
Abstract
Autophagy is a highly regulated cellular pathway for degrading long-lived proteins and is the only known pathway for clearing cytoplasmic organelles. Autophagy is a major contributor to maintain cellular homeostasis and metabolism. The quality control of mitochondria is essential to maintain cell energy and this process appears to be achieved via autophagy. Warburg hypothesized that cancer growth is caused by the fact that tumor cells mainly generate energy by the non-oxidative breakdown of glucose. This cellular behavior relies on a respiratory impairment, characterized by a mitochondrial dysfunction, which results in a switch to glycolysis. Moreover, epithelial cancer cells may induce the Warburg effect in neighboring stromal fibroblasts in which autophagy was activated. Here, we introduce the autophagy process, its regulation, the selective pathways, and its role in cancer cell metabolism. We define the Warburg effect and the “reverse” hypothesis and we discuss the potential value of modulating autophagy. The association of the Warburg effect in tumor and stromal cells to cancer-related autophagy is of significant relevance in experimental therapeutics.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Autophagy
- Cancer
- Warburg effect
- Mitochondria
- Caveolin
- Mitophagy
- Cancer cell metabolism
- Metformin
- Rapamycin
- Chloroquine
- mTOR
- PI3K
- VMP1
- BECN1
- ROS
- RAGE
6.1 Autophagy, a Self-Eating Cellular Process
Autophagy is an evolutionarily conserved and highly regulated lysosomal pathway that degrades macromolecules (e.g., proteins, glycogen, lipids, and nucleotides) and cytoplasmic organelles [1–3]. This catabolic process is involved in the turnover of long-lived proteins and other cellular macromolecules, and it might play a protective role in development, aging, cell death, and defense against intracellular pathogens [4, 5]. By morphological studies, autophagy has been linked to a variety of pathological processes, such as neurodegenerative diseases and tumorigenesis, which highlights its biological and medical importance [6, 7] .
Autophagy consists of several sequential steps, which are: induction, autophagosome formation, and autophagosome–lysosome fusion and degradation. Although autophagy was first identified in mammalian liver upon glucagon treatment approximately 50 years ago, its molecular understanding started only in the past decade, largely based on the discovery of the autophagy-related genes (ATGs) by genetic analyses in yeast.
Depending on the delivery route of the cytoplasmic material to the lysosome, there are three major types of autophagy in eukaryotes: (1) chaperone-mediated autophagy (CMA), (2) microautophagy, and (3) macroautophagy, hereafter referred to as autophagy [8]. CMA allows the direct lysosomal import of unfolded, soluble proteins that contain a particular pentapeptide motif. In microautophagy, cytoplasmic material is directly engulfed into the lysosome at the surface of the lysosome by membrane rearrangement. Finally, autophagy involves the sequestration of cytoplasm into a double-membrane cytosolic vesicle, referred to as an autophagosome that subsequently fuses with a lysosome to form an autolysosome for the degradation by lysosomal hydrolases [9] .
6.1.1 The Process of Autophagy
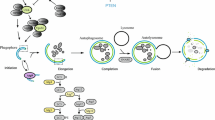
Autophagy is characterized by sequestration of bulk cytoplasm and organelles in double-membrane vesicles called autophagosomes, which eventually acquire lysosomal-like features [9, 10]. The autophagic process is described in Fig. 6.1 . An isolation membrane forms, invaginates, and sequesters cytoplasmic components. The edges of the membrane fuse to form the autophagosome. The outer membrane of the autophagosome fuses with the lysosome to deliver the inner membrane vesicle to the lumen of the digestive compartment forming the autolysosome.

During autophagy, an isolation membrane forms as a pre-autophagosomal structure, invaginates and sequesters cytoplasmic constituents. The edges of the membrane fuse to form a double-membrane vesicle known as the autophagosome. The outer membrane of the autophagosome fuses with the lysosome to deliver the inner vesicle with the contents to the lumen of the degradative compartment, the autolysosome
Autophagy is mediated by a set of evolutionarily conserved gene products (termed the ATG proteins) originally discovered in yeast [11]. In mammalian cells, BECN1 [2, 12–14] promotes autophagosome formation when it functions as part of a complex with the class III phosphatidylinositol 3-kinase (PI3K) mediating the localization of other autophagic proteins to the autophagosomal membrane [15]. However, despite the advances in understanding autophagy, autophagosome formation in mammalian cells is a complex process, and neither the molecular mechanisms nor all the implicated genes involved in its formation are fully elucidated.
