
Abstract
Feeding is a vital function that provides nutritional and energy metabolism needs for animals. To ensure feeding, mammalian brains possess several interrelated neuronal systems that regulate different aspects of feeding behaviors. These neuronal circuits controlling food intake are strongly regulated by peripheral signals that contribute to the fine regulation of the energy homeostasis, such as metabolites and hormones. Among the signals regulating food intake, the stomach-derived hormone ghrelin and its receptor [named ghrelin receptor or the growth hormone secretagogue receptor type 1a (ghrelin receptor 1a)] play a major role. Ghrelin is the only mammalian peptide hormone able to increase food intake. Ghrelin stimulates appetite by affecting both food intake itself and also the rewarding aspects of feeding. As discussed below, the central distribution of ghrelin receptor 1a supports the concept that ghrelin regulates both homeostatic and hedonic aspects of feeding, and evidence from different studies confirms that ghrelin promotes food intake via diverse mechanisms. Of note, derangements in the ghrelin/ghrelin receptor 1a system have been reported in several eating disorders, including obesity, anorexia nervosa, bulimia nervosa, binge eating disorders, cachexia, and Prader-Willi syndrome. Here, the potential pathways by which ghrelin receptor 1a regulates feeding, with a special focus on hedonic aspects of eating, are delineated. Also, recent evidence suggesting a role of the ghrelin system in disorders with alterations of food intake is briefly reviewed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
Homeostatic and Hedonic Feeding Circuits
Feeding regulation involves an integrated regulatory system in which homeostatic brain circuits, that drive food intake depending on energy store levels, interact with the hedonic circuits that drive consumption based on rewarding properties of foods (Berthoud 2011; Saper et al. 2002). The homeostatic circuits provide a means by which signals of energy availability, including ghrelin, modulate food intake (Williams and Elmquist 2012; Schwartz et al. 2000). Thus, homeostatic-driven feeding occurs under negative energy balance conditions, when fuel stores are depleted and plasma ghrelin is elevated (Williams and Elmquist 2012; Schwartz et al. 2000). In contrast, hedonic-driven feeding refers to the involvement of cognitive, reward, and emotional factors that lead to the consumption of pleasurable foods even when extra calories are not necessary (Berthoud 2011; Saper et al. 2002). Neuronal systems controlling homeostatic feeding are located mainly in the brainstem and hypothalamus while neuronal systems controlling hedonic feeding are primarily related to cortico-limbic structures (Berthoud 2011; Saper et al. 2002; Williams and Elmquist 2012; Schwartz et al. 2000). Importantly, both homeostatic and hedonic brain circuits driving food intake are regulated by peripheral signals.
The hypothalamus contains several nuclei involved in food intake regulation, including the arcuate nucleus (ARC), the paraventricular nucleus (PVN), the lateral hypothalamic area (LHA), the ventromedial nucleus (VMN), and the dorsomedial nucleus (DMN) (Williams and Elmquist 2012; Schwartz et al. 2000; Suzuki et al. 2010). The ARC has become a major focus for energy balance research because circulating factors, such as ghrelin, have increased accessibility to this nucleus, where receptors for peripheral signals are highly expressed (Williams and Elmquist 2012; Schwartz et al. 2000; Suzuki et al. 2010). The ARC contains a key set of neurons that express the potent orexigenic neuropeptides agouti-gene-related protein (AgRP) and neuropeptide Y (NPY), and also the neurotransmitter γ-aminobutyric acid (GABA) (Williams and Elmquist 2012; Schwartz et al. 2000; Suzuki et al. 2010). To explain homeostatic food intake, initial emphasis has been placed on a simple model in which ARC neurons act as first-order neurons that sense peripheral factors and then regulate second-order neurons of the PVN, VMN, DMH, and LHA (Williams and Elmquist 2012; Schwartz et al. 2000; Suzuki et al. 2010). Recent evidence shows that another target of ARC neurons is the parabrachial nucleus (PBN), which is located in the hindbrain and inhibits feeding (Wu and Palmiter 2011; Atasoy et al. 2012). Second-order neurons project then to other brain areas, including the dorsal vagal complex in the brainstem, which comprises the nucleus tractus solitarius (NTS), the area postrema (AP), and the dorsomotor nucleus of the vagus (DMV), and plays a major role regulating food intake in concert with the ARC (Williams and Elmquist 2012; Schwartz et al. 2000; Suzuki et al. 2010). The dorsal vagal complex senses peripheral hormones directly and also integrates neuronal inputs from the hypothalamic and peripheral centers. In particular, the NTS is a termination site of the vagal afferent fibers that transmit visceral sensory information, including gastric distension and gut factors, from cell bodies located in the nodose ganglia (Williams and Elmquist 2012; Schwartz et al. 2000; Suzuki et al. 2010). Thus, homeostatic adjustments of food intake integrate not only hypothalamic systems governing intake on a meal-to-meal basis but also brainstem systems regulating meal size and/or meal frequency.
A key element of neuronal circuits regulating food reward behaviors is the dopaminegic pathway emanating from the midbrain ventral tegmental area (VTA) (Berthoud 2011; Saper et al. 2002; DiLeone et al. 2012; Hyman et al. 2006). Dopaminergic VTA neurons project to the nucleus accumbens (NAc) in the ventral striatum and other areas such as the amygdala, medial prefrontal cortex (mPFC), hippocampus, and hypothalamus (DiLeone et al. 2012; Hyman et al. 2006). The VTA receives projections from many brain nuclei, including the above-mentioned areas that receive projections from the VTA and cholinergic neurons of the laterodorsal tegmental area (LDTg) (Dickson et al. 2010). In addition, the VTA receives taste information via afferent sensory fibers that have two brainstem relays, in the NTS and in the PBN (DiLeone et al. 2012; Hyman et al. 2006). Dopamine release in the NAc potently augments the drive to obtain food rewards (Palmiter 2007). The shell part of the NAc is particularly important for eating behaviors since it sends projections to the LHA neurons controlling food intake (Stratford and Kelley 1999; Zheng et al. 2007). Orexigenic LHA neurons seem to be under a tonic inhibition that can be relieved by activation of reward pathways (Stratford and Kelley 1999; Zheng et al. 2007). In addition, LHA orexin neurons send projections to the VTA, where they activate dopaminergic neurons (Nakamura et al. 2000; Korotkova et al. 2003). Thus, LHA orexin neurons have been proposed as a potential link between homeostatic and hedonic circuits regulating food intake (Mahler et al. 2012).
Ghrelin and Ghrelin Receptor 1a in Feeding Centers
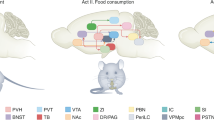
The ghrelin receptor 1a is present in and regulates both homeostatic and hedonic feeding centers (Perello and Zigman 2012; Skibicka and Dickson 2011; Zigman et al. 2006; Guan et al. 1997). Initially, ghrelin was shown to stimulate food intake by acting on homeostatic hypothalamic circuits (Nakazato et al. 2001; Briggs and Andrews 2011). Ghrelin effects on homeostatic eating likely involve the NPY/AgRP/GABA neurons of the ARC that express high levels of ghrelin receptor 1a (Nakazato et al. 2001; Briggs and Andrews 2011; Kageyama et al. 2010; Willesen et al. 1999). Ghrelin-induced food intake also seems to depend on orexin neurons of the LHA, where ghrelin receptor 1a is expressed (Toshinai et al. 2003; Olszewski et al. 2003). Additionally, some evidence indicates that the vagus nerve integrity is required for ghrelin-induced food intake (Date 2012; Date et al. 2002). According to this possibility, ghrelin receptor 1a is expressed in vagal afferent neurons of nodose ganglia and in the dorsal vagal complex (Zigman et al. 2006; Sakata et al. 2003). The presence of ghrelin receptor 1a in dopaminergic VTA neurons supports the possibility that ghrelin can regulate hedonic aspects of eating (Abizaid et al. 2006; Zigman et al. 2006; Chuang et al. 2011). Ghrelin may also regulate mesolimbic circuits indirectly via the cholinergic neurons of the LDTg, which express ghrelin receptor 1a (Dickson et al. 2010; Jerlhag et al. 2008). Ghrelin’s action on food reward requires intact orexin signaling; however, the neuronal circuits by which ghrelin recruits the LHA orexin neurons are still unknown (Perello et al. 2010). Ghrelin presumably affects eating behaviors by also acting on the hippocampus, a brain structure involved in memory and decision making that expresses ghrelin receptor 1a (Zigman et al. 2006; Diano et al. 2006). Figure 1 summarizes the ghrelin targets and the potential neuronal circuits controlling homeostatic and hedonic aspects of food intake affected by ghrelin.

Model of ghrelin action on neuronal circuits controlling homeostatic and hedonic eating. Cartoons represent sagittal slices of rodent brain depicting brain circuits implicated in ghrelin’s regulation of the homeostatic (upper panel) or hedonic (lower panel) aspects of eating. Black areas represent brain nuclei involved in each circuit, and arrows indicate probable connections between those brain nuclei. Stars label brain nuclei where GHSR is expressed. Abbreviations: Amyg amygdala, AP area postrema, ARC arcuate nucleus, DMN dorsomedial nucleus, Hipp hippocampus, LDTg laterodorsal tegmental area, LHA lateral hypothalamic area, mPFC medial prefrontal cortex, NAc nucleus accumbens, NTS nucleus tractus solitaries, PBN parabrachial nucleus, PVN paraventricular nucleus of the hypothalamus, VMN ventromedial nucleus, VTA ventral tegmental area
The ability of ghrelin to act in the brain and increase food intake depends on the accessibility of circulating ghrelin to the above-mentioned brain areas. Circulating ghrelin cannot freely cross the blood–brain barrier, and it is currently unclear how this hormone enters the brain (Fry and Ferguson 2010). In mice, ghrelin can be transported from the brain for circulation via a saturable transport system; however, no such system has been identified for blood to brain transport (Banks 2008). It is frequently assumed that circulating ghrelin is able to access to the ARC, where blood–brain barrier is presumably weaker; however, this possibility is still under debate (Fry and Ferguson 2010; Rodriguez et al. 2010; Schaeffer et al. 2013). Another possibility is that circulating ghrelin gains access to the brain through the sensory circumventricular organs, which are specialized areas with fenestrated capillaries. The median eminence, located in close apposition to the ARC, is a circumventricular organ where plasma ghrelin can easily diffuse to reach neuronal ghrelin receptor 1a (Schaeffer et al. 2013). The AP is another circumventricular organ also known to participate in food intake regulation and that expresses ghrelin receptor 1a (Fry and Ferguson 2007, 2010; Zigman et al. 2006). Thus, circulating ghrelin could directly act on AP neurons, which then innervate several hypothalamic and brainstem feeding centers (Fry and Ferguson 2007). Some evidence does suggest that ghrelin-induced feeding depends on intact signaling at the AP (Gilg and Lutz 2006; Date et al. 2006).
The relevance of the expression of ghrelin receptor 1a in brain areas without access to circulating ghrelin is unclear. It has been proposed that ghrelin can be centrally produced; however, evidence about the source and physiological significance of centrally produced ghrelin is inconsistent (Cowley et al. 2003; Sakata et al. 2009; Furness et al. 2011). Ghrelin receptor 1a mainly signals through Gαq/11, phospholipase C, inositol phosphate, and calcium mobilization from intracellular stores; although it also activates other signaling pathways (Cong et al. 2010). An interesting feature of ghrelin receptor 1a is its strong constitutive activity that makes it capable to signal in a ghrelin-independent manner (Mokrosinski and Holst 2010; Damian et al. 2012). Thus, the increase of ghrelin receptor 1a expression would accordingly increase activation of the downstream signaling pathways affecting, as a consequence, food intake and body weight regulation (Petersen et al. 2009). Additionally, it has been proposed that an alternative mechanism by which ghrelin receptor 1a regulates food intake involves its dimerization with other G protein-coupled receptors. The ghrelin receptor 1a has been shown to heterodimerize with the melanocortin 3 receptor, the serotonin 2C receptor, and the dopamine receptors, all involved in food intake and food reward regulation (Schellekens et al. 2013; Kern et al. 2012; Jiang et al. 2006; Rediger et al. 2011). Heterodimerization could serve to modulate specific functions of the ghrelin receptor 1a, such as signaling pathways, or to act as an allosteric mechanism to regulate signaling pathways of the other receptors, independently of ghrelin binding (Schellekens et al. 2013; Kern et al. 2012; Jiang et al. 2006; Rediger et al. 2011).
Modulation of Hedonic Aspects of Eating in Rodent Models by Ghrelin
Evidence from Studies Using Pharmacological Manipulations of the Ghrelin System
Evidence shows that ghrelin enhances preference for pleasurable, sweet, and fatty foods. In this regard, ghrelin administration shifts food preference toward a high-fat diet (HFD) (Shimbara et al. 2004). Ghrelin administration also increases intake of palatable saccharin solution and preference for saccharin-flavored foods in mice (Disse et al. 2010). Similarly, rats treated with a ghrelin receptor 1a antagonist consume less peanut butter and the liquid nutritional supplement Ensure®, but do not change intake of regular chow in a free choice protocol (Egecioglu et al. 2010). Likewise, treatment with a ghrelin receptor 1a antagonist selectively decreases intake of sucrose solution in rats and saccharin solution self-administration in mice (Landgren et al. 2011).
Ghrelin also enhances the motivation to obtain preferred foods, as evaluated by operant lever-pressing or operant nose-poking behavioral tasks in progressive ratio paradigms. Ghrelin administration increases operant lever-pressing for sucrose, peanut butter-flavored sucrose or HFD pellets in rodents (Perello et al. 2010; Finger et al. 2012; Skibicka et al. 2011; Overduin et al. 2012). Conversely, treatment with a ghrelin receptor 1a antagonist reduces operant responding for sucrose solution (Landgren et al. 2011). In addition, ghrelin increases food anticipatory activity, which is characterized by increased arousal, increased locomotor activity, and an elevated body temperature in anticipation of a predicted meal (Merkestein et al. 2012; Jerlhag et al. 2006). Also, ghrelin secreted in anticipation of a meal correlates to anticipatory locomotor activity, and administration of ghrelin increases locomotor activity and foraging-like activities in rodents (Blum et al. 2009; Keen-Rhinehart and Bartness 2005; Jerlhag et al. 2007). On the other hand, ghrelin receptor 1a antagonists decrease anticipatory behavior for a palatable meal (Merkestein et al. 2012).
Ghrelin can also affect more complex, reward-related eating behaviors such as those that take place in a food conditioned place preference (CPP) test. In the food CPP test, animals are conditioned to associate one chamber of the CPP apparatus with regular chow and a second, visually and texturally distinct chamber with an equal-calorie amount of a more pleasurable food, such as HFD. After conditioning, animals have free access to both chambers in the absence of food, and conditioned place preference for HFD is demonstrated by animals spending more time in the chamber associated with the more rewarding food. Food CPP studies performed in mice reveal that both administration of ghrelin and physiological increases in plasma ghrelin induced by caloric restriction enable acquisition of CPP for HFD (Perello et al. 2010; Disse et al. 2011). Similarly, treatment with a ghrelin receptor 1a antagonist blocks CPP for chocolate pellets in satiated rats (Egecioglu et al. 2010). Of note, the assessment of the ghrelin effect on the hedonic valuation per se by monitoring the avidity of ingestion of a liquid food via lickometry has suggested that ghrelin does not affect food palatability (Overduin et al. 2012).
The dopaminergic VTA neurons are important for ghrelin’s effects on hedonic aspects of eating. Exogenous ghrelin releases dopamine in the NAc from VTA neuronal terminals, and ghrelin increases action potential frequency in dopaminergic VTA neurons (Abizaid et al. 2006; McCallum et al. 2011; Jerlhag 2008; Jerlhag et al. 2006, 2007). Acute intra-VTA administration of ghrelin increases intake of regular food, intake of peanut butter over regular chow, and operant lever-pressing for sucrose and banana-flavored pellets (Abizaid et al. 2006; Naleid et al. 2005; Egecioglu et al. 2010; Skibicka et al. 2011; Weinberg et al. 2011). In addition, pretreatment with a dopamine D1 receptor antagonist eliminates ghrelin-induced increases in lever pressing in rats, without compromising generalized motor control, indicating a role for dopamine signaling in ghrelin’s motivational feeding effects (Overduin et al. 2012). On the other hand, intra-VTA administration of ghrelin receptor 1a antagonists decreases food intake in response to peripherally administrated ghrelin, intake of a more preferred HFD, and fasting-induced operant lever pressing for sucrose pellets (Abizaid et al. 2006; Naleid et al. 2005; King et al. 2011; Skibicka et al. 2011). Chronic intra-VTA administration of ghrelin also dose-dependently increases intake of regular chow (King et al. 2011), and VTA-lesioned rats spend less time than control rats exploring tubes containing peanut butter in response to centrally administrated ghrelin (Egecioglu et al. 2010). Similar effects are observed in food-restricted rats, in which chronic intra-VTA administration of ghrelin enhances while chronic intra-VTA delivery of a ghrelin receptor 1a antagonist blunts operant responding for chocolate-flavored pellets (King et al. 2011). Furthermore, intra-VTA administration of ghrelin fails to affect operant lever-pressing for food rewards in animals with dopamine depletion induced by delivery of the neurotoxin 6-hydroxydopamine in the VTA (Weinberg et al. 2011). Ghrelin administration into the VTA also stimulates locomotor activity via an increase in the extracellular concentration of dopamine in the NAc (Jerlhag et al. 2007).
The rest of the neuronal circuit recruited by ghrelin to regulate hedonic aspects of eating is just starting to be elucidated. Ghrelin action on food reward requires intact orexin signaling, as evidenced by the failure of orexin-knockout mice or wild-type (WT) mice given an orexin receptor antagonist to manifest ghrelin-induced effects on HFD reward (Perello et al. 2010). Other signals that likely mediate ghrelin actions on food intake are the endocannabinoids, which regulate both homeostatic and hedonic aspects of eating (Harrold and Williams 2003). Central injection of ghrelin to endocannabinoid receptor type 1 knockout mice fails to increase food intake, suggesting that the endocannabinoid signaling is necessary for ghrelin’s orexigenic effect (Kola et al. 2008). Moreover, the ghrelin-induced enhancement of food CPP seems to be partially mediated by the cholinergic pathway (Disse et al. 2011). In this regard, nicotinic receptor signaling seems to play a role in ghrelin’s actions on food reward since administration of a selective antagonist of the α3β4 nicotinic receptor blocks both ghrelin-induced increase of sucrose intake and dopamine release in the NAc following intra-VTA administration of ghrelin (McCallum et al. 2011). The stimulatory effect of ghrelin on dopaminergic neurons of the VTA also appears to depend on the excitatory glutamatergic inputs (Abizaid et al. 2006). In fact, the ability of ghrelin to activate the dopaminergic VTA system and the locomotor activity is suppressed by pharmacological blockade of glutamatergic N-methyl-D-aspartate (NMDA) receptors but not by blockade of opioid or orexin receptors (Jerlhag et al. 2011).
Evidence from Studies Using Genetic Manipulations of the Ghrelin System
Mouse models with genetic manipulations of the ghrelin system have been instrumental in order to establish the mechanisms underlying ghrelin’s actions on eating behaviors. These models include mice over-expressing ghrelin and mice with deletion of the genes encoding ghrelin, ghrelin receptor 1a, or the enzyme that octanoylates ghrelin [ghrelin O-acyltransferase (GOAT)]. In addition, a conditional ghrelin receptor 1a null mouse model in which ghrelin receptor 1a transcription is globally blocked but can be cell-specifically reactivated in a Cre recombinase-mediated fashion has been generated.
Most mouse models overexpressing or lacking bioactive ghrelin show minor alterations on food intake behaviors. Transgenic mice with increased brain and circulating bioactive ghrelin do not differ from WT controls in food intake or body weight (Reed et al. 2008). In contrast, chronic overproduction of bioactive ghrelin in the stomach increases food intake but does not alter long-term body weight gain due to a paradoxical increase in energy expenditure (Bewick et al. 2009). The double-transgenic mice overexpressing both human ghrelin and GOAT genes in the liver have decreased energy expenditure and increased body weight without food intake alterations only when fed on HFD rich in medium-chain triglycerides (Kirchner et al. 2009). Similarly, ghrelin-deficient mice show normal food intake and body weight, as compared to WT mice. (De Smet et al. 2006; Wortley et al. 2005; Sun et al. 2003; Dezaki et al. 2006; Sato et al. 2008). In addition, no differences are observed when some other aspects of eating behaviors of ghrelin-deficient mice are evaluated, including post-fasting hyperphagia or forced dark cycle induced eating (Wortley et al. 2005; Sun et al. 2003; Pfluger et al. 2008; Sato et al. 2008; De Smet et al. 2006). Of note, ghrelin-deficient mice show some alterations in their food intake behaviors under particular experimental settings. For instance, they lack anticipatory eating response failing to match the increase in food intake observed in WT type controls during 6 h food intake following repeated overnight fasts (Abizaid et al. 2006). Studies where ghrelin-deficient mice were chronically fed with HFD failed to show any reduction of food intake (Dezaki et al. 2006; Wortley et al. 2005; Sun et al. 2003). Only one of these studies was able to detect that ghrelin deficiency results in reduced body weight and fat mass, among other beneficial effects (Wortley et al. 2005). On the other hand, the GOAT-deficient mice, which lack plasma bioactive ghrelin, do not differ from WT controls in food intake or body weight, when fed with regular chow (Kirchner et al. 2009; Zhao et al. 2010). One study showed that GOAT deficiency results in decreased body weight when animals were fed on HFD rich in medium-chain triglycerides (Kirchner et al. 2009), but this body weight phenotype was not observed by other researchers (Zhao et al. 2010). GOAT-deficient mice display an attenuated motivation for HFD in an operant responding model and also a decreased hedonic feeding response examined in a “dessert effect” protocol, in which the intake of a palatable HFD pellet “dessert” is assessed in calorically sated mice (Davis et al. 2012).
The use of ghrelin receptor 1a deficient mice has shown an obligatory role of ghrelin signaling in certain hedonic aspects of eating that are separated from eating associated with body weight homeostasis. Ghrelin receptor 1a deficient mice show a subtle but significant decrease in body weight without food intake alterations when they have free access to regular chow diet (Abizaid et al. 2006; Zigman et al. 2005; Sun et al. 2004). Interestingly, ghrelin receptor 1a null mice are resistant to HFD-induced body weight gain, if they are exposed to HFD early in their life (Zigman et al. 2005; Perello et al. 2012). However, no differences in HFD-induced body weight gain are observed if mice are exposed to HFD during adulthood (Sun et al. 2008). Additionally, ghrelin receptor 1a deficient mice show an improvement of aging-associated obesity due mainly to a reduced adiposity and increased thermogenesis (Lin et al. 2011; Ma et al. 2011). Ghrelin/ghrelin receptor 1a double knockout mice exhibit decreased body weight when placed on a standard chow diet (Pfluger et al. 2008). Ghrelin receptor 1a deficient mice are protected from the weight gain induced by exposure to HFD although no reduction in HFD intake is observed (Zigman et al. 2005; Perello et al. 2012). Importantly, ghrelin receptor 1a deficient mice have a reduced intake of the more rewarding food in a free choice paradigm and a reduced dopamine release in the NAc induced by rewarding foods (Egecioglu et al. 2010). Also, ghrelin receptor 1a null mice also fail to enhance feeding in response to a light cue used as positive-conditioned stimulus as compared to WT mice (Walker et al. 2012).
The significance of ghrelin signaling on hedonic eating regulation becomes more evident in situations in which plasma ghrelin is physiologically elevated, such as fasting, caloric restriction, or stress (Perello and Zigman 2012). In this regard, ghrelin receptor 1a deficient mice show important eating behavior alterations under specific experimental conditions. For instance, WT mice subjected to prolonged caloric restriction show enhanced-CPP for HFD while ghrelin receptor 1a deficient mice lack such response (Perello et al. 2010; Disse et al. 2011). Moreover, ghrelin receptor 1a deficient mice in response to scheduled meals have both attenuated anticipatory hyperlocomotion and reduced expression of the marker of cellular activation c-fos in the mesolimbic pathway (Lamont et al. 2012; Blum et al. 2009). Similarly, ghrelin receptor 1a deficient mice do not anticipate food when exposed to an activity-based anorexia model, in which mice are given free access to a running wheel and fed once per day for 2 h (Verhagen et al. 2011). The chronic social defeat stress (CSDS) procedure, which subjects mice to daily bouts of social defeat by aggressive male mice, has been also used to study the physiological effect of ghrelin on feeding behaviors (Lutter et al. 2008; Patterson et al. 2013). WT mice exposed to CSDS increase their plasma ghrelin concentration and regular chow intake during and for at least 1 month after the defeat period. In contrast, ghrelin receptor 1a null mice fail to show CSDS-induced hyperphagia (Lutter et al. 2008; Patterson et al. 2013). In WT mice, CSDS also increases CPP for HFD while such a stress-induced food reward response is not observed in CSDS-exposed ghrelin receptor 1a null mice (Chuang et al. 2011). In contrast to these findings, a chronic unpredictable stress model that also elevates plasma ghrelin decreases food intake and body weight gain in WT mice, while similarly treated ghrelin receptor 1a deficient mice lack these changes (Patterson et al. 2010). Thus, further work is needed to clarify the role of ghrelin on food intake among different rodent models of stress.
The mouse model with reactivable genetic deletion of ghrelin receptor 1a has been very valuable to establish the physiological roles of some of ghrelin’s brain targets. In this nontraditional mouse model, ghrelin receptor 1a gene expression is disrupted by a transcriptional blocking cassette flanked by loxP sites that enable Cre recombinase-mediated ghrelin receptor 1a gene re-expression (Zigman et al. 2005). Thus, the ghrelin receptor 1a transcription is globally blocked in ghrelin receptor 1a null mice, but it can be cell-specifically reactivated in a Cre-mediated fashion (Zigman et al. 2005). Using this strategy, mice expressing ghrelin receptor 1a selectively in tyrosine hydroxylase-containing cells, including a subset of VTA dopaminergic neurons, was generated (Chuang et al. 2011). These mice show a significant, albeit reduced, response to the orexigenic effects of ghrelin (Chuang et al. 2011). Interestingly, mice with re-expression of ghrelin receptor 1a selectively in tyrosine hydroxylase-containing neurons show full CPP for HFD when treated with exogenous ghrelin or exposed to a CSDS protocol (Chuang et al. 2011). This study suggests that expression of ghrelin receptor 1a in dopaminergic neurons is sufficient for ghrelin’s actions on both food intake and food reward. Of note, mice with re-expression of ghrelin receptor 1a in specific hindbrain nuclei, including the NTS, DMV, AP, nucleus ambiguous, and facial motor nucleus, fail to show ghrelin-induced food intake (Scott et al. 2012). Thus, direct action of circulating ghrelin on ghrelin receptor 1a expressing hindbrain neurons is not sufficient to mediate acute orexigenic effects of ghrelin.
Relevance of Ghrelin Effects on Hedonic Aspects of Eating for Humans
Many studies suggest that ghrelin signaling is relevant for human food intake regulation. Human beings have a preprandial rise and a postprandial decline in plasma ghrelin levels suggesting that ghrelin recapitulates in humans its physiological role in hunger and/or meal initiation observed in rodents (Cummings 2006; Cummings et al. 2001). The preprandial ghrelin surge occurs as many times per day as meals are provided to subjects exposed to habituated feeding schedules (Cummings 2006; Cummings et al. 2001). Importantly, ghrelin levels also rise preprandially initiating meals voluntarily in the absence of cues related to time or food, and the temporal profiles of plasma ghrelin levels and hunger scores tightly overlap in this setting (Cummings 2006; Cummings et al. 2001). The postprandial ghrelin decrease seems to be critical for satiety sensation and, accordingly, it decreases proportionally to meal calorie content (le Roux et al. 2005). Of note, postprandial ghrelin decrease is impaired after high-fat meals likely contributing to reduce satiety and causing overeating (Yang et al. 2009). The mechanisms involved in the control of pre and postprandial ghrelin regulation in humans are currently unclear.
Most studies show that intravenous bolus or continuous administration of ghrelin stimulates hunger sensations and food intake in healthy individuals (Akamizu et al. 2008; Adachi et al. 2010; Schmid et al. 2005; Levin et al. 2006; Wren et al. 2001; Falken et al. 2010; Druce et al. 2005). It is interesting to note that some of these studies have used ghrelin doses that result in supra-physiological increases in plasma hormone levels. Also, administration of exogenous ghrelin cannot mimic the postprandial decrease of the hormone levels that occur in physiological conditions. Despite these considerations, it is normally accepted that exogenous ghrelin can regulate meal initiation and food intake of human beings (Cummings 2006). Functional magnetic resonance imaging studies indicate that ghrelin increases the neural response in brain centers implicated in hedonic feeding of human subjects (Goldstone et al. 2009; Malik et al. 2008; Neary and Batterham 2010). Fasting-induced increases of plasma ghrelin enhance both the appeal of high-calorie more than low-calorie foods and the reward-related brain centers’ response to pictures of high-calorie over low-calorie foods (Goldstone et al. 2009). Also, ghrelin administration to human subjects increases the activation of some hedonic feeding-related brain centers, including the substance nigra and the VTA, in response to tempting food pictures (Malik et al. 2008; Neary and Batterham 2010). Thus, ghrelin seems to have a significant role in food reward behavior and appetite regulation in humans.
Role of Ghrelin and Ghrelin Receptor 1a on Disorders with Alterations of Food Intake
Obesity. Obesity is defined as an excessive fat accumulation that presents a risk to health. Obesity is a heterogeneous disorder with several potential etiologies including genetic and environmental factors. Little association has been found between obesity and ghrelin or ghrelin receptor 1a mutations in humans (Gueorguiev et al. 2009; Liu et al. 2011). However, the ghrelin system appears relevant for human obesity (Hillman et al. 2011). Most obese patients have chronically low levels of circulating ghrelin and a blunting of the nocturnal plasma ghrelin increase compared to normal subjects (Hillman et al. 2011; Tschop et al. 2001). Similarly, plasma ghrelin is decreased in diet-induced obesity mouse models, where a resistance to ghrelin-induced food intake and ghrelin-induced motivation to obtain food rewards is observed (Finger et al. 2012; Perreault et al. 2004; Briggs et al. 2010). Still, obese people seem to be fully sensitive to the orexigenic effects of exogenous ghrelin (Druce et al. 2005). Several studies show that obese people have a blunted postprandial decrease of plasma ghrelin, which likely increases the time they feel hungry and participates in the pathophysiology of obesity (le Roux et al. 2005; Yang et al. 2009; Morpurgo et al. 2003; English et al. 2002). Also, ghrelin levels rise in obese individuals after weight loss induced by dieting, and such increase of plasma ghrelin likely contributes to the rebound weight gain commonly observed in dieters (Cummings et al. 2002b). In addition, the marked and prolonged weight loss observed in obese individuals who undergo Roux-en-Y gastric bypass surgery is thought to be enhanced by postsurgery reductions in circulating ghrelin (Cummings and Shannon 2003; Beckman et al. 2010). These clinical studies, among others (Schellekens et al. 2012), support the concept that pharmacological manipulations of ghrelin signaling may be a potential strategy to reduce food intake and ultimately body weight in obese patients (See “Ghrelin Receptors a Novel Target for Obesity” for details).
Prader-Willi syndrome (PWS). PWS is a genetic obesity syndrome caused by a defect in the chromosome 15 (q11–13). Children with PWS display growth hormone deficiency, rapid weight gain, and voracious appetite. Hyperphagia of PWS seems to involve alterations of hedonic aspects of feeding, since functional magnetic resonance imaging in these patients shows enhanced activation of the mesolimbic system areas following regular meals intake, when high-calorie foods are offered or even when food pictures are displayed to them (Miller et al. 2007; Holsen et al. 2006; Dimitropoulos and Schultz 2008). Of note, most PWS patients have several-fold higher ghrelin levels compared to weight-matched controls (Cummings et al. 2002a; DelParigi et al. 2002; Haqq et al. 2003a). In some PWS patients, the hyperphagia is related to high plasma ghrelin as hyperghrelinemia precedes obesity and plasma ghrelin levels positively correlate with their feelings of hunger (Haqq et al. 2003a; Purtell et al. 2011; Feigerlova et al. 2008). Of note, not all young PWS patients have elevated plasma ghrelin levels (Haqq et al. 2008). In addition, intervention studies suppressing ghrelin levels in PWS patients have failed to reduce appetite or compulsive eating (Tan et al. 2004; De Waele et al. 2008; Haqq et al. 2003b). Thus, the role of the ghrelin system in the pathogenesis of this disorder is still unclear.
Anorexia Nervosa. Anorexia nervosa is an eating disorder of unknown etiology characterized by refusal to maintain a minimally required healthy weight, intense fear of gaining weight, and misinterpretation of body shape. Anorexia nervosa can be divided into a restrictive type, with reduced food intake, and a binge eating/purging type, with binge eating/purging episodes during anorexia phases. Most studies report that fasted anorexia nervosa patients show high ghrelin levels, which normalize after food intake or body weight recovery (Ogiso et al. 2011). Patients with binging/purging anorexia nervosa type have higher ghrelin levels (Tanaka et al. 2003, 2004). Also, single nucleotide polymorphisms in ghrelin gene are specifically associated with binging/purging anorexia nervosa type (Dardennes et al. 2007). However, these findings have not been fully reproduced by other studies (Cardona Cano et al. 2012). Thus, the pathophysiological implications of high plasma ghrelin in anorexia nervosa are currently unclear. It has been proposed that administration of ghrelin (or ghrelin agonists) could increase food intake and hunger in these patients and thus promote weight gain. Until now, three studies have evaluated the effect of ghrelin administration on anorexia nervosa patients (Miljic et al. 2006; Broglio et al. 2004; Hotta et al. 2009). In one study, anorexia nervosa patients felt significantly less hungry compared to the thin control subjects, suggesting that anorexia nervosa patients are resistant to the orexigenic effects of ghrelin (Miljic et al. 2006). However, other studies found increased hunger sensation and increased food intake after ghrelin administration in some patients with anorexia nervosa (Broglio et al. 2004; Hotta et al. 2009). Thus, further studies are needed to determine if ghrelin treatment is a therapeutic option for this disorder.
Bulimia nervosa. Bulimia nervosa is a psychiatric disorder characterized by repetitive episodes of consumption of large amounts of food followed by compensatory behaviors in order to prevent weight gain, including self-induced vomiting, laxative abuse, and excessive exercising. As discussed in a recent review, findings from many studies that have investigated the potential pathophysiological role of ghrelin in the bulimia nervosa are inconsistent, and it is currently unclear whether the ghrelin system dysfunctions are relevant in this eating disorder (Cardona Cano et al. 2012).
Binge eating disorders. In contrast to bulimia nervosa, patients who suffer binge eating disorders engage in bouts of binge eating with no compensatory behavior afterwards that increases the risk for obesity. Some patients with binge eating disorders have an altered ghrelin dynamics, characterized by less postprandial decrease of ghrelin with a longer time to nadir compared with obese subjects, that could contribute to larger meals as seen during binge episodes (Geliebter et al. 2005, 2008). However, other studies have shown that fasting plasma ghrelin levels do not correlate with the frequency and severity of binging (Monteleone et al. 2005). Interestingly, a single nucleotide polymorphism of the ghrelin gene has been associated with binge eating disorders (Monteleone et al. 2007). As for other eating disorders, further studies are necessary to establish a link between binge eating disorders and ghrelin.
Cachexia. Cachexia or wasting syndrome is defined as unintentional appetite and body mass loss that cannot be reversed nutritionally. Lean body mass is lost even when the affected patient eats more calories, indicating that body mass loss is due to another primary pathology taking place. Cachexia is seen in patients with cancer, acquired immunodeficiency syndrome, chronic obstructive pulmonary disease, chronic renal insufficiency, congestive heart failure, tuberculosis, among others. Total plasma ghrelin levels are elevated in patients with cachexia, as expected for a chronic state of energy deficiency (DeBoer 2008). Despite the elevated plasma ghrelin concentrations, patients with cachexia remain sensitive to the orexigenic effects of ghrelin. Clinical studies have shown that administration of ghrelin or ghrelin receptor 1a agonists increased both food intake and body weight in patients with cachexia secondary to congestive heart failure, chronic obstructive pulmonary disease, or chronic renal insufficiency (Nagaya et al. 2004, 2005; Wynne et al. 2005; Deboer et al. 2008; Ashby et al. 2009). In addition, several trials have demonstrated the efficacy and safety of ghrelin or ghrelin receptor 1a agonists to increase food intake and body weight in patients with cancer-associated cachexia (Neary et al. 2004; Strasser et al. 2008; Garcia et al. 2013). Thus, ghrelin system may be a potential pharmacological target in the treatment of cachexia (Argiles and Stemmler 2013).
Concluding Remarks
Recent studies have started to reveal the complex neuronal circuits and mechanisms by which ghrelin promotes food intake. Ghrelin not only acts on neuronal circuits that regulate homeostatic intake of food but also on neuronal circuits that affect hedonic aspects of eating including preference for palatable foods, motivation to obtain preferred foods, food anticipatory locomotor activity, rewarding value of preferred foods, and acquisition of food CPP. Thus, ghrelin modulates a variety of key aspects of hedonic eating that directly impact on feeding behaviors. Of note, other peripheral signals from adipose tissue (e.g., leptin), pancreas (e.g., insulin), and the gastrointestinal tract (e.g., peptide YY, glucagon-like peptide-1, cholecystokinin) also regulate central circuits controlling food intake. However, ghrelin is the only known peptide hormone that causes an acute and potent increase of food intake when administrated in small doses to animals or human beings. This unique feature makes the ghrelin system exceptionally attractive for the development of specific pharmacological therapies to treat eating disorders.
References
Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschop MH, Gao XB, Horvath TL (2006) Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest 116(12):3229–3239. doi:10.1172/JCI29867
Adachi S, Takiguchi S, Okada K, Yamamoto K, Yamasaki M, Miyata H, Nakajima K, Fujiwara Y, Hosoda H, Kangawa K, Mori M, Doki Y (2010) Effects of ghrelin administration after total gastrectomy: a prospective, randomized, placebo-controlled phase II study. Gastroenterology 138(4):1312–1320. doi:10.1053/j.gastro.2009.12.058, S0016-5085(10)00010-7 [pii]
Akamizu T, Iwakura H, Ariyasu H, Hosoda H, Murayama T, Yokode M, Teramukai S, Seno H, Chiba T, Noma S, Nakai Y, Fukunaga M, Kangawa K (2008) Repeated administration of ghrelin to patients with functional dyspepsia: its effects on food intake and appetite. Eur J Endocrinol 158(4):491–498. doi:10.1530/EJE-07-0768, 158/4/491 [pii]
Argiles JM, Stemmler B (2013) The potential of ghrelin in the treatment of cancer cachexia. Expert Opin Biol Ther 13(1):67–76. doi:10.1517/14712598.2013.727390
Ashby DR, Ford HE, Wynne KJ, Wren AM, Murphy KG, Busbridge M, Brown EA, Taube DH, Ghatei MA, Tam FW, Bloom SR, Choi P (2009) Sustained appetite improvement in malnourished dialysis patients by daily ghrelin treatment. Kidney Int 76(2):199–206. doi:10.1038/ki.2009.114, ki2009114 [pii]
Atasoy D, Betley JN, Su HH, Sternson SM (2012) Deconstruction of a neural circuit for hunger. Nature 488(7410):172–177. doi:10.1038/nature11270, nature11270 [pii]
Banks WA (2008) The blood-brain barrier: connecting the gut and the brain. Regul Pept 149(1–3):11–14. doi:10.1016/j.regpep.2007.08.027, S0167-0115(08)00061-X [pii]
Beckman LM, Beckman TR, Earthman CP (2010) Changes in gastrointestinal hormones and leptin after Roux-en-Y gastric bypass procedure: a review. J Am Diet Assoc 110(4):571–584. doi:10.1016/j.jada.2009.12.023, S0002-8223(09)02093-8 [pii]
Berthoud HR (2011) Metabolic and hedonic drives in the neural control of appetite: who is the boss? Curr Opin Neurobiol 21(6):888–896. doi:10.1016/j.conb.2011.09.004, S0959-4388(11)00148-6[pii]
Bewick GA, Kent A, Campbell D, Patterson M, Ghatei MA, Bloom SR, Gardiner JV (2009) Mice with hyperghrelinemia are hyperphagic and glucose intolerant and have reduced leptin sensitivity. Diabetes 58(4):840–846. doi:10.2337/db08-1428, db08-1428 [pii]
Blum ID, Patterson Z, Khazall R, Lamont EW, Sleeman MW, Horvath TL, Abizaid A (2009) Reduced anticipatory locomotor responses to scheduled meals in ghrelin receptor deficient mice. Neuroscience 164(2):351–359. doi:10.1016/j.neuroscience.2009.08.009, S0306-4522(09)01292-5 [pii]
Briggs DI, Andrews ZB (2011) Metabolic status regulates ghrelin function on energy homeostasis. Neuroendocrinology 93(1):48–57. doi:10.1159/000322589, 000322589 [pii]
Briggs DI, Enriori PJ, Lemus MB, Cowley MA, Andrews ZB (2010) Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology 151(10):4745–4755. doi:10.1210/en.2010-0556, en.2010-0556, [pii]
Broglio F, Gianotti L, Destefanis S, Fassino S, Abbate Daga G, Mondelli V, Lanfranco F, Gottero C, Gauna C, Hofland L, Van der Lely AJ, Ghigo E (2004) The endocrine response to acute ghrelin administration is blunted in patients with anorexia nervosa, a ghrelin hypersecretory state. Clin Endocrinol (Oxf) 60(5):592–599. doi:10.1111/j.1365-2265.2004.02011.x, CEN2011 [pii]
Cardona Cano S, Merkestein M, Skibicka KP, Dickson SL, Adan RA (2012) Role of ghrelin in the pathophysiology of eating disorders: implications for pharmacotherapy. CNS Drugs 26(4):281–296. doi:10.2165/11599890-000000000-00000, 1 [pii]
Chuang JC, Perello M, Sakata I, Osborne-Lawrence S, Savitt JM, Lutter M, Zigman JM (2011) Ghrelin mediates stress-induced food-reward behavior in mice. J Clin Invest 121(7):2684–2692. doi:10.1172/JCI57660, 57660 [pii]
Cong WN, Golden E, Pantaleo N, White CM, Maudsley S, Martin B (2010) Ghrelin receptor signaling: a promising therapeutic target for metabolic syndrome and cognitive dysfunction. CNS Neurol Disord Drug Targets 9(5):557–563. doi:BSP/CDTCNSND/E-Pub/00055, [pii]
Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, Horvath TL (2003) The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37(4):649–661. doi:S0896627303000631, [pii]
Cummings DE (2006) Ghrelin and the short- and long-term regulation of appetite and body weight. Physiol Behav 71:71–84
Cummings DE, Clement K, Purnell JQ, Vaisse C, Foster KE, Frayo RS, Schwartz MW, Basdevant A, Weigle DS (2002a) Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med 8(7):643–644. doi:10.1038/nm0702-643, nm0702-643 [pii]
Cummings DE, Purnell JQ, Frayo RS, Schmidova K, Wisse BE, Weigle DS (2001) A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50(8):1714–1719
Cummings DE, Shannon MH (2003) Ghrelin and gastric bypass: is there a hormonal contribution to surgical weight loss? J Clin Endocrinol Metab 88(7):2999–3002
Cummings DE, Weigle DS, Frayo RS, Breen PA, Ma MK, Dellinger EP, Purnell JQ (2002b) Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 346(21):1623–1630. doi:10.1056/NEJMoa012908, 346/21/1623 [pii]
Damian M, Marie J, Leyris JP, Fehrentz JA, Verdie P, Martinez J, Baneres JL, Mary S (2012) High constitutive activity is an intrinsic feature of ghrelin receptor protein: a study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J Biol Chem 287(6):3630–3641. doi:10.1074/jbc.M111.288324, M111.288324 [pii]
Dardennes RM, Zizzari P, Tolle V, Foulon C, Kipman A, Romo L, Iancu-Gontard D, Boni C, Sinet PM, Therese Bluet M, Estour B, Mouren MC, Guelfi JD, Rouillon F, Gorwood P, Epelbaum J (2007) Family trios analysis of common polymorphisms in the obestatin/ghrelin, BDNF and AGRP genes in patients with Anorexia nervosa: association with subtype, body-mass index, severity and age of onset. Psychoneuroendocrinology 32(2):106–113. doi:10.1016/j.psyneuen.2006.11.003, S0306-4530(06)00197-1 [pii]
Date Y (2012) Ghrelin and the vagus nerve. Methods Enzymol 514:261–269. doi:10.1016/B978-0-12-381272-8.00016-7, B978-0-12-381272-8.00016-7 [pii]
Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M (2002) The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123(4):1120–1128. doi:S0016508502002172, [pii]
Date Y, Shimbara T, Koda S, Toshinai K, Ida T, Murakami N, Miyazato M, Kokame K, Ishizuka Y, Ishida Y, Kageyama H, Shioda S, Kangawa K, Nakazato M (2006) Peripheral ghrelin transmits orexigenic signals through the noradrenergic pathway from the hindbrain to the hypothalamus. Cell Metab 4(4):323–331. doi:10.1016/j.cmet.2006.09.004, S1550-4131(06)00303-2 [pii]
Davis JF, Perello M, Choi DL, Magrisso IJ, Kirchner H, Pfluger PT, Tschoep M, Zigman JM, Benoit SC (2012) GOAT induced ghrelin acylation regulates hedonic feeding. Horm Behav 62(5):598–604. doi:10.1016/j.yhbeh.2012.08.009, S0018-506X(12)00202-4 [pii]
De Smet B, Depoortere I, Moechars D, Swennen Q, Moreaux B, Cryns K, Tack J, Buyse J, Coulie B, Peeters TL (2006) Energy homeostasis and gastric emptying in ghrelin knockout mice. J Pharmacol Exp Ther 316(1):431–439. doi:10.1124/jpet.105.091504, jpet.105.091504 [pii]
De Waele K, Ishkanian SL, Bogarin R, Miranda CA, Ghatei MA, Bloom SR, Pacaud D, Chanoine JP (2008) Long-acting octreotide treatment causes a sustained decrease in ghrelin concentrations but does not affect weight, behaviour and appetite in subjects with Prader-Willi syndrome. Eur J Endocrinol 159(4):381–388. doi:10.1530/EJE-08-0462, EJE-08-0462 [pii]
DeBoer MD (2008) Emergence of ghrelin as a treatment for cachexia syndromes. Nutrition 24(9):806–814. doi:10.1016/j.nut.2008.06.013, S0899-9007(08)00291-8 [pii]
Deboer MD, Zhu X, Levasseur PR, Inui A, Hu Z, Han G, Mitch WE, Taylor JE, Halem HA, Dong JZ, Datta R, Culler MD, Marks DL (2008) Ghrelin treatment of chronic kidney disease: improvements in lean body mass and cytokine profile. Endocrinology 149(2):827–835. doi:10.1210/en.2007-1046, en.2007-1046 [pii]
DelParigi A, Tschop M, Heiman ML, Salbe AD, Vozarova B, Sell SM, Bunt JC, Tataranni PA (2002) High circulating ghrelin: a potential cause for hyperphagia and obesity in prader-willi syndrome. J Clin Endocrinol Metab 87(12):5461–5464
Dezaki K, Sone H, Koizumi M, Nakata M, Kakei M, Nagai H, Hosoda H, Kangawa K, Yada T (2006) Blockade of pancreatic islet-derived ghrelin enhances insulin secretion to prevent high-fat diet-induced glucose intolerance. Diabetes 55(12):3486–3493. doi:10.2337/db06-0878, 55/12/3486 [pii]
Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA, Morley JE, Pinto S, Sherwin RS, Xu L, Yamada KA, Sleeman MW, Tschop MH, Horvath TL (2006) Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci 9(3):381–388. doi:10.1038/nn1656, nn1656 [pii]
Dickson SL, Hrabovszky E, Hansson C, Jerlhag E, Alvarez-Crespo M, Skibicka KP, Molnar CS, Liposits Z, Engel JA, Egecioglu E (2010) Blockade of central nicotine acetylcholine receptor signaling attenuate ghrelin-induced food intake in rodents. Neuroscience 171(4):1180–1186. doi:10.1016/j.neuroscience.2010.10.005, S0306-4522(10)01335-7 [pii]
DiLeone RJ, Taylor JR, Picciotto MR (2012) The drive to eat: comparisons and distinctions between mechanisms of food reward and drug addiction. Nat Neurosci 15(10):1330–1335. doi:10.1038/nn.3202, nn.3202 [pii]
Dimitropoulos A, Schultz RT (2008) Food-related neural circuitry in Prader-Willi syndrome: response to high- versus low-calorie foods. J Autism Dev Disord 38(9):1642–1653. doi:10.1007/s10803-008-0546-x
Disse E, Bussier AL, Deblon N, Pfluger PT, Tschop MH, Laville M, Rohner-Jeanrenaud F (2011) Systemic ghrelin and reward: effect of cholinergic blockade. Physiol Behav 102(5):481–484. doi:10.1016/j.physbeh.2010.12.006, S0031-9384(10)00453-1 [pii]
Disse E, Bussier AL, Veyrat-Durebex C, Deblon N, Pfluger PT, Tschop MH, Laville M, Rohner-Jeanrenaud F (2010) Peripheral ghrelin enhances sweet taste food consumption and preference, regardless of its caloric content. Physiol Behav 101(2):277–281. doi:10.1016/j.physbeh.2010.05.017, S0031-9384(10)00234-9 [pii]
Druce MR, Wren AM, Park AJ, Milton JE, Patterson M, Frost G, Ghatei MA, Small C, Bloom SR (2005) Ghrelin increases food intake in obese as well as lean subjects. Int J Obes (Lond) 29(9):1130–1136. doi:10.1038/sj.ijo.0803001, 0803001 [pii]
Egecioglu E, Jerlhag E, Salome N, Skibicka KP, Haage D, Bohlooly YM, Andersson D, Bjursell M, Perrissoud D, Engel JA, Dickson SL (2010) Ghrelin increases intake of rewarding food in rodents. Addict Biol 15(3):304–311. doi:10.1111/j.1369-1600.2010.00216.x, ADB216 [pii]
English PJ, Ghatei MA, Malik IA, Bloom SR, Wilding JP (2002) Food fails to suppress ghrelin levels in obese humans. J Clin Endocrinol Metab 87(6):2984
Falken Y, Hellstrom PM, Sanger GJ, Dewit O, Dukes G, Gryback P, Holst JJ, Naslund E (2010) Actions of prolonged ghrelin infusion on gastrointestinal transit and glucose homeostasis in humans. Neurogastroenterol Motil 22(6):e192–200. doi:10.1111/j.1365-2982.2009.01463.x, NMO1463 [pii]
Feigerlova E, Diene G, Conte-Auriol F, Molinas C, Gennero I, Salles JP, Arnaud C, Tauber M (2008) Hyperghrelinemia precedes obesity in Prader-Willi syndrome. J Clin Endocrinol Metab 93(7):2800–2805. doi:10.1210/jc.2007-2138, jc.2007-2138 [pii]
Finger BC, Dinan TG, Cryan JF (2012) Diet-induced obesity blunts the behavioural effects of ghrelin: studies in a mouse-progressive ratio task. Psychopharmacology 220(1):173–181. doi:10.1007/s00213-011-2468-0
Fry M, Ferguson AV (2007) The sensory circumventricular organs: brain targets for circulating signals controlling ingestive behavior. Physiol Behav 91(4):413–423. doi:10.1016/j.physbeh.2007.04.003, S0031-9384(07)00130-8 [pii]
Fry M, Ferguson AV (2010) Ghrelin: central nervous system sites of action in regulation of energy balance. Int J Pept 2010. doi:10.1155/2010/616757, 616757 [pii]
Furness JB, Hunne B, Matsuda N, Yin L, Russo D, Kato I, Fujimiya M, Patterson M, McLeod J, Andrews ZB, Bron R (2011) Investigation of the presence of ghrelin in the central nervous system of the rat and mouse. Neuroscience 193:1–9. doi:10.1016/j.neuroscience.2011.07.063, S0306-4522(11)00889-X [pii]
Garcia JM, Friend J, Allen S (2013) Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study. Support Care Cancer 21(1):129–137. doi:10.1007/s00520-012-1500-1
Geliebter A, Gluck ME, Hashim SA (2005) Plasma ghrelin concentrations are lower in binge-eating disorder. J Nutr 135(5):1326–1330. doi:135/5/1326, [pii]
Geliebter A, Hashim SA, Gluck ME (2008) Appetite-related gut peptides, ghrelin, PYY, and GLP-1 in obese women with and without binge eating disorder (BED). Physiol Behav 94(5):696–699. doi:10.1016/j.physbeh.2008.04.013, S0031-9384(08)00114-5 [pii]
Gilg S, Lutz TA (2006) The orexigenic effect of peripheral ghrelin differs between rats of different age and with different baseline food intake, and it may in part be mediated by the area postrema. Physiol Behav 87(2):353–359. doi:10.1016/j.physbeh.2005.10.015, S0031-9384(05)00492-0 [pii]
Goldstone AP, Prechtl de Hernandez CG, Beaver JD, Muhammed K, Croese C, Bell G, Durighel G, Hughes E, Waldman AD, Frost G, Bell JD (2009) Fasting biases brain reward systems towards high-calorie foods. Eur J Neurosci 30(8):1625–1635. doi:10.1111/j.1460-9568.2009.06949.x, EJN6949 [pii]
Guan XM, Yu H, Palyha OC, McKee KK, Feighner SD, Sirinathsinghji DJ, Smith RG, Van der Ploeg LH, Howard AD (1997) Distribution of mRNA encoding the growth hormone secretagogue receptor in brain and peripheral tissues. Brain Res Mol Brain Res 48(1):23–29. doi:S0169328X97000715, [pii]
Gueorguiev M, Lecoeur C, Meyre D, Benzinou M, Mein CA, Hinney A, Vatin V, Weill J, Heude B, Hebebrand J, Grossman AB, Korbonits M, Froguel P (2009) Association studies on ghrelin and ghrelin receptor gene polymorphisms with obesity. Obesity (Silver Spring) 17(4):745–754. doi:10.1038/oby.2008.589, oby2008589 [pii]
Haqq AM, Farooqi IS, O’Rahilly S, Stadler DD, Rosenfeld RG, Pratt KL, LaFranchi SH, Purnell JQ (2003a) Serum ghrelin levels are inversely correlated with body mass index, age, and insulin concentrations in normal children and are markedly increased in Prader-Willi syndrome. J Clin Endocrinol Metab 88(1):174–178
Haqq AM, Grambow SC, Muehlbauer M, Newgard CB, Svetkey LP, Carrel AL, Yanovski JA, Purnell JQ, Freemark M (2008) Ghrelin concentrations in Prader-Willi syndrome (PWS) infants and children: changes during development. Clin Endocrinol (Oxf) 69(6):911–920. doi:10.1111/j.1365-2265.2008.03385.x, CEN3385 [pii]
Haqq AM, Stadler DD, Rosenfeld RG, Pratt KL, Weigle DS, Frayo RS, LaFranchi SH, Cummings DE, Purnell JQ (2003b) Circulating ghrelin levels are suppressed by meals and octreotide therapy in children with Prader-Willi syndrome. J Clin Endocrinol Metab 88(8):3573–3576
Harrold JA, Williams G (2003) The cannabinoid system: a role in both the homeostatic and hedonic control of eating? Br J Nutr 90(4):729–734. doi:S000711450300179X, [pii]
Hillman JB, Tong J, Tschop M (2011) Ghrelin biology and its role in weight-related disorders. Discov Med 11(61):521–528
Holsen LM, Zarcone JR, Brooks WM, Butler MG, Thompson TI, Ahluwalia JS, Nollen NL, Savage CR (2006) Neural mechanisms underlying hyperphagia in Prader-Willi syndrome. Obesity (Silver Spring) 14(6):1028–1037. doi:10.1038/oby.2006.118, 14/6/1028 [pii]
Hotta M, Ohwada R, Akamizu T, Shibasaki T, Takano K, Kangawa K (2009) Ghrelin increases hunger and food intake in patients with restricting-type anorexia nervosa: a pilot study. Endocr J 56(9):1119–1128. doi:JST.JSTAGE/endocrj/K09E-168, [pii]
Hyman SE, Malenka RC, Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29:565–598. doi:10.1146/annurev.neuro.29.051605.113009
Jerlhag E (2008) Systemic administration of ghrelin induces conditioned place preference and stimulates accumbal dopamine. Addict Biol 13(3–4):358–363. doi:10.1111/j.1369-1600.2008.00125.x, ADB125 [pii]
Jerlhag E, Egecioglu E, Dickson SL, Andersson M, Svensson L, Engel JA (2006) Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol 11(1):45–54. doi:10.1111/j.1369-1600.2006.00002.x, ADB002 [pii]
Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA (2007) Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol 12(1):6–16. doi:10.1111/j.1369-1600.2006.00041.x, ADB041 [pii]
Jerlhag E, Egecioglu E, Dickson SL, Engel JA (2011) Glutamatergic regulation of ghrelin-induced activation of the mesolimbic dopamine system. Addict Biol 16(1):82–91. doi:10.1111/j.1369-1600.2010.00231.x, ADB231 [pii]
Jerlhag E, Egecioglu E, Dickson SL, Svensson L, Engel JA (2008) Alpha-conotoxin MII-sensitive nicotinic acetylcholine receptors are involved in mediating the ghrelin-induced locomotor stimulation and dopamine overflow in nucleus accumbens. Eur Neuropsychopharmacol 18(7):508–518. doi:10.1016/j.euroneuro.2008.02.006. S0924-977X(08)00051-5 [pii]
Jiang H, Betancourt L, Smith RG (2006) Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol Endocrinol 20(8):1772–1785. doi:10.1210/me.2005-0084, me.2005-0084 [pii]
Kageyama H, Takenoya F, Shiba K, Shioda S (2010) Neuronal circuits involving ghrelin in the hypothalamus-mediated regulation of feeding. Neuropeptides 44(2):133–138. doi:10.1016/j.npep.2009.11.010, S0143-4179(09)00139-5 [pii]
Keen-Rhinehart E, Bartness TJ (2005) Peripheral ghrelin injections stimulate food intake, foraging, and food hoarding in Siberian hamsters. Am J Physiol Regul Integr Comp Physiol 288(3):R716–722. doi:10.1152/ajpregu.00705.2004, 00705.2004 [pii]
Kern A, Albarran-Zeckler R, Walsh HE, Smith RG (2012) Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 73(2):317–332. doi:10.1016/j.neuron.2011.10.038, S0896-6273(11)01087-7 [pii]
King SJ, Isaacs AM, O’Farrell E, Abizaid A (2011) Motivation to obtain preferred foods is enhanced by ghrelin in the ventral tegmental area. Horm Behav 60(5):572–580. doi:10.1016/j.yhbeh.2011.08.006, S0018-506X(11)00188-7 [pii]
Kirchner H, Gutierrez JA, Solenberg PJ, Pfluger PT, Czyzyk TA, Willency JA, Schurmann A, Joost HG, Jandacek RJ, Hale JE, Heiman ML, Tschop MH (2009) GOAT links dietary lipids with the endocrine control of energy balance. Nat Med 15(7):741–745. doi:10.1038/nm.1997, nm.1997 [pii]
Kola B, Farkas I, Christ-Crain M, Wittmann G, Lolli F, Amin F, Harvey-White J, Liposits Z, Kunos G, Grossman AB, Fekete C, Korbonits M (2008) The orexigenic effect of ghrelin is mediated through central activation of the endogenous cannabinoid system. PLoS ONE 3(3):e1797. doi:10.1371/journal.pone.0001797
Korotkova TM, Sergeeva OA, Eriksson KS, Haas HL, Brown RE (2003) Excitation of ventral tegmental area dopaminergic and nondopaminergic neurons by orexins/hypocretins. J Neurosci 23(1):7–11. doi:23/1/7, [pii]
Lamont EW, Patterson Z, Rodrigues T, Vallejos O, Blum ID, Abizaid A (2012) Ghrelin-deficient mice have fewer orexin cells and reduced cFOS expression in the mesolimbic dopamine pathway under a restricted feeding paradigm. Neuroscience 218:12–19. doi:10.1016/j.neuroscience.2012.05.046, S0306-4522(12)00534-9 [pii]
Landgren S, Simms JA, Thelle DS, Strandhagen E, Bartlett SE, Engel JA, Jerlhag E (2011) The ghrelin signalling system is involved in the consumption of sweets. PLoS ONE 6(3):e18170. doi:10.1371/journal.pone.0018170
le Roux CW, Patterson M, Vincent RP, Hunt C, Ghatei MA, Bloom SR (2005) Postprandial plasma ghrelin is suppressed proportional to meal calorie content in normal-weight but not obese subjects. J Clin Endocrinol Metab 90(2):1068–1071. doi:10.1210/jc.2004-1216, jc.2004-1216 [pii]
Levin F, Edholm T, Schmidt PT, Gryback P, Jacobsson H, Degerblad M, Hoybye C, Holst JJ, Rehfeld JF, Hellstrom PM, Naslund E (2006) Ghrelin stimulates gastric emptying and hunger in normal-weight humans. J Clin Endocrinol Metab 91(9):3296–3302. doi:10.1210/jc.2005-2638, jc.2005-2638 [pii]
Lin L, Saha PK, Ma X, Henshaw IO, Shao L, Chang BH, Buras ED, Tong Q, Chan L, McGuinness OP, Sun Y (2011) Ablation of ghrelin receptor reduces adiposity and improves insulin sensitivity during aging by regulating fat metabolism in white and brown adipose tissues. Aging Cell 10(6):996–1010. doi:10.1111/j.1474-9726.2011.00740.x
Liu B, Garcia EA, Korbonits M (2011) Genetic studies on the ghrelin, growth hormone secretagogue receptor (GHSR) and ghrelin O-acyl transferase (GOAT) genes. Peptides 32(11):2191–2207. doi:10.1016/j.peptides.2011.09.006, S0196-9781(11)00372-X [pii]
Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ, Zigman JM (2008) The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci 11(7):752–753. doi:10.1038/nn.2139, nn.2139 [pii]
Ma X, Lin L, Qin G, Lu X, Fiorotto M, Dixit VD, Sun Y (2011) Ablations of ghrelin and ghrelin receptor exhibit differential metabolic phenotypes and thermogenic capacity during aging. PLoS ONE 6(1):e16391. doi:10.1371/journal.pone.0016391
Mahler SV, Smith RJ, Moorman DE, Sartor GC, Aston-Jones G (2012) Multiple roles for orexin/hypocretin in addiction. Prog Brain Res 198:79–121. doi:10.1016/B978-0-444-59489-1.00007-0, B978-0-444-59489-1.00007-0 [pii]
Malik S, McGlone F, Bedrossian D, Dagher A (2008) Ghrelin modulates brain activity in areas that control appetitive behavior. Cell Metab 7(5):400–409. doi:10.1016/j.cmet.2008.03.007, S1550-4131(08)00078-8 [pii]
McCallum SE, Taraschenko OD, Hathaway ER, Vincent MY, Glick SD (2011) Effects of 18-methoxycoronaridine on ghrelin-induced increases in sucrose intake and accumbal dopamine overflow in female rats. Psychopharmacology 215(2):247–256. doi:10.1007/s00213-010-2132-0
Merkestein M, Brans MA, Luijendijk MC, de Jong JW, Egecioglu E, Dickson SL, Adan RA (2012) Ghrelin mediates anticipation to a palatable meal in rats. Obesity (Silver Spring) 20(5):963–971. doi:10.1038/oby.2011.389, oby2011389 [pii]
Miljic D, Pekic S, Djurovic M, Doknic M, Milic N, Casanueva FF, Ghatei M, Popovic V (2006) Ghrelin has partial or no effect on appetite, growth hormone, prolactin, and cortisol release in patients with anorexia nervosa. J Clin Endocrinol Metab 91(4):1491–1495. doi:10.1210/jc.2005-2304, jc.2005-2304 [pii]
Miller JL, James GA, Goldstone AP, Couch JA, He G, Driscoll DJ, Liu Y (2007) Enhanced activation of reward mediating prefrontal regions in response to food stimuli in Prader-Willi syndrome. J Neurol Neurosurg Psychiatry 78(6):615–619. doi:10.1136/jnnp.2006.099044jnnp.2006.099044 [pii]
Mokrosinski J, Holst B (2010) Modulation of the constitutive activity of the ghrelin receptor by use of pharmacological tools and mutagenesis. Methods Enzymol 484:53–73. doi:10.1016/B978-0-12-381298-8.00003-4, B978-0-12-381298-8.00003-4 [pii]
Monteleone P, Fabrazzo M, Tortorella A, Martiadis V, Serritella C, Maj M (2005) Circulating ghrelin is decreased in non-obese and obese women with binge eating disorder as well as in obese non-binge eating women, but not in patients with bulimia nervosa. Psychoneuroendocrinology 30(3):243–250. doi:10.1016/j.psyneuen.2004.07.004, S0306-4530(04)00125-8 [pii]
Monteleone P, Tortorella A, Castaldo E, Di Filippo C, Maj M (2007) The Leu72Met polymorphism of the ghrelin gene is significantly associated with binge eating disorder. Psychiatr Genet 17(1):13–16. doi:10.1097/YPG.0b013e328010e2c3, 00041444-200702000-00007 [pii]
Morpurgo PS, Resnik M, Agosti F, Cappiello V, Sartorio A, Spada A (2003) Ghrelin secretion in severely obese subjects before and after a 3-week integrated body mass reduction program. J Endocrinol Invest 26(8):723–727. doi:5567, [pii]
Nagaya N, Itoh T, Murakami S, Oya H, Uematsu M, Miyatake K, Kangawa K (2005) Treatment of cachexia with ghrelin in patients with COPD. Chest 128(3):1187–1193. doi:10.1378/chest.128.3.1187, 128/3/1187 [pii]
Nagaya N, Moriya J, Yasumura Y, Uematsu M, Ono F, Shimizu W, Ueno K, Kitakaze M, Miyatake K, Kangawa K (2004) Effects of ghrelin administration on left ventricular function, exercise capacity, and muscle wasting in patients with chronic heart failure. Circulation 110(24):3674–3679. doi:10.1161/01.CIR.0000149746.62908.BB, 01.CIR.0000149746.62908.BB [pii]
Nakamura T, Uramura K, Nambu T, Yada T, Goto K, Yanagisawa M, Sakurai T (2000) Orexin-induced hyperlocomotion and stereotypy are mediated by the dopaminergic system. Brain Res 873(1):181–187. doi:S0006-8993(00)02555-5, [pii]
Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S (2001) A role for ghrelin in the central regulation of feeding. Nature 409(6817):194–198
Naleid AM, Grace MK, Cummings DE, Levine AS (2005) Ghrelin induces feeding in the mesolimbic reward pathway between the ventral tegmental area and the nucleus accumbens. Peptides 26(11):2274–2279. doi:10.1016/j.peptides.2005.04.025, S0196-9781(05)00350-5 [pii]
Neary MT, Batterham RL (2010) Gaining new insights into food reward with functional neuroimaging. Forum Nutr 63:152–163. doi:10.1159/000264403, 000264403 [pii]
Neary NM, Small CJ, Wren AM, Lee JL, Druce MR, Palmieri C, Frost GS, Ghatei MA, Coombes RC, Bloom SR (2004) Ghrelin increases energy intake in cancer patients with impaired appetite: acute, randomized, placebo-controlled trial. J Clin Endocrinol Metab 89(6):2832–2836. doi:10.1210/jc.2003-031768, 89/6/2832[pii]
Ogiso K, Asakawa A, Amitani H, Inui A (2011) Ghrelin and anorexia nervosa: a psychosomatic perspective. Nutrition 27(10):988–993. doi:10.1016/j.nut.2011.05.005, S0899-9007(11)00155-9 [pii]
Olszewski PK, Li D, Grace MK, Billington CJ, Kotz CM, Levine AS (2003) Neural basis of orexigenic effects of ghrelin acting within lateral hypothalamus. Peptides 24(4):597–602. doi:S0196978103001050, [pii]
Overduin J, Figlewicz DP, Bennett-Jay J, Kittleson S, Cummings DE (2012) Ghrelin increases the motivation to eat, but does not alter food palatability. Am J Physiol Regul Integr Comp Physiol 303(3):R259–269. doi:10.1152/ajpregu.00488.2011, ajpregu.00488.2011 [pii]
Palmiter RD (2007) Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci 30(8):375–381. doi:10.1016/j.tins.2007.06.004, S0166-2236(07)00133-6 [pii]
Patterson ZR, Ducharme R, Anisman H, Abizaid A (2010) Altered metabolic and neurochemical responses to chronic unpredictable stressors in ghrelin receptor-deficient mice. Eur J Neurosci 32(4):632–639. doi:10.1111/j.1460-9568.2010.07310.x, EJN7310 [pii]
Patterson ZR, Khazall R, Mackay H, Anisman H, Abizaid A (2013) Central ghrelin signaling mediates the metabolic response of C57BL/6 male mice to chronic social defeat stress. Endocrinology 154(3):1080–1091. doi:10.1210/en.2012-1834, en.2012-1834 [pii]
Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, Woloszyn J, Yanagisawa M, Lutter M, Zigman JM (2010) Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry 67(9):880–886. doi:10.1016/j.biopsych.2009.10.030, S0006-3223(09)01318-3 [pii]
Perello M, Scott MM, Sakata I, Lee CE, Chuang JC, Osborne-Lawrence S, Rovinsky SA, Elmquist JK, Zigman JM (2012) Functional implications of limited leptin receptor and ghrelin receptor coexpression in the brain. J Comp Neurol 520(2):281–294. doi:10.1002/cne.22690
Perello M, Zigman JM (2012) The role of ghrelin in reward-based eating. Biol Psychiatry 72(5):347–353. doi:10.1016/j.biopsych.2012.02.016, S0006-3223(12)00143-6 [pii]
Perreault M, Istrate N, Wang L, Nichols AJ, Tozzo E, Stricker-Krongrad A (2004) Resistance to the orexigenic effect of ghrelin in dietary-induced obesity in mice: reversal upon weight loss. Int J Obes Relat Metab Disord 28(7):879–885. doi:10.1038/sj.ijo.0802640, 0802640 [pii]
Petersen PS, Woldbye DP, Madsen AN, Egerod KL, Jin C, Lang M, Rasmussen M, Beck-Sickinger AG, Holst B (2009) In vivo characterization of high Basal signaling from the ghrelin receptor. Endocrinology 150(11):4920–4930. doi:10.1210/en.2008-1638, en.2008-1638 [pii]
Pfluger PT, Kirchner H, Gunnel S, Schrott B, Perez-Tilve D, Fu S, Benoit SC, Horvath T, Joost HG, Wortley KE, Sleeman MW, Tschop MH (2008) Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am J Physiol Gastrointest Liver Physiol 294(3):G610–618. doi:10.1152/ajpgi.00321.2007, 00321.2007 [pii]
Purtell L, Sze L, Loughnan G, Smith E, Herzog H, Sainsbury A, Steinbeck K, Campbell LV, Viardot A (2011) In adults with Prader-Willi syndrome, elevated ghrelin levels are more consistent with hyperphagia than high PYY and GLP-1 levels. Neuropeptides 45(4):301–307. doi:10.1016/j.npep.2011.06.001, S0143-4179(11)00046-1 [pii]
Rediger A, Piechowski CL, Yi CX, Tarnow P, Strotmann R, Gruters A, Krude H, Schoneberg T, Tschop MH, Kleinau G, Biebermann H (2011) Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J Biol Chem 286(45):39623–39631. doi:10.1074/jbc.M111.287607, M111.287607 [pii]
Reed JA, Benoit SC, Pfluger PT, Tschop MH, D’Alessio DA, Seeley RJ (2008) Mice with chronically increased circulating ghrelin develop age-related glucose intolerance. Am J Physiol Endocrinol Metab 294(4):E752–760. doi:10.1152/ajpendo.00463.2007, 00463.2007 [pii]
Rodriguez EM, Blazquez JL, Guerra M (2010) The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: the former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 31(4):757–776. doi:10.1016/j.peptides.2010.01.003, S0196-9781(10)00023-9 [pii]
Sakata I, Nakano Y, Osborne-Lawrence S, Rovinsky SA, Lee CE, Perello M, Anderson JG, Coppari R, Xiao G, Lowell BB, Elmquist JK, Zigman JM (2009) Characterization of a novel ghrelin cell reporter mouse. Regul Pept 155(1–3):91–98. doi:10.1016/j.regpep.2009.04.001, S0167-0115(09)00077-9 [pii]
Sakata I, Yamazaki M, Inoue K, Hayashi Y, Kangawa K, Sakai T (2003) Growth hormone secretagogue receptor expression in the cells of the stomach-projected afferent nerve in the rat nodose ganglion. Neurosci Lett 342(3):183–186. doi:S0304394003002945, [pii]
Saper CB, Chou TC, Elmquist JK (2002) The need to feed: homeostatic and hedonic control of eating. Neuron 36(2):199–211. doi:S0896627302009698 [pii]
Sato T, Kurokawa M, Nakashima Y, Ida T, Takahashi T, Fukue Y, Ikawa M, Okabe M, Kangawa K, Kojima M (2008) Ghrelin deficiency does not influence feeding performance. Regul Pept 145(1–3):7–11. doi:10.1016/j.regpep.2007.09.010, S0167-0115(07)00186-3 [pii]
Schaeffer M, Langlet F, Lafont C, Molino F, Hodson DJ, Roux T, Lamarque L, Verdie P, Bourrier E, Dehouck B, Baneres JL, Martinez J, Mery PF, Marie J, Trinquet E, Fehrentz JA, Prevot V, Mollard P (2013) Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. Proc Natl Acad Sci U S A 110(4):1512–1517. doi:10.1073/pnas.1212137110, 1212137110 [pii]
Schellekens H, Finger BC, Dinan TG, Cryan JF (2012) Ghrelin signalling and obesity: at the interface of stress, mood and food reward. Pharmacol Ther 135(3):316–326. doi:10.1016/j.pharmthera.2012.06.004, S0163-7258(12)00122-2 [pii]
Schellekens H, van Oeffelen WE, Dinan TG, Cryan JF (2013) Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signaling. J Biol Chem 288(1):181–191. doi:10.1074/jbc.M112.382473, M112.382473 [pii]
Schmid DA, Held K, Ising M, Uhr M, Weikel JC, Steiger A (2005) Ghrelin stimulates appetite, imagination of food, GH, ACTH, and cortisol, but does not affect leptin in normal controls. Neuropsychopharmacology 30(6):1187–1192. doi:10.1038/sj.npp.1300670, 1300670 [pii]
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404(6778):661–671. doi:10.1038/35007534
Scott MM, Perello M, Chuang JC, Sakata I, Gautron L, Lee CE, Lauzon D, Elmquist JK, Zigman JM (2012) Hindbrain ghrelin receptor signaling is sufficient to maintain fasting glucose. PLoS One 7(8):e44089. doi:10.1371/journal.pone.0044089, PONE-D-12-16911 [pii]
Shimbara T, Mondal MS, Kawagoe T, Toshinai K, Koda S, Yamaguchi H, Date Y, Nakazato M (2004) Central administration of ghrelin preferentially enhances fat ingestion. Neurosci Lett 369(1):75–79. doi:10.1016/j.neulet.2004.07.060, S0304-3940(04)00943-7 [pii]
Skibicka KP, Dickson SL (2011) Ghrelin and food reward: the story of potential underlying substrates. Peptides 32(11):2265–2273. doi:10.1016/j.peptides.2011.05.016, S0196-9781(11)00208-7 [pii]
Skibicka KP, Hansson C, Alvarez-Crespo M, Friberg PA, Dickson SL (2011) Ghrelin directly targets the ventral tegmental area to increase food motivation. Neuroscience 180:129–137. doi:10.1016/j.neuroscience.2011.02.016, S0306-4522(11)00157-6 [pii]
Strasser F, Lutz TA, Maeder MT, Thuerlimann B, Bueche D, Tschop M, Kaufmann K, Holst B, Brandle M, von Moos R, Demmer R, Cerny T (2008) Safety, tolerability and pharmacokinetics of intravenous ghrelin for cancer-related anorexia/cachexia: a randomised, placebo-controlled, double-blind, double-crossover study. Br J Cancer 98(2):300–308. doi:10.1038/sj.bjc.6604148, 6604148 [pii]
Stratford TR, Kelley AE (1999) Evidence of a functional relationship between the nucleus accumbens shell and lateral hypothalamus subserving the control of feeding behavior. J Neurosci 19(24):11040–11048
Sun Y, Ahmed S, Smith RG (2003) Deletion of ghrelin impairs neither growth nor appetite. Mol Cell Biol 23(22):7973–7981
Sun Y, Butte NF, Garcia JM, Smith RG (2008) Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology 149(2):843–850. doi:10.1210/en.2007-0271, en.2007-0271 [pii]
Sun Y, Wang P, Zheng H, Smith RG (2004) Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc Natl Acad Sci U S A 101(13):4679–4684. doi:10.1073/pnas.0305930101, 0305930101 [pii]
Suzuki K, Simpson KA, Minnion JS, Shillito JC, Bloom SR (2010) The role of gut hormones and the hypothalamus in appetite regulation. Endocr J 57(5):359–372. doi:JST.JSTAGE/endocrj/K10E-077, [pii]
Tan TM, Vanderpump M, Khoo B, Patterson M, Ghatei MA, Goldstone AP (2004) Somatostatin infusion lowers plasma ghrelin without reducing appetite in adults with Prader-Willi syndrome. J Clin Endocrinol Metab 89(8):4162–4165. doi:10.1210/jc.2004-0835, 89/8/4162 [pii]
Tanaka M, Nakahara T, Kojima S, Nakano T, Muranaga T, Nagai N, Ueno H, Nakazato M, Nozoe S, Naruo T (2004) Effect of nutritional rehabilitation on circulating ghrelin and growth hormone levels in patients with anorexia nervosa. Regul Pept 122(3):163–168. doi:10.1016/j.regpep.2004.06.015, S0167011504002058 [pii]
Tanaka M, Naruo T, Yasuhara D, Tatebe Y, Nagai N, Shiiya T, Nakazato M, Matsukura S, Nozoe S (2003) Fasting plasma ghrelin levels in subtypes of anorexia nervosa. Psychoneuroendocrinology 28(7):829–835. doi:S0306453002000665, [pii]
Toshinai K, Date Y, Murakami N, Shimada M, Mondal MS, Shimbara T, Guan JL, Wang QP, Funahashi H, Sakurai T, Shioda S, Matsukura S, Kangawa K, Nakazato M (2003) Ghrelin-induced food intake is mediated via the orexin pathway. Endocrinology 144(4):1506–1512
Tschop M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML (2001) Circulating ghrelin levels are decreased in human obesity. Diabetes 50(4):707–709
Verhagen LA, Egecioglu E, Luijendijk MC, Hillebrand JJ, Adan RA, Dickson SL (2011) Acute and chronic suppression of the central ghrelin signaling system reveals a role in food anticipatory activity. Eur Neuropsychopharmacol 21(5):384–392. doi:10.1016/j.euroneuro.2010.06.005, S0924-977X(10)00118-5 [pii]
Walker AK, Ibia IE, Zigman JM (2012) Disruption of cue-potentiated feeding in mice with blocked ghrelin signaling. Physiol Behav 108:34–43. doi:10.1016/j.physbeh.2012.10.003, S0031-9384(12)00327-7 [pii]
Weinberg ZY, Nicholson ML, Currie PJ (2011) 6-Hydroxydopamine lesions of the ventral tegmental area suppress ghrelin’s ability to elicit food-reinforced behavior. Neurosci Lett 499(2):70–73. doi:10.1016/j.neulet.2011.05.034, S0304-3940(11)00648-3 [pii]
Willesen MG, Kristensen P, Romer J (1999) Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 70(5):306–316. doi:nen70306, [pii]
Williams KW, Elmquist JK (2012) From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci 15(10):1350–1355. doi:10.1038/nn.3217, nn.3217 [pii]
Wortley KE, del Rincon JP, Murray JD, Garcia K, Iida K, Thorner MO, Sleeman MW (2005) Absence of ghrelin protects against early-onset obesity. J Clin Invest 115(12):3573–3578. doi:10.1172/JCI26003
Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR (2001) Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 86(12):5992
Wu Q, Palmiter RD (2011) GABAergic signaling by AgRP neurons prevents anorexia via a melanocortin-independent mechanism. Eur J Pharmacol 660(1):21–27. doi:10.1016/j.ejphar.2010.10.110, S0014-2999(10)01275-6 [pii]
Wynne K, Giannitsopoulou K, Small CJ, Patterson M, Frost G, Ghatei MA, Brown EA, Bloom SR, Choi P (2005) Subcutaneous ghrelin enhances acute food intake in malnourished patients who receive maintenance peritoneal dialysis: a randomized, placebo-controlled trial. J Am Soc Nephrol 16(7):2111–2118. doi:10.1681/ASN.2005010039, ASN.2005010039 [pii]
Yang N, Liu X, Ding EL, Xu M, Wu S, Liu L, Sun X, Hu FB (2009) Impaired ghrelin response after high-fat meals is associated with decreased satiety in obese and lean Chinese young adults. J Nutr 139(7):1286–1291. doi:10.3945/jn.109.104406, jn.109.104406 [pii]
Zhao TJ, Liang G, Li RL, Xie X, Sleeman MW, Murphy AJ, Valenzuela DM, Yancopoulos GD, Goldstein JL, Brown MS (2010) Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc Natl Acad Sci U S A 107(16):7467–7472. doi:10.1073/pnas.1002271107, 1002271107 [pii]
Zheng H, Patterson LM, Berthoud HR (2007) Orexin signaling in the ventral tegmental area is required for high-fat appetite induced by opioid stimulation of the nucleus accumbens. J Neurosci 27(41):11075–11082. doi:10.1523/JNEUROSCI.3542-07.2007, 27/41/11075 [pii]
Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK (2006) Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol 494(3):528–548. doi:10.1002/cne.20823
Zigman JM, Nakano Y, Coppari R, Balthasar N, Marcus JN, Lee CE, Jones JE, Deysher AE, Waxman AR, White RD, Williams TD, Lachey JL, Seeley RJ, Lowell BB, Elmquist JK (2005) Mice lacking ghrelin receptors resist the development of diet-induced obesity. J Clin Invest 115(12):3564–3572. doi:10.1172/JCI26002
Acknowledgments
This manuscript was supported by grants from the National Agency of Scientific and Technological Promotion of Argentina (PICT2010-1954 and PICT2011-2142 to MP, and PICT2010-1589 and PICT2011-1816 to JR). We would like to thank Nicolas De Francesco and Agustina Cabral for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Perello, M., Raingo, J. (2014). Central Ghrelin Receptors and Food Intake. In: Portelli, J., Smolders, I. (eds) Central Functions of the Ghrelin Receptor. The Receptors, vol 25. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-0823-3_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-0823-3_5
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-0822-6
Online ISBN: 978-1-4939-0823-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)