
Abstract
Despite effective antimicrobial therapy, case-fatality rates and neurologic sequelae of bacterial meningitis remain unacceptably high. Adverse outcomes are related primarily to neurologic complications occurring secondary to meningitis. These complications are mainly a consequence of a hyper-inflammatory reaction to bacterial infection of the subarachnoid space. The harmful inflammatory response is initiated by the recognition of bacterial products through pattern recognition receptors such as toll-like receptors. Their activation leads to a MyD88-dependent production of multiple pro-inflammatory factors like cytokines of the interleukin-1 family or terminal complement products. Subsequently, huge numbers of neutrophils are recruited to the site of infection where they release their antimicrobial arsenal, e.g., oxidants. This can cause collateral damage to brain tissue, resulting in the liberation of endogenous danger molecules. Their presence is also recognized by host pattern recognition receptors and, in consequence, mediates an aggravation and propagation of the hyper-inflammatory response. Based on this knowledge, the most promising targets for adjunctive therapy of bacterial meningitis seem to be limiting the release of bacterial products and interfering with the generation of key pro-inflammatory host factors.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pneumococcal meningitis
- Toll-like receptors
- Interleukin
- Anaphylatoxin
- High-mobility group box 1 protein
- Pneumolysin
- Daptomycin
1 Introduction
Bacterial meningitis remains a serious threat to global health. Every year, meningococcal meningitis epidemics threaten millions of people in the African meningitis belt. In this area, close to 1,000,000 cases were reported in the last 20 years. Of these cases, approximately 100,000 died, with another 100,000–200,000 developing neurological sequelae [1]. Aside from epidemics, at least 1.2 million cases of endemic bacterial meningitis are estimated to occur worldwide each year with 135,000 deaths [2]. This makes bacterial meningitis one of the top ten infectious causes of death on Earth. Three species, Streptococcus pneumoniae, Neisseria meningitidis, and S. agalactiae, are responsible for most cases of bacterial meningitis. Among these bacteria, only N. meningitidis is able to generate epidemics. S. agalactiae is the predominant pathogen among newborns, N. meningitidis among children 2–18 years old, and S. pneumoniae among adults [3, 4]. Pneumococcal meningitis has the worst prognosis: even with the best medical care and the use of modern antibiotics (plus adjuvant dexamethasone therapy), still about 15 % of the patients with pneumococcal meningitis die of the disease and up to one-third of survivors remain with neurologic deficits [4–7]. Unfortunate courses of the disease are mainly due to intracranial complications occurring secondary to meningitis, notably cerebrovascular alterations such as vasculitis, vasospasm, or venous thrombosis as well as hydrocephalus and brain edema [8–10]. These alterations result in hypoperfusion and increased intracranial pressure, frequently leading to cerebral ischemia and/or herniation [11, 12]. Nearly 60 years ago, the hypothesis that these complications occur predominantly as a consequence of a hyper-inflammatory reaction within the central nervous system was formulated [13]. This hypothesis became the rationale for treating patients suffering from bacterial meningitis with immunosuppressive corticosteroids. Nowadays, dexamethasone is recommended for adjunctive therapy in selected patients, namely, in adults who suffer from pneumococcal meningitis and have not yet received antimicrobial treatment. Adjunctive dexamethasone is, however, far from giving complete protection. It only halves mortality and has only marginal effects on neurologic sequelae. Furthermore, a positive effect of dexamethasone was not found in studies performed in low-income countries (for review, see [14, 15]). Thus, there still is the urgent need for additional treatment strategies which can further reduce the adverse outcome of the disease. It is likely that the key will lie in more pathophysiologically targeted approaches. The scope of this article is to summarize the current knowledge on the pathophysiology of bacterial meningitis, using the example of pneumococcal meningitis which is the experimentally best characterized subtype, and to provide an outlook on promising therapeutic approaches.
2 Pathophysiology of Pneumococcal Meningitis
2.1 Survival of Pathogens in the CSF
Pneumococcal meningitis typically develops when bacteria enter the subarachnoid space from the blood compartment (hematogenous meningitis; predominant route in neonates and children) or through continuous spread of infection from a nearby focus (the mostly used route in adults). The subarachnoid space is the space between the arachnoid mater and the pia mater, which contains cerebrospinal fluid (CSF). From an immunological point of view, the subarachnoid space is a special compartment of the body. The subarachnoid space lacks a fully organized drainage by lymphatic vessels [16]. Moreover, soluble pattern recognition receptors (PRRs) like complement factors that perceive the presence of bacteria and mediate their uptake by phagocytes are virtually absent [17, 18]. Additionally, highly specialized blood–CSF barriers seclude the subarachnoid space from the blood circulation and impede the entry of most blood components like soluble PRRs into the CSF [19]. Even in the presence of bacterial meningitis, which is regularly associated with damage to blood–CSF barriers, concentrations of soluble PRRs remain far below those found in serum [20]. In contrast to this humoral deficit, functionally active macrophages, dendritic cells, and mast cells are present in tissues lining the CSF, namely, the choroid plexus, the perivascular spaces, and the leptomeninges [21, 22]. These cells are potential candidates for sensing the invasion of bacteria into the CSF through their cellular PRRs [21, 22]. The PRRs are expressed on the surface (like toll-like receptor (TLR) 2), within endosomes (like TLR9), and in the cytoplasm nucleotide-binding oligomerization domain (Nod)-like receptors (NLRs) of these cells. Activation of PRRs can initiate an inflammatory response by activating specific transcription factors (like nuclear factor (NF)-κB) and subsequently stimulating the synthesis and release of a variety of cytokines. However, the reactivity of the immune cells is probably restricted by diverse immunosuppressive factors that are constitutively expressed in the CSF, like members of the transforming growth factor family, cystatin C, or tumor necrosis factor (TNF)-related apoptosis-inducing ligand [23–26]. As a consequence, when bacteria reach the subarachnoid space, they can multiply easily, reaching similar high titers (up to 109 colony-forming units (CFU)/ml) as under bacterial culture conditions [27]. Bacteria like S. pneumoniae undergo autolysis when they are injured by a hostile environment or attain the stationary phase of growth. Hence, pneumococcal degradation products are liberated into the extracellular milieu. Their recognition by PRRs is the starting shot for the host inflammatory reaction. All in all, the CSF space exhibits a defective humoral (but not cellular) immunity which allows bacteria to prosper. This leads to the generation of large quantities of bacterial products and, as a result, a massive inflammatory reaction in the subarachnoid space.
2.2 Initiation of the Immune Response
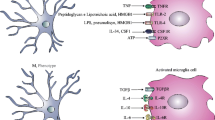
In landmark experiments in the 1980s, the major pneumococcal cell wall components peptidoglycan (PG) and lipoteichoic acid (LTA) were indicated to be the key activators of the host immune response during meningitis (Fig. 1) [28, 29]. In a rabbit model, intracisternal injection of pneumococcal PG or LTA was sufficient to induce meningeal inflammation and to cause clinical symptoms of meningitis [30]. Accordingly, in patients with pneumococcal meningitis, mortality and morbidity from the disease were significantly associated with high LTA concentrations in the CSF [31]. In the last 15 years, the mechanisms underlying immune activation by pneumococcal cell wall components have been clarified to a great extent. First insight came from overexpression assays in cell lines: when the pattern recognition receptor TLR2 was ectopically expressed in fibroblast cell lines, the cells became responsive to pneumococcal degradation products or live S. pneumoniae [32, 33]. In further experimental series, the pneumococcal cell wall components PG and LTA were identified as the key ligands for TLR2 [34]. Moreover, TLR4, also located at the cell surface, was reported to interact with the pneumococcal toxin pneumolysin (PLY) [35, 36], but this finding was questioned later by other groups [37, 38]. In addition, TLR9 was described to sense the presence of genomic DNA from S. pneumoniae [39]. More recently, TLR13 was implicated in the recognition of pneumococcal RNA [40]. Investigations on isolated macrophages of gene-deficient mice demonstrated that single deficiencies of TLR2, TLR4, or TLR9 had no significant impact on pneumococci-induced macrophage activation. The combined loss of TLR2, TLR4, and TLR9, however, resulted in a marked reduction in cytokine production by macrophages upon exposure to S. pneumoniae [41]. It is also noteworthy that macrophages become nearly unresponsive to Gram-positive bacteria when they have defects in endosomal TLR signalling in addition to the lack of TLR2 and TLR4 [40]. Besides endosomal TLRs, NLRs likely contribute to the immune activation in response to internalized pneumococci. This is supported by the following observations: (1) Opitz et al. [42] reported that viable S. pneumoniae are capable of invading human fibroblasts. (2) Genetic complementation studies in human fibroblasts revealed that NF-κB activation induced by S. pneumoniae depends on the NLR Nod2. (3) By using primary cells from gene-deficient mice, NOD2 was found necessary for mounting a maximal inflammatory responses of microglial cells and astrocytes to live S. pneumoniae [43]. (4) Apart from NOD2, the NLR family, pyrin domain-containing protein 3 (NLRP3) was implicated as a sensor for pneumolysin and was capable of mediating interleukin (IL)-1β production by macrophages following challenge with pneumolysin or viable S. pneumoniae that express pneumolysin [37, 38, 44]. Collectively, these in vitro findings suggest that S. pneumoniae is sensed by immunocompetent cells through TLRs and NLRs in a cooperative manner. Thereby, extracellular bacterial components are sensed in a synergistic fashion by TLR2 and TLR4, while internalized bacterial fragments are recognized by endosomal TLRs and NOD2.

A simplified model for the recognition of Streptococcus pneumoniae by pattern recognition receptors. See text for details. Lyt A N-acetylmuramoyl-l-alanine amidase (autolysin), LTA lipoteichoic acid, PG peptidoglycan, PLY pneumolysin, MDP muramyl dipeptide, TLR toll-like receptor, Nod2 nucleotide-binding oligomerization domain (Nod)-like receptor 2, Nlrp3 Nod-like receptor family, pyrin domain-containing protein 3, NF nuclear factor
Substantial in vivo evidence for the involvement of TLRs in pneumococcal meningitis came from studies in mice lacking functional MyD88 which is kind of a bottleneck in the signalling cascades of all TLRs except TLR3. In an adult mouse model of pneumococcal meningitis, MyD88-deficient mice exhibited a defective host immune response inside the CSF, as evidenced by a substantial abrogation of the expression of pro-inflammatory cytokines (e.g., IL-1β), chemokines, and complement factors in the brain and, hence, an insufficient neutrophil infiltration [45]. By utilizing mice with single or combined deficiencies of cell surface and endosomal TLRs, our group was able to demonstrate that TLR2, TLR4, and TLR11, TLR12 or TLR13 (but not TLR3, TLR7, and TLR9) are crucial for mounting an immune response in the CSF in pneumococcal meningitis [41] (unpublished data). This is deduced from the following constellation of findings: first, infected TLR2-TLR4-double-deficient mice showed a 50 % reduction in CSF leukocyte counts and a selective cytokine production, whereas the single deficiency of neither TLR2 nor TLR4 had any substantial impact on meningeal inflammation. Moreover, additional TLR9 or TLR3-TLR7-TLR9 deficiency did not result in a significant further attenuation of the inflammatory reaction as observed in TLR2-TLR4-double-deficient mice [41] (unpublished data). In addition, infected Unc93b1 mutant mice that lack endosomal TLR signalling (TLR3, TLR7, TLR9, TLR11, TLR12, TLR13) exhibited an inflammatory phenotype comparable to that of TLR2-TLR4-double-deficient mice. Finally, the combined loss of endosomal TLR signalling, TLR2, and TLR4 was accompanied by a reduction of CSF pleocytosis by about 75 % (unpublished data). This reduction is quite similar to that observed in MyD88-deficient mice [45]. The fact that MyD88 deficiency was paralleled by a strong but incomplete inhibition of the host immune response argues for the presence of additional PRRs in the recognition of S. pneumoniae in vivo. Genetic association studies showed an increased risk for pneumococcal infections in humans with complement (C) deficiencies [46]. Moreover, studies in animal models demonstrated the importance of an intact C system for a successful host defense against systemic pneumococcal infections like pneumonia and sepsis [47]. Accordingly, in a mouse model of pneumococcal meningitis, mice lacking the complement factors C1q or C3 displayed an enhanced bacterial outgrowth in the brain, which was associated with an attenuated innate immune response [48]. As mentioned above, complement concentrations are rather low in the CSF under normal conditions but increased substantially during the course of meningitis. The increase in C concentrations, however, occurs in a MyD88-dependent manner, arguing against a role of C factors as initial sensors of pneumococcal infection. Other potential sensors for pneumococcal infection of the subarachnoid space include NOD2 and NLRP3, as indicated by in vitro data [43]. This concept is strengthened by findings in mouse models where (1) increases in brain levels of the inflammatory cytokine TNFα and the chemokine CCL3 that were observed after intracerebral S. pneumoniae inoculation were virtually absent in NOD2-deficient mice and (2) the infiltration of leukocytes into the subarachnoid space following intracisternal pneumococcal infection was significantly lower in NLRP3-deficient mice than in wild-type mice [43, 44].
All in all, the presence of S. pneumoniae in the subarachnoid space seems to be initially recognized by TLR2, TLR4, a currently unidentified endosomal TLR (possibly TLR13) as well as other PRRs like NOD2 and NLRP3. Their engagement leads to the activation of transcription factors like NF-κB [49] and, as a consequence, the production of pro-inflammatory cytokines and complement factors [45, 48]. These host mediators, in turn, seem to be critical for the amplification of inflammation in pneumococcal meningitis, as described in the following section.
2.3 Amplification and Propagation of the Immune Response
2.3.1 Role of IL-1 Family Cytokines in Pneumococcal Meningitis
Among the cytokines that have been implicated in the amplification and perpetuation of meningeal inflammation, IL-1 family cytokines are prominent (Fig. 2) (for detailed information about this cytokine family, see [50]). Elevated concentrations of IL-1β and IL-18 were observed in CSF samples withdrawn from patients with bacterial meningitis on hospital admission. High CSF IL-1β (but not IL-18) levels were found to be significantly associated with high CSF leukocyte numbers and an adverse clinical outcome [51, 52]. In animal models, intracisternal injection of recombinant IL-1β was sufficient for inducing meningitis [53, 54], and antibodies directed against IL-1β attenuated meningeal inflammation after intracisternal pneumococcal infection [55]. In line with the latter finding, mice lacking the IL-1 receptor type 1 exhibited less profound inflammatory infiltrates in the leptomeninges and lower brain cytokines levels than wild-type mice in a mouse model of hematogenous pneumococcal meningitis [56]. In this model, IL-18-deficient mice also showed a suppressed inflammatory response, as evidenced by a less profound inflammatory infiltrate around the meninges as well as lower brain cytokine and chemokine concentrations [57]. Accordingly, using a mouse model in which S. pneumoniae is instilled directly into the CSF, we observed that (1) IL-1 receptor blockade (by anakinra) significantly attenuated meningeal inflammation and (2) IL-18 neutralization (using recombinant IL-18 binding protein) in addition to IL-1 receptor blockade resulted in a further reduction of CSF pleocytosis [44]. Moreover, mice lacking caspase-1 which is crucial for the generation of active IL-1β and IL-18 showed a strongly diminished inflammatory host response, and treatment of rats with a broad spectrum caspase inhibitor resulted in a marked attenuation of meningeal inflammation [58]. Similarly, Braun et al. reported that this inhibitor also attenuated leukocyte influx into the CSF in rabbits with pneumococcal meningitis [59].

Schematic diagram of the key steps in the pathophysiological cascade of pneumococcal meningitis. See text for details. CSF cerebrospinal fluid space, PAMPs pathogen-associated molecular patterns, DAMPs danger-associated molecular patterns, HMGB1 high-mobility group box 1 protein, IL interleukin, C5a complement component C5a, CXCLs chemokines
Combined, these data provide substantial evidence for IL-1 family cytokines as key regulators of inflammation. On the one hand, they may boost TLR-induced inflammation by forming a positive feedback loop involving IL-1 receptors and MyD88, the adapter molecule shared by IL-1β, IL-18, and most TLRs. On the other hand, IL-1 family cytokines may contribute to the perpetuation of inflammation. This hypothesis is deduced from our observation that treatment with IL-1β-neutralizing antibodies when started at 21 h after pneumococcal infection (in combination with the antibiotic ceftriaxone) is still effective in reducing meningeal inflammation. In contrast, co-application of ceftriaxone with antibodies directed against both TLR2 and TLR4 had no impact on the host immune response, arguing against a major role of these signalling receptors in established pneumococcal meningitis [60].
2.3.2 Role of the Anaphylatoxin C5a in Pneumococcal Meningitis
Apart from complement opsonins (like C1q or C3b), the complement activation cascade yields soluble factors known as anaphylatoxins (like C3a and C5a) and ends in the formation of the terminal complement complex (C5b-9) [61, 62]. The anaphylatoxins are generally considered pro-inflammatory polypeptides. Their effector functions include chemotaxis and activation of granulocytes, mast cells, and macrophages [63]. C5b-9 causes cytolysis through the formation of the membrane attack complex (MAC), and sub-lytic MAC and soluble C5b-9 also possess a multitude of non-cytolytic immune functions [64]. Markedly elevated C5a and C5b-9 concentrations were detected in the CSF of meningitis patients and in brain lysates of infected mice that correlated with CSF leukocyte counts [60]. In rabbits, intracisternal injection of C5a caused a rapid influx of leukocytes into the CSF [65]. Similarly, intracerebroventricular application of C5b-9 resulted in the production of cytokines (like IL-1β), chemokines, and subsequent accumulation of neutrophils in the CSF [66]. Moreover, the chemotactic activity of CSF samples obtained from rabbits with pneumococcal meningitis could be inhibited by treatment with antibodies to native human C5 [67]. In experiments using mouse strains with selected complement deficiencies, C5a was singled out to be crucial for the propagation of the inflammatory response in pneumococcal meningitis. The deficiency of the receptor for C5a (but not the receptor for C3a or of C6) was associated with a profound reduction of brain cytokine/chemokine expression and in CSF pleocytosis [60]. In addition, treatment of mice with neutralizing antibodies directed against C5, irrespective if started prior or 24 h after infection, dampened the host immune response, suggesting that C5a acts both as an early and late mediator of inflammation in pneumococcal meningitis.
All in all, as a consequence of release of IL-1 family cytokines and C5a, large numbers of neutrophils are recruited into the subarachnoid space. The recruitment of neutrophils to sites of infection is required for an effective host defense against invading pathogens. However, their defense mechanisms that destroy or digest pathogens can also be deleterious to host tissue.
2.4 Maintenance of the Immune Response
Stressed or injured cells can release alarm signals (so-called danger-associated molecular patterns, DAMPs) that can orchestrate inflammation [68, 69]. Well-known DAMPs include heat shock proteins, S100 proteins, and high-mobility group box 1 protein (HMGB1) [70]. HMGB1 is a ubiquitously expressed, highly conserved nuclear protein with multiple intracellular functions including stabilizing nucleosome structure and facilitating DNA bending [71]. It can be actively secreted by macrophages upon stimulation with PAMPs or pro-inflammatory cytokines (through a nonconventional pathway which requires inflammasome assembly and caspase-1 activation [72]) or passively released from dying cells following nuclear and cell membrane disintegration (for review, see [71, 73, 74]). Extracellular HMGB1 behaves much like a cytokine. HMGB1, by itself and/or by forming complexes with exogenous or endogenous pro-inflammatory molecules, can induce and enhance the production of cytokines and chemokines. In addition, HMGB1 can promote chemotaxis and accumulation of granulocytes at inflammatory sites (for review, see [71, 73, 74]). Recently, two case studies reported that HMGB1 levels were significantly elevated in CSF samples from children with bacterial meningitis as compared to those from children with no or aseptic meningitis [75, 76]. Correspondingly, we detected large quantities of HMGB1 in the CSF of adult patients with pneumococcal meningitis as well as in mice subjected to pneumococcal meningitis [77]. In the mouse model, we further observed a substantial rise in CSF HMGB1 between 24 and 45 h after infection, pointing at a possible role of this protein in advanced rather than in early stages of the disease. Accordingly, in the mouse model, treatment with the HMGB1 antagonists, ethyl pyruvate or Box A protein, had no effect on the development of meningitis but led to better resolution of inflammation during antibiotic therapy. Additional experiments using gene-deficient mice and murine neutrophils provided evidence that HMGB1 acts as a chemoattractant for neutrophils in a RAGE (receptor for advanced glycosylation end products)-dependent fashion. Moreover, by using macrophages, we observed that the release of HMGB1 from these cells upon challenge with S. pneumoniae is passive in nature. All in all, these data suggest that HMGB1, presumably released from dying cells, acts as a propagator of inflammation in pneumococcal meningitis. This may provide an explanation for the empiric observation that inflammation can persist over days even though antibiotic therapy sterilizes the CSF quickly and is paralleled by a fast reduction in CSF pneumococcal fragments (within hours) [78–82]. This delay in the resolution of inflammation may be the consequence of a vicious cycle in which inflammation-induced cell injury leads to the release of endogenous DAMPs that drive the inflammatory response, causing further damage.
3 Mechanisms of Brain Injury in Pneumococcal Meningitis
Accumulation of neutrophils at sites of infection is required for an effective host defense. However, activated neutrophils secrete a large arsenal of cytotoxic agents which can also damage host cells. Over 50 years ago, Johnson and colleagues [13] were the first to hypothesize that, in pneumococcal meningitis, the inflammatory response does more harm than good. The validity of this hypothesis was established by studies in animal models of the disease. First strong evidence for a harmful role of neutrophils in bacterial meningitis came from studies with antibodies against adhesion-promoting receptors of neutrophils. In a rabbit model, intravenous injection of anti-CD18 antibodies was reported to effectively block the development of pleocytosis in the CSF of animals challenged intracisternally with living S. pneumoniae or pneumococcal cell wall components [30]. This effect was associated with protection from blood–brain barrier injury. Therapy with anti-CD18 antibodies also prevented development of brain edema and death in animals infected with a lethal dose of S. pneumoniae [30]. Similarly, in a mouse model, parenteral treatment with anti-CD18 antibodies effectively inhibited leukocyte recruitment to the CSF and attenuated hippocampal injury 24 h after instillation of pneumococcal cell wall components into the lumbar spinal channel [26]. These findings were strengthened by results of mouse studies in which neutrophils were depleted by cell-specific antibodies [26, 60, 83]. The elimination of neutrophils resulted in a dramatic reduction of meningeal inflammation, as indicated by markedly lower CSF leukocyte numbers and brain cytokine concentrations. This was paralleled by significant reductions in intracranial pressure, blood–brain barrier breaching, and intracerebral bleeding (due to vasculitis) [44, 60, 83]. Combined, these studies implicated neutrophils to be major effector cells of brain injury in pneumococcal meningitis.
Among the effector molecules in the neutrophils’ arsenal are strong oxidants like peroxynitrite and proteolytic enzymes such as matrix metalloproteinase (MMP). Oxygen radicals can exert a vast variety of cytotoxic effects, e.g., through lipid peroxidation, DNA strand breakage, or mitochondrial damage. Oxidative alterations to vital macromolecules such as membrane phospholipids, DNA, or proteins were detected in brain samples obtained from both patients who died from meningitis and in animal models of meningitis (for review, see [84]). In humans with bacterial meningitis, high-grade oxidative stress as indicated by high CSF levels of biomarkers of oxidative stress such as nitrotyrosine was significantly associated with an adverse outcome of the disease [85]. Accordingly, studies in rodent models provided substantial evidence that antioxidant therapy can be protective against meningitis-associated brain injury (for review, see [84]). Besides oxygen radicals, proteolytic enzymes like MMP are released from activated neutrophils. Abnormal production and activation of these proteases can result in blood–brain barrier breaching and neuronal cell death [86]. High concentrations of MMP-9 were found in CSF samples of patients and animals with bacterial meningitis (for review, see [87]). Thereby, CSF MMP-9 concentrations were significantly higher in patients who developed neurologic sequelae than those who fully recovered. Moreover, experimental studies conducted in animal models of pneumococcal meningitis showed that MMP inhibitors (like GM6001 and BB-1101) are capable of reducing brain damage, neurologic sequelae, and mortality from pneumococcal meningitis.
Taken together, meningitis-associated brain damage is predominantly due to the massive accumulation of neutrophils inside the central nervous system whose antimicrobial weapons, namely, oxidants and MMPs, cause collateral damage to host cells.
4 Potential Targets for Therapy
New ideas for adjunctive therapy have emerged from studies on the mechanisms underlying meningitis-associated brain pathology. The principle behind novel treatment strategies is to reduce CNS inflammation by interfering at critical steps of the inflammation cascade which compromise (1) release of inflammatory bacterial products (PAMPs), (2) recognition of these PAMPs, (3) amplification and perpetuation of the immune response, as well as, (4) generation and release of cytotoxic agents (see also reviews [15, 88]). The following section will highlight two promising approaches for adjunctive therapy of pneumococcal meningitis, namely, the coadministration of non-bacteriolytic antibiotics as well as neutralizing antibodies directed against C5.
4.1 Limiting the Release of Inflammatory Bacterial Products
During conventional treatment of pneumococcal meningitis with β-lactam antibiotics, large amounts of pneumococcal cell wall degradation products are liberated into the CSF. As a consequence, the inflammatory host reaction is boosted, potentially causing additional harm to host tissues. Therefore, non-bacteriolytic antibiotics like daptomycin may represent a promising option for meningitis therapy. Daptomycin appears to insert into the cell membrane of Gram-positive cells, leading to pore formation and cellular depolarization, resulting in an arrest of DNA, RNA, and protein synthesis, and subsequently in non-lytic cell death [89]. In a rabbit model of pneumococcal meningitis, daptomycin monotherapy was superior to ceftriaxone monotherapy and was highly efficacious in sterilizing the CSF [82]. Administration of dexamethasone prior to daptomycin affected the antibacterial activity of daptomycin only marginally, either as monotherapy or combined with ceftriaxone, although the penetration of daptomycin into inflamed meninges was significantly reduced by two-thirds [90, 91]. In an infant rat model of pneumococcal meningitis, daptomycin monotherapy was demonstrated to clear pneumococci more rapidly from the CSF than ceftriaxone, to attenuate CSF inflammation, and to prevent the development of cortical injury [92, 93]. Since daptomycin (due to a lack of efficacy in pneumococcal pneumonia) is not a candidate for monotherapy of pneumococcal meningitis, supplementary studies assessed whether combining daptomycin with ceftriaxone is superior to ceftriaxone monotherapy. In an infant rat model, the combination therapy was accompanied with reduced inflammation, less brain damage, and improved hearing capacity [94]. The neuroprotective efficacy of this therapeutic approach was recently confirmed by our group in an adult mouse model (unpublished data). Open questions like the comparison of the antibiotic co-treatment with co-therapies consisting of dexamethasone and ceftriaxone (current standard therapy) as well as of daptomycin, dexamethasone, and ceftriaxone underline the need of further experimental investigations before clinical trials can be attempted. Moreover, human data on the CSF penetration of daptomycin are scarce. A recent study reported that mean concentrations of daptomycin in the CSF after a single intravenous dose (10 mg/kg) were significantly lower in patients than that previously reported in animal studies [95]. In order to better characterize the CSF penetration of this drug, additional pharmacokinetic studies evaluating multiple and/or higher dosages of daptomycin are necessary in humans, especially in those suffering from pneumococcal meningitis.
4.2 Neutralizing Endogenous C5a Activity
The anaphylatoxin C5a was identified to be a key player in the inflammatory cascade of pneumococcal meningitis. Both genetic deficiency of the receptor of C5a and pharmacologic neutralization of C5 resulted in a marked reduction of meningeal inflammation but also of meningitis-associated neuropathologic alterations like blood–brain barrier disruption or cerebral hemorrhages [60]. Interestingly, in adults with bacterial meningitis (including pneumococcal meningitis), high CSF C5a levels were associated with death and an unfavorable outcome [60]. Therefore, C5a was hypothesized to be a promising target for adjunctive therapy in pneumococcal meningitis. In line with this hypothesis, adjuvant treatment with a monoclonal antibody directed against C5 was completely protective against death due to pneumococcal meningitis in an adult mouse model. Moreover, this treatment strategy was effective in dampening meningitis-induced neuropathologic alterations. Its efficacy was clearly superior to that of adjuvant dexamethasone [60]. Since anti-C5 antibodies are already licensed for clinical use (e.g., in patients with paroxysmal nocturnal hemoglobinuria), adjuvant therapy with anti-C5 antibodies may be a promising therapeutic approach for patients with bacterial meningitis. However, this treatment approach still needs to be evaluated in meningitis models that measure neurologic (long-term) sequelae and/or use other meningitis pathogens (especially Neisseria meningitidis which can be killed by the membrane attack complex whose formation is blocked by anti-C5 antibodies). Moreover, data on its efficacy in combination with dexamethasone are lacking.
5 Conclusion
During the past two decades, great progress has been made in our understanding on the immunopathogenesis of pneumococcal meningitis. Mechanisms of immune activation, amplification, and perpetuation just as well as causes of meningitis-associated brain damage have been largely unveiled. This knowledge provides the basis for the development of novel strategies for treatment of this disease. Two novel therapeutic approaches have been recently evaluated in animal models of pneumococcal meningitis, namely, killing bacteria without lysing them and blocking the pro-inflammatory activity of C5a in combination with antibiotic therapy. Early results from experimental studies are very encouraging. However, there is still a difficult way to go until the ultimate goal of helping patients with meningitis is reached. First of all, animal studies are needed to assess the efficacy of these strategies when applied together with steroids—a prerequisite for clinical trials as steroids are part of the standard therapy for bacterial meningitis. Moreover, it has to be investigated whether and how novel therapeutic strategies affect the outcome of meningitis due to pathogens other than pneumococci as well as in special patient groups. This appears necessary in consideration of the observation that steroids are ineffective in less developed countries or in patients suffering from meningitis due to pathogens other than S. pneumoniae. Additionally, different pharmacologic issues, like the CSF penetration of daptomycin in humans, have to be clarified. It has also to be checked whether a successful treatment of bacterial meningitis requires the simultaneous targeting of multiple steps of the pathophysiologic cascade, like killing bacteria softly and blocking critical steps in the inflammatory cascade. Even when a therapeutic approach has been proven highly beneficial in animal models, its translation into the clinical practice will be challenging since the recruitment of sufficient number of patients requires a multicenter study design. Moreover, patient cohorts are usually relatively heterogeneous (with regard to the causative agent or the degree and type of comorbidities). However, the promising data from animal models, coupled with the still unfavorable prognosis in humans, should be incentive enough for researchers and physicians to follow this path all the way to the end.
Abbreviations
- C:
-
Complement
- CSF:
-
Cerebrospinal fluid
- DAMP:
-
Danger-associated molecular pattern
- HMGB1:
-
High-mobility group box 1 protein
- IL:
-
Interleukin
- LTA:
-
Lipoteichoic acid
- MAC:
-
Membrane attack complex
- MMP:
-
Matrix metalloproteinase
- MyD88:
-
Myeloid differentiation primary response gene 88 protein
- NF:
-
Nuclear factor
- NLR:
-
NOD-like receptor
- NLRP3:
-
The NLR family, pyrin domain-containing protein 3
- NOD:
-
Nucleotide-binding oligomerization domain
- PG:
-
Peptidoglycan
- PLY:
-
Pneumolysin
- PRR:
-
Pattern recognition receptor
- RAGE:
-
Receptor for advanced glycosylation end products
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
References
World Health Organization. Global Health Observatory: Number of suspected meningitis cases and deaths reported. http://www.who.int/gho/epidemic_diseases/meningitis/suspected_cases_deaths_text/en/2013.
World Health Organization. Epidemic meningococcal disease. WHO Fact Sheet 105, 1998.
Schuchat A, Robinson K, Wenger JD, Harrison LH, Farley M, Reingold AL, et al. Bacterial meningitis in the United States in 1995. Active Surveillance Team. N Engl J Med. 1997;337:970–6.
Thigpen MC, Whitney CG, Messonnier NE, Zell ER, Lynfield R, Hadler JL, et al. Bacterial meningitis in the United States, 1998–2007. N Engl J Med. 2011;364:2016–25.
De Gans J, Van de Beek D. Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347:1549–56.
Schmidt H, Heimann B, Djukic M, Mazurek C, Fels C, Wallesch CW, et al. Neuropsychological sequelae of bacterial and viral meningitis. Brain. 2006;129:333–45.
Ramakrishnan M, Ulland AJ, Steinhardt LC, Moisi JC, Were F, Levine OS. Sequelae due to bacterial meningitis among African children: a systematic literature review. BMC Med. 2009;7:47.
Kastenbauer S, Pfister HW. Pneumococcal meningitis in adults: spectrum of complications and prognostic factors in a series of 87 cases. Brain. 2003;126:1015–25.
Van de Beek D, De Gans J, Spanjaard L, Weisfelt M, Reitsma JB, Vermeulen M. Clinical features and prognostic factors in adults with bacterial meningitis. N Engl J Med. 2004;351:1849–59.
Katchanov J, Siebert E, Endres M, Klingebiel R. Focal parenchymal lesions in community-acquired bacterial meningitis in adults: a clinico-radiological study. Neuroradiology. 2009;51: 723–9.
Pfister HW, Feiden W, Einhäupl KM. Spectrum of complications during bacterial meningitis in adults. Arch Neurol. 1993;505:575–81.
Schut ES, Lucas MJ, Brouwer MC, Vergouwen MD, van der Ende A, van de Beek D. Cerebral infarction in adults with bacterial meningitis. Neurocrit Care. 2011;16:421–7.
Perry FE, Elson CJ, Greenham LW, Catterall JR. Interference with the oxidative response of neutrophils by Streptococcus pneumoniae. Thorax. 1993;48:364–9.
Brouwer MC, McIntyre P, de Gans J, Prasad K, van de Beek D. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. 2010;9, CD004405.
Van de Beek D, Brouwer MC, Thwaites GE, Tunkel AR. Advances in treatment of bacterial meningitis. Lancet. 2012;380:1693–702.
Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res. 2004;1:2.
Dujardin BC, Driedijk PC, Roijers AF, Out TA. The determination of the complement components C1q, C4 and C3 in serum and cerebrospinal fluid by radioimmunoassay. J Immunol Methods. 1985;80:227–37.
Stahel PF, Nadal D, Pfister HW, Paradisis PM, Barnum SR. Complement C3 and factor B cerebrospinal fluid concentrations in bacterial and aseptic meningitis. Lancet. 1997;349: 1886–7.
Pachter JS, De Vries HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropathol Exp Neurol. 2003;62:593–604.
Smith H, Bannister B, O’Shea MJ. Cerebrospinal fluid immunoglobulins in meningitis. Lancet. 1977;2:591–3.
Pashenkov M, Link H. Dendritic cells and immune responses in the central nervous system. Trends Immunol. 2002;23:69–70.
Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–97.
Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006;7:354–9.
Gordon LB, Nolan SC, Ksander BR, Knopf PM, Harling-Berg CJ. Normal cerebrospinal fluid suppresses the in vitro development of cytotoxic T cells: role of the brain microenvironment in CNS immune regulation. J Neuroimmunol. 1998;88:77–84.
Nagai A, Terashima M, Sheikh AM, Notsu Y, Shimode K, Yamaguchi S, et al. Involvement of cystatin C in pathophysiology of CNS diseases. Front Biosci. 2008;13:3470–9.
Hoffmann O, Priller J, Prozorovski T, Schulze-Topphoff U, Baeva N, Lunemann JD, et al. TRAIL limits excessive host immune responses in bacterial meningitis. J Clin Invest. 2007;117:2004–13.
Small PM, Tauber MG, Hackbarth CJ, Sande MA. Influence of body temperature on bacterial growth rates in experimental pneumococcal meningitis in rabbits. Infect Immun. 1986;52:484–7.
Tuomanen E, Tomasz A, Hengstler B, Zak O. The relative role of bacterial cell wall and capsule in the induction of inflammation in pneumococcal meningitis. J Infect Dis. 1985;151: 535–40.
Tuomanen E, Hengstler B, Zak O, Tomasz A. Induction of meningeal inflammation by diverse bacterial cell walls. Eur J Clin Microbiol. 1986;5:682–4.
Tuomanen EI, Saukkonen K, Sande S, Cioffe C, Wright SD. Reduction of inflammation, tissue damage, and mortality in bacterial meningitis in rabbits treated with monoclonal antibodies against adhesion-promoting receptors of leukocytes. J Exp Med. 1989;170:959–69.
Schneider O, Michel U, Zysk G, Dubuis O, Nau R. Clinical outcome in pneumococcal meningitis correlates with CSF lipoteichoic acid concentrations. Neurology. 1999;53:1584–7.
Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5.
Koedel U, Angele B, Rupprecht T, Wagner H, Roggenkamp A, Pfister HW, et al. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J Immunol. 2003;170:438–44.
Schroder NW, Morath S, Alexander C, Hamann L, Hartung T, Zahringer U, et al. Lipoteichoic acid (LTA) of S. pneumoniae and S. aureus activates immune cells via toll-like receptor (TLR)-2, LPS binding protein (LBP) and CD14 while TLR-4 and MD-2 are not involved. J Biol Chem. 2003;278:15587–94.
Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A. 2003;100:1966–71.
Shoma S, Tsuchiya K, Kawamura I, Nomura T, Hara H, Uchiyama R, et al. Critical involvement of pneumolysin in production of IL-1α and caspase-1-dependent cytokines in infection with Streptococcus pneumoniae in vitro: a novel function of pneumolysin in caspase-1 activation. Infect Immun. 2008;76:1547–57.
McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, et al. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 2010;6:e1001191.
Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–40.
Mogensen TH, Paludan SR, Kilian M, Ostergaard L. Live Streptococcus pneumoniae, Haemophilus influenzae, and Neisseria meningitidis activate the inflammatory response through Toll-like receptors 2, 4, and 9 in species-specific patterns. J Leukoc Biol. 2006;80:267–77.
Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science. 2012;337:1111–5.
Klein M, Angele B, Pfister HW, Wagner H, Koedel U, Kirschning CJ. Detection of pneumococcal infection of the central nervous system depends upon TLR2 and TLR4. J Infect Dis. 2008;198:1028–36.
Opitz B, Puschel A, Schmeck B, Hocke AC, Rosseau S, Hammerschmidt S, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem. 2004;279:36426–32.
Liu X, Chauhan VS, Young AB, Marriott I. NOD2 mediates inflammatory responses of primary murine glia to Streptococcus pneumoniae. Glia. 2010;58:839–47.
Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, et al. The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol. 2011;187:5440–51.
Koedel U, Rupprecht T, Angele B, Heesemann J, Wagner H, Pfister HW, et al. MyD88 is required for mounting a robust host immune response to Streptococcus pneumoniae in the CNS. Brain. 2004;127:1437–45.
Brouwer MC, de Gans J, Heckenberg SG, Zwinderman AH, van der Poll T, van de Beek D. Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. Lancet Infect Dis. 2009;9:31–44.
Bogaert D, Thompson CM, Trzcinski K, Malley R, Lipsitch M. The role of complement in innate and adaptive immunity to pneumococcal colonization and sepsis in a murine model. Vaccine. 2010;28:681–5.
Rupprecht TA, Angele B, Klein M, Heesemann J, Pfister HW, Botto M, et al. Complement C1q and C3 are critical for the innate immune response to Streptococcus pneumoniae in the central nervous system. J Immunol. 2007;178:1861–9.
Koedel U, Bayerlein I, Paul R, Sporer B, Pfister HW. Pharmacological interference with NF-B activation attenuates central nervous system complications in experimental pneumococcal meningitis. J Infect Dis. 2000;182:1437–45.
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50.
Mustafa MM, Lebel MH, Ramilo O, Olsen KD, Reisch JS, Beutler B, et al. Correlation of interleukin-1 beta and cachectin concentrations in cerebrospinal fluid and outcome from bacterial meningitis. J Pediatr. 1989;115:208–13.
Fassbender K, Mielke O, Bertsch T, Muehlhauser F, Hennerici M, Kurimoto M, et al. Interferon-gamma-inducing factor (IL-18) and interferon-gamma in inflammatory CNS diseases. Neurology. 1999;53:1104–6.
Quagliarello VJ, Wispelwey B, Long WJJ, Scheld WM. Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. J Clin Invest. 1991;87:1360–6.
Ramilo O, Saez-Llorens X, Mertsola J, Jafari H, Olsen KD, Hansen EJ, et al. Tumor necrosis factor alpha/cachectin and interleukin 1 beta initiate meningeal inflammation. J Exp Med. 1990;172:497–507.
Saukkonen K, Sande S, Cioffe C, Wolpe S, Sherrry B, Cerami A, et al. The role of cytokines in the generation of inflammation and tissue damage in experimental pneumococcal meningitis. J Exp Med. 1990;171:439–48.
Zwijnenburg PJ, Van der Poll T, Florquin S, Roord JJ, Van Furth AM. IL-1 receptor type 1 gene-deficient mice demonstrate an impaired host defense against pneumococcal meningitis. J Immunol. 2003;170:4724–30.
Zwijnenburg PJ, Van der Poll T, Florquin S, Akira S, Takeda K, Roord JJ, et al. Interleukin-18 gene-deficient mice show enhanced defense and reduced inflammation during pneumococcal meningitis. J Neuroimmunol. 2003;138:31–7.
Koedel U, Winkler F, Angele B, Fontana A, Flavell RA, Pfister HW. Role of caspase-1 in experimental pneumococcal meningitis: evidence from pharmacologic caspase inhibition and caspase-1-deficient mice. Ann Neurol. 2002;51:319–29.
Braun JS, Novak R, Herzog K-H, Bodner SM, Cleveland JL, Tuomanen EI. Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat Med. 1999;5:298–302.
Woehrl B, Brouwer MC, Murr C, Heckenberg SG, Baas F, Pfister HW, et al. Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis. J Clin Invest. 2011;121:3943–53.
Ehrnthaller C, Ignatius A, Gebhard F, Huber-Lang M. New insights of an old defense system: structure, function, and clinical relevance of the complement system. Mol Med. 2011;17:317–29.
Trouw LA, Daha MR. Role of complement in innate immunity and host defense. Immunol Lett. 2011;138:35–7.
Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46:2753–66.
Woodruff TM, Nandakumar KS, Tedesco F. Inhibiting the C5-C5a receptor axis. Mol Immunol. 2011;48:1631–42.
Kadurugamuwa JL, Hengstler B, Bray MA, Zak O. Inhibition of complement-factor-C5a-induced inflammatory reactions by prostaglandin E2 in experimental meningitis. J Infect Dis. 1989;160:715–9.
Casarsa C, De Luigi A, Pausa M, De Simoni MG, Tedesco F. Intracerebroventricular injection of the terminal complement complex causes inflammatory reaction in the rat brain. Eur J Immunol. 2003;33:1260–70.
Ernst JD, Hartiala KT, Goldstein IM, Sande MA. Complement (C5)-derived chemotactic activity accounts for accumulation of polymorphonuclear leukocytes in cerebrospinal fluid of rabbits with pneumococcal meningitis. Infect Immun. 1984;46:81–6.
Matzinger P. Friendly and dangerous signals: is the tissue in control? Nat Immunol. 2007;8:11–3.
Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89.
Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5.
Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62.
Lamkanfi M, Sarkar A, Vande WL, Vitari AC, Amer AO, Wewers MD, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J Immunol. 2010;185:4385–92.
Bianchi ME. HMGB1 loves company. J Leukoc Biol. 2009;86:573–6.
Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8:195–202.
Tang D, Kang R, Cao L, Zhang G, Yu Y, Xiao W, et al. A pilot study to detect high mobility group box 1 and heat shock protein 72 in cerebrospinal fluid of pediatric patients with meningitis. Crit Care Med. 2008;36:291–5.
Asano T, Ichiki K, Koizumi S, Kaizu K, Hatori T, Mashiko K, et al. High mobility group box 1 in cerebrospinal fluid from several neurological diseases at early time points. Int J Neurosci. 2011;121:480–4.
Höhne C, Wenzel M, Angele B, Hammerschmidt S, Häcker H, Klein M, et al. High mobility group box 1 prolongs inflammation and worsens disease in pneumococcal meningitis. Brain. 2013. doi:10.1093/brain/awt064.
Blazer S, Berant M, Alon U. Bacterial meningitis. Effect of antibiotic treatment on cerebrospinal fluid. Am J Clin Pathol. 1983;80:386–7.
Viallon A, Guyomarc’h P, Guyomarc’h S, Tardy B, Robert F, Marjollet O, et al. Decrease in serum procalcitonin levels over time during treatment of acute bacterial meningitis. Crit Care. 2005;9:R344–50.
Kanegaye JT, Soliemanzadeh P, Bradley JS. Lumbar puncture in pediatric bacterial meningitis: defining the time interval for recovery of cerebrospinal fluid pathogens after parenteral antibiotic pretreatment. Pediatrics. 2001;108:1169–74.
Gerber J, Pohl K, Sander V, Bunkowski S, Nau R. Rifampin followed by ceftriaxone for experimental meningitis decreases lipoteichoic acid concentrations in cerebrospinal fluid and reduces neuronal damage in comparison to ceftriaxone alone. Antimicrob Agents Chemother. 2003;47:1313–7.
Stucki A, Cottagnoud M, Winkelmann V, Schaffner T, Cottagnoud P. Daptomycin produces an enhanced bactericidal activity compared to ceftriaxone, measured by [3H]choline release in the cerebrospinal fluid, in experimental meningitis due to a penicillin-resistant pneumococcal strain without lysing its cell wall. Antimicrob Agents Chemother. 2007;51:2249–52.
Koedel U, Frankenberg T, Kirschnek S, Obermaier B, Hacker H, Paul R, et al. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog. 2009;5:e1000461.
Klein M, Koedel U, Pfister HW. Oxidative stress in pneumococcal meningitis: a future target for adjunctive therapy? Prog Neurobiol. 2006;80:269–80.
Kastenbauer S, Koedel U, Becker BF, Pfister HW. Oxidative stress in bacterial meningitis in humans. Neurology. 2002;58:186–91.
Rosenberg GA. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009;8:205–16.
Leppert D, Lindberg RL, Kappos L, Leib SL. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res Brain Res Rev. 2001;36:249–57.
Woehrl B, Klein M, Grandgirard D, Koedel U, Leib S. Bacterial meningitis: current therapy and possible future treatment options. Expert Rev Anti Infect Ther. 2011;9:1053–65.
Straus SK, Hancock RE. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: comparison with cationic antimicrobial peptides and lipopeptides. Biochim Biophys Acta. 2006;1758:1215–23.
Egermann U, Stanga Z, Ramin A, Acosta F, Stucki A, Gerber P, et al. Combination of daptomycin plus ceftriaxone is more active than vancomycin plus ceftriaxone in experimental meningitis after addition of dexamethasone. Antimicrob Agents Chemother. 2009;53:3030–3.
Mook-Kanamori BB, Rouse MS, Kang CI, van de Beek D, Steckelberg JM, Patel R. Daptomycin in experimental murine pneumococcal meningitis. BMC Infect Dis. 2009;9:50.
Grandgirard D, Schurch C, Cottagnoud P, Leib SL. Prevention of brain injury by the nonbacteriolytic antibiotic daptomycin in experimental pneumococcal meningitis. Antimicrob Agents Chemother. 2007;51:2173–8.
Grandgirard D, Oberson K, Buhlmann A, Gaumann R, Leib SL. Attenuation of cerebrospinal fluid inflammation by the non-bacteriolytic antibiotic daptomycin vs. ceftriaxone in experimental pneumococcal meningitis. Antimicrob Agents Chemother. 2010;54:1323–6.
Ramos TN, Wohler JE, Barnum SR. Deletion of both the C3a and C5a receptors fails to protect against experimental autoimmune encephalomyelitis. Neurosci Lett. 2009;467:234–6.
Kullar R, Chin JN, Edwards DJ, Parker D, Coplin WM, Rybak MJ. Pharmacokinetics of single-dose daptomycin in patients with suspected or confirmed neurological infections. Antimicrob Agents Chemother. 2011;55:3505–9.
Acknowledgment
The authors’ research is funded by the German Research Foundation, the German Ministry for Research and Education, the Else Kroener-Fresenius-Foundation, the Research and Education Program of the University of Munich, and Novartis Pharmaceuticals.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Koedel, U., Klein, M., Pfister, HW. (2014). Immunopathogenesis of Bacterial Meningitis. In: Peterson, P., Toborek, M. (eds) Neuroinflammation and Neurodegeneration. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1071-7_18
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1071-7_18
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1070-0
Online ISBN: 978-1-4939-1071-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)