Although autophagy has been studied in mammals since the 1960s, only since 2000 has yeast genetics allowed us to understand this process at a molecular level. More than 30 highly conserved genes that are involved in autophagy have been identified so far [16]. Among these, a core molecular machinery has defined and is composed of four subgroups: first, the ATG1/unc−51-like kinase (ULK) complex; second, the class III phosphatidylinositol 3 kinase (PtdIns3K)/Vps34 complex I; third, two ubiquitin-like proteins ATG12 and ATG8 (LC3) conjugation systems; and four, two transmembrane proteins, ATG9/mATG9 (and associated proteins involved in its movement such as ATG18/WIPI-1) and VMP1 (whose expression triggers autophagy) [17–19]. Basal autophagy in unstressed cells is kept down by the action of the mammalian target of rapamycin complex 1 (mTORC1). Key upstream regulators of mTORC1 include the class I phosphoinositide 3-kinase (PI3K)-Akt pathway, which keeps mTORC1 active in cells with sufficient growth factors, and the adenosine monophosphate (AMP)-activated protein kinase (AMPK) pathway that inhibits mTORC1 upon starvation and calcium signals [20, 21] .
6.1.2 Regulation of Autophagy Induction Through mTOR
Under stress conditions such as amino acid starvation, autophagy is strongly induced in many types of cultured cells. The effects of individual amino acids differ in their abilities to regulate autophagy. Amino acids including Leu, Tyr, Phe, Gln, Pro, His, Trp, Met, and Ala suppress autophagy in an ex vivo-perfused liver [22]. However, such profiles depend on cell types showing their different amino acid metabolisms in tissues. The questions on how cells sense amino acid concentration and physiological significance of autophagy regulation by amino acid starvation are not fully understood yet. It has been demonstrated that amino acid signaling pathways exist, which involve the activation of serine/threonine kinase mTOR and the subsequent regulation of the class III PI3K . The mTOR is involved in the control of multiple cell processes in response to changes in nutrient conditions [23]. Especially, mTORC1 requires Rag GTPase, Rheb, and Vps34 for its activation and subsequent inhibition of autophagy in response to amino acids [24, 25]. Energy levels are primarily sensed by AMP-activated protein kinase (AMPK), a key factor for cellular energy homeostasis. In low energy states, AMPK is activated and the activated AMPK then inactivates mTORC1 through TSC1/TSC2 and Rheb protein [26].
Thus, the inactivation of mTORC1 is essential for the induction of autophagy and plays a central role in autophagy. In addition to amino acid signaling, hormones, growth factors, and many other factors, including bcl-2 [27], reactive oxygen species (ROS) [28], calcium [29], BNIP3 [30], p19ARF [31], DRAM [32], calpain [33], TRAIL [34], FADD [35] and myo-inositol-1,4,5-triphosphate (IP3) [36], have also been reported to regulate autophagy. But, not all autophagy signals are transduced through mTOR signaling. A recent study showed that small-molecule enhancers of the cytostatic effects of rapamycin (called SMERs) induce autophagy independently of mTOR [37]. Activities of the ULK1 kinase complex are regulated by mTOR, depending on nutrient conditions. Under growing and high-nutrient conditions, the active mTORC1 interacts with the ULK1 kinase complex (ULK1–mATG13–FIP200–ATG101) and phosphorylates ULK1 and mATG13, and thus inhibits the membrane targeting of the ULK1 kinase complex. During starvation conditions, on the other hand, the inactivated mTORC1 dissociates from the ULK1 kinase complex and results in the ULK1 kinase complex, free to phosphorylate components, such as mATG13 and FIP200, in the ULK1 kinase complex, leading to autophagy induction [38].
The pancreatitis-associated protein named vacuole membrane protein 1 (VMP1) is a transmembrane protein with no known homologs in yeast. VMP1 expression induces autophagosome formation, even under nutrient-replete conditions while remaining an integrated autophagosomal membrane protein in mammalian cells [39]. VMP1 expression is induced by hyperstimulation of Gq-coupled cholecystokinin (CCK) receptor in pancreatic acinar cells during acute pancreatitis [40] and by mutated KRas in pancreatic cancer cells [41]. VMP1 interacts with Beclin 1/ATG6 through its hydrophilic C-terminal region (VMP1-ATG domain), which is necessary for early steps of autophagosome formation [39, 42]. Besides, EPG-3/VMP1 is one of three essential autophagy genes conserved from worms to mammals, which regulates early steps of the autophagic pathway in Caenorhabditis elegans [43]. VMP1 along with ULK1 and ATG14 localizes in the endoplasmic reticulum-associated autophagosome formation sites in a PI3K activity-independent manner, confirming the key role of expression is induced by hyperstimulat VMP1 in the formation of autophagosomes [18]. Interestingly, Dictyostelium cells lacking the VMP1 gene showed accumulation of huge ubiquitin-positive protein aggregates containing the autophagy marker ATG8/LC3 and p. 62 homolog [44]. Moreover, the knockdown of VMP1 expression abolishes starvation and rapamycin -induced autophagosome formation [39], as well as autophagy induced by hyperstimulation of the Gq-coupled CCK receptor in pancreatic acinar cells [40] or by chemotherapy in pancreatic tumor cells [45]. Furthermore, VMP1 is the only human disease-inducible ATG protein described so far.
6.1.3 The Class III PI3K Complex in Autophagosome Nucleation
The autophagosome formation process is composed of isolation membrane nucleation, elongation, and completion steps. In mammals, the class III PI3K complex plays an essential role in isolation membrane nucleation during autophagy [46], while the class I PI3K pathway is also involved in autophagy regulation through the insulin signaling cascade to activate mTOR and PKB [3]. The class III PI3K (Vps34) is associated with Beclin1 (ATG6) and p150, the homolog of Vps15 (phosphoinositide-3- kinase, regulatory subunit 4), to form the class III PI3K core complex.
As the first step of autophagosome formation, the autophagosome nucleation system includes ATG12–ATG5–ATG16 L, which is essential for the formation of pre-autophagosomes. ATG12 is a 186-amino acid protein and is conjugated to ATG5 [47]. The carboxy-terminal glycine residue of ATG12 is activated by E1-like ATG7 through a high-energy thioester bond in an ATP-dependent manner [48–51]. ATG12 is then transferred to E2-like ATG10 [52] and finally attached to lysine 149 of ATG5 via an isopeptide bond [48]. The ATG12–ATG5 conjugate further interacts with ATG16L1 to form a ~ 350 kDa multimeric ATG12–ATG5–ATG16 protein complex through the homo-oligomerization of ATG16 [53].
Once the autophagosome formation is completed, ATG proteins are released back to the cytoplasm by an uncharacterized mechanism. The second ubiquitin-like protein conjugation system is the modification of LC3 (a mammalian homolog of ATG8) by the phospholipid phosphatidylethanolamine (PE) [54], an essential process for the formation of autophagosomes. LC3 is cleaved by cysteine protease ATG4 and then conjugated with PE by ATG7 and ATG3, a second E2-like enzyme. This lipidated LC3-II then associates with newly forming autophagosome membranes. LC3-II remains on mature autophagosomes until its fusion with lysosomes [55]. The conversion of LC3 to LC3-II is thus well known as a marker of autophagy induction (Fig. 6.2). However, the increase of LC3-II alone is not enough to show autophagy activation because the inhibition of LC3-II degradation in the lysosome by the impaired autophagy flux can also cause its accumulation.

During autophagy the cytosolic form of LC3 (LC3-I) undergoes C-terminal proteolytic and lipid modifications (LC3-II) and translocates to the autophagosomal membrane. LC3 is currently used as a specific marker of autophagy
While the origin of autophagic vacuoles remains disputable, several hypotheses have been proposed for the source of autophagosomal membrane during autophagosome formation. The first hypothesis is the “de novo” formation of autophagosome by ATG9 reservoirs [56]. In the second hypothesis, various organelles such as endoplasmic reticulum (ER) [57], mitochondria [58], and plasma membrane [59] are used as an origin for the formation of the phagophore. Recently, cup-shaped structures called omegasome, a discrete region of the ER, were identified as a platform for autophagosome formation [60]. The ATG5 complex, LC3, and ULK1 have been shown to recruit into the omegasome after starvation, and ATG5- and LC3-positive membranes seem to emerge from the omegasome. It was also observed that omegasomes form in close proximity to the Vps34-containing vesicles which may synthesize phosphatidylinositol 3-phosphate (PI(3)P). This hypothesis is also supported by the notion of a physical association between the ER and early autophagic membranes [57].
Recent studies have identified new regulators of autophagosome maturation and degradation, including activating molecule in Beclin 1-regulated autophagy (AMBRA1) [61], ultraviolet radiation resistance-associated gene (UVRAG) [62], Rubicon [63], and VMP1 [42]. VMP1, along with Ulk1 and ATG14, localizes to the sites where autophagosomes are formed independently of the other ATG proteins [18]. The 20 amino acids of the C-terminal hydrophilic domain of VMP1, the VMP1 autophagy-related domain (VMP1-ATGD) [39], bind with the BH3 domain of Beclin 1 promoting the displacement of Bcl-2, a negative regulator of autophagy , and driving BECN1 to the autophagic pathway. This interaction leads the formation of a VMP1-BECN1-hVps34 complex and the subsequent association of ATG16L1 to the autophagosomal membranes, providing a model describing one of the key steps in the peripheral anterior synechia (PAS) formation and autophagy regulation in mammalian cells (Fig. 6.3) [42, 64].

VMP1 is a transmembrane protein whose expression triggers autophagy interacting with BECN1 and regulating the autophagy-specific PI3K complex in mammalian cells
6.1.4 Vesicle Completion, Autophagosome—Lysosome Fusion, and Degradation
Autophagosome then fuses with lysosomes/vacuoles, which is an essential process for completion of the autophagy pathway. The sequestration of cytoplasm into a double-membrane cytosolic vesicle is followed by the fusion of the vesicle with a late endosome or lysosome to form an autophagolysosome (or autolysosome). Then, the inner membrane of the autophagosome and autophagosome-containing cytoplasm-derived materials are degraded by lysosomal/vacuolar hydrolases inside the autophagosome. The molecular mechanisms underlying the transport and fusion of autophagosomes are just beginning to be understood, and through active investigations, several major events involved in the process have recently been clarified. In mammalian cells, autophagosome maturation is a prior step for the fusion between autophagosomes and lysosomes. The degradation products, including macromolecules, