7 Aug 2013
FDA (1996) FDA Advisory Committee for Pharmaceutical Science Meeting, August 1996, Gaithersburg
FDA (1997) FDA Advisory Committee for Pharmaceutical Science Meeting, May 1997, Gaithersburg
FDA (1998) FDA Advisory Committee for Pharmaceutical Science Meeting, October 1998, Gaithersburg
FDA (1999a) FDA Advisory Committee for Pharmaceutical Science Meeting, September 1999, Gaithersburg
FDA (1999b) Average, population, and individual approaches to establishing bioequivalence. http://www.fda.gov/OHRMS/DOCKETS/98fr/3657gd1.pdf. Accessed 8 Aug 2013
FDA (2000a) FDA Advisory Committee for Pharmaceutical Science Meeting, November 2000, Gaithersburg
FDA (2000b) Guidance for industry: waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070246.pdf. Accessed 12 July 2013
FDA (2003a) Guidance for industry bioavailability and bioequivalence studies for orally administered drug products general considerations. http://www.fda.gov/downloads/Drugs/…/Guidances/ucm070124.pdf. Accessed 26 Aug 2013
FDA (2003b) Guidance for industry: bioavailability and bioequivalence studies for nasal aerosols and nasal sprays for local action. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm070111.pdf. Accessed 31 July 2013
FDA (2004) Background information for advisory committee meeting on bioequivalence requirements for highly variable drugs and drug products. http://www.fda.gov/ohrms/dockets/ac/04/briefing/4034B1_07_Bioequivalence%20Requirments-Draft.doc. Accessed 21 Aug 2013
FDA (2008a) Draft guidance on vancomycin hydrochloride capsule. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM082278.pdf. Accessed 26 Aug 2013
FDA (2008b) Meeting of the advisory committee for pharmaceutical science and clinical pharmacology. FDA. http://www.fda.gov/ohrms/dockets/ac/08/briefing/2008-4370b1-01-FDA.pdf. Accessed 21 Aug 2013
FDA (2009a) Draft guidance on acarbose tablet. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM170242.pdf. Accessed 26 Aug 2013
FDA (2009b) Draft guidance on calcium acetate capsule. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM148185.pdf. Accessed 26 Aug 2013
FDA (2010a) Draft guidance on doxorubicin hydrochloride liposome injection. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatory%20Information/Guidances/UCM199635.pdf. Accessed 15 Aug 2013
FDA (2010b) Draft guidance on lubiprostone capsule. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM224220.pdf. Accessed 26 Aug 2013
FDA (2011a) Draft guidance on lanthanum carbonate tablet. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM270541.pdf. Accessed 26 Aug 2013
FDA (2011b) Draft guidance on sevelamer carbonate tablet. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm089620.pdf. Accessed 26 Aug 2013
FDA (2011c) Guidance on zolpidem tablet. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM175029.pdf. Accessed 26 Aug 2013
FDA (2011d) July 26th Topic 1: Bioequivalence (BE) and quality standards for narrow therapeutic index (NTI) drug products. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM263465.pdf. Accessed 13 Aug 2013
FDA (2012a) Draft guidance on cholestyramine power. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM273910.pdf. Accessed 26 Aug 2013
FDA (2012b) Draft guidance on mesalamine tablet. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM320002.pdf. Accessed 26 Aug 2013
FDA (2012c) Draft guidance on methylphenidate hydrochloride tablet. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM320007.pdf. Accessed 26 Aug 2013
FDA (2012d) Draft guidance on rifaximin tablet. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291392.pdf. Accessed 26 Aug 2013
FDA (2013a) Approved drug products with therapeutic equivalence evaluations. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/UCM071436.pdf. Accessed 31 July 2013
FDA (2013b) Draft guidance on fluticasone propionate: salmeterol xinafoate power/inhalation. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm081320.htm. Accessed 20 Oct 2013
Federal Register (1977) Bioavailability and bioequivalence requirements
Fleisher D, Li C, Zhou Y, Pao LH, Karim A (1999) Drug, meal and formulation interactions influencing drug absorption after oral administration. Clinical implications. Clin Pharmacokinet 36:233–254
Fourie Zirkelbach J, Jackson AJ, Wang Y, Schuirmann DJ (2013) Use of partial AUC (PAUC) to evaluate bioequivalence—a case study with complex absorption: methylphenidate. Pharm Res 30:191–202
Glazko AJ, Kinkel AW, Alegnani WC, Holmes EL (1968) An evaluation of the absorption characteristics of different chloramphenicol preparations in normal human subjects. Clin Pharmacol Ther 9:472–483
Grbic S, Parojcic J, Ibric S, Djuric Z (2011) In vitro-in vivo correlation for gliclazide immediate-release tablets based on mechanistic absorption simulation. AAPS PharmSciTech 12:165–171
Haidar SH, Davit B, Chen ML, Conner D, Lee L, Li QH, Lionberger R, Makhlouf F, Patel D, Schuirmann DJ, Yu LX (2008) Bioequivalence approaches for highly variable drugs and drug products. Pharm Res 25:237–241
Hauck WW, Anderson S (1984) A new statistical procedure for testing equivalence in two-group comparative bioavailability trials. J Pharmacokinet Biopharm 12:83–91
Hauck WW, Anderson S (1994) Measuring switchability and prescribability: when is average bioequivalence sufficient? J Pharmacokinet Biopharm 22:551–564
Hauck WW, Hyslop T, Chen ML, Patnaik R, Williams RL (2000) Subject-by-formulation interaction in bioequivalence: conceptual and statistical issues. FDA Population/Individual Bioequivalence Working Group. Food and Drug Administration. Pharm Res 17:375–380
Haynes JD (1981) Statistical simulation study of new proposed uniformity requirement for bioequivalency studies. J Pharm Sci 70:673–675
Heald D (2010) Conventional bioequivalence criteria may not ensure clinical equivalence and, therefore, interchangeability for products with complex pharmacokinetic profiles. FDA. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/AdvisoryCommitteeforPharmaceuticalScienceandClinicalPharmacology/UCM209322.pdf. Accessed 13 Aug 2013
Huang W, Lee SL, Yu LX (2009) Mechanistic approaches to predicting oral drug absorption. AAPS J 11:217–224
Jiang W, Lionberger R, Yu LX (2011) In vitro and in vivo characterizations of PEGylated liposomal doxorubicin. Bioanalysis 3:333–344
Lee SL, Adams WP, Li BV, Conner DP, Chowdhury BA, Yu LX (2009) In vitro considerations to support bioequivalence of locally acting drugs in dry powder inhalers for lung diseases. AAPS J 11:414–423
Li BV, Jin F, Lee SL, Bai T, Chowdhury B, Caramenico HT, Conner DP (2013) Bioequivalence for locally acting nasal spray and nasal aerosol products: standard development and generic approval. AAPS J 15:875–883
Lindenbaum J, Mellow MH, Blackstone MO, Butler VP Jr (1971) Variation in biologic availability of digoxin from four preparations. N Engl J Med 285:1344–1347
Lionberger R (2004) Bioequivalence of locally acting GI drugs. http://www.fda.gov/ohrms/dockets/ac/04/slides/2004-4078S2_11_Lionberger.ppt. Accessed 21 Aug 2013
Lionberger, R. 2008. Bioequivalence of Poorly Soluble Locally Acting GI Drugs [Online]. FDA. Available: http://www.fda.gov/ohrms/dockets/ac/08/slides/2008-4370s2-02-fda-lionberger.ppt. Accessed 21 Aug 2013
Lionberger RA, Raw AS, Kim SH, Zhang X, Yu LX (2012) Use of partial AUC to demonstrate bioequivalence of zolpidem tartrate extended release formulations. Pharm Res 29:1110–1120
Midha KK, Rawson MJ, Hubbard JW (2005) The bioequivalence of highly variable drugs and drug products. Int J Clin Pharmacol Ther 43:485–498
Molimard M, Raherison C, Lignot S, Depont F, Abouelfath A, Moore N (2003) Assessment of handling of inhaler devices in real life: an observational study in 3811 patients in primary care. J Aerosol Med 16:249–254
OTA (1974a) Drug bioequivalence. Recommendations from the Drug Bioequivalence Study Panel to the Office of Technology Assessment, Congress of the United States. J Pharmacokinet Biopharm 2:433–466
OTA (1974b) Drug bioequivalence: a report of the Office of Technology Assessment, Drug Bioequivalence Study Panel, Washington, The Office: for sale by the Superintendent of Documents, U.S. Government Printing Office
Patterson S, James B (2005) Bioequivalence and statistics in clinical pharmacology. CRC Press, Boca Raton
Schuirmann DJ (1987) A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm 15:657–680
Scott RC (2008) Targeting immunoliposomes containing pro-angiogenic compounds to the infarcted rat heart. Philadelphia: Temple University
Skelly JP (1976) Bioavailability and bioequivalence. J Clin Pharmacol 16:539–545
Skelly JP (2010) A history of biopharmaceutics in the Food and Drug Administration 1968–1993. AAPS J 12:44–50
Stier EM, Davit BM, Chandaroy P, Chen ML, Fourie-Zirkelbach J, Jackson A, Kim S, Lionberger R, Mehta M, Uppoor RS, Wang Y, Yu L, Conner DP (2012) Use of partial area under the curve metrics to assess bioequivalence of methylphenidate multiphasic modified release formulations. AAPS J 14:925–926
Thompson T (2011) The clinical significance of drug transporters in drug disposition and drug interactions. In: Bonate PL, Howard DR (eds) Pharmacokinetics in drug development. Springer, New York
Van Petten GR, Feng H, Withey RJ, Lettau HF (1971) The physiologic availability of solid dosage forms of phenylbutazone. I. In vivo physiologic availability and pharmacologic considerations. J Clin Pharmacol New Drugs 11:177–186
Vitti TG, Banes D, Byers TE (1971) Bioavailability of digoxin. N Engl J Med 285:1433–1434
Wagner JG, Christensen M, Sakmar E, Blair D, Yates JD, Willis PW 3rd, Sedman AJ, Stoll RG (1973) Equivalence lack in digoxin plasma levels. JAMA 224:199–204
Wu CY, Benet LZ (2005) Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a biopharmaceutics drug disposition classification system. Pharm Res 22:11–23
Yu LX (1999) An integrated model for determining causes of poor oral drug absorption. Pharm Res 16:1883–1887
Yu LX (2008) Bioequivalence of locally acting gastrointestinal drugs: an overview. FDA. http://www.fda.gov/ohrms/dockets/ac/08/slides/2008-4370s2-01-FDA-YU.ppt. Accessed 21 Aug 2013
Yu LX (2011) Quality and bioequivalence standards for narrow therapeutic index drugs. FDA. http://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/AbbreviatedNewDrugApplicationANDAGenerics/UCM292676.pdf. Accessed 13 Aug 2013
Yu LX (2013) Scientific and regulatory considerations for the new requirements for demonstrating bioequivalence of NTI drugs in US. AAPS
Yu LX, Amidon GL (1998a) Characterization of small intestinal transit time distribution in humans. Int J Pharm 171:157–163
Yu LX, Amidon GL (1998b) Saturable small intestinal drug absorption in humans: modeling and interpretation of cefatrizine data. Eur J Pharm Biopharm 45:199–203
Yu LX, Crison JR, Amidon GL (1996a) Compartmental transit and dispersion model analysis of small intestinal transit flow in humans. Int J Pharm 140:111–118
Yu LX, Lipka E, Crison JR, Amidon GL (1996b) Transport approaches to the biopharmaceutical design of oral drug delivery systems: prediction of intestinal absorption. Adv Drug Deliv Rev 19:359–376
Zhang X, Lionberger RA, Davit BM, Yu LX (2011) Utility of physiologically based absorption modeling in implementing Quality by Design in drug development. AAPS J 13:59–71
Zhang X, Zheng N, Lionberger RA, Yu LX (2013) Innovative approaches for demonstration of bioequivalence: the US FDA perspective. Ther Deliv 4:725–740
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 The United States Government
About this chapter
Cite this chapter
Yu, A., Sun, D., Li, B.V., Yu, L.X. (2014). Bioequivalence History. In: Yu, L., Li, B. (eds) FDA Bioequivalence Standards. AAPS Advances in the Pharmaceutical Sciences Series, vol 13. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1252-0_1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1252-0_1
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1251-3
Online ISBN: 978-1-4939-1252-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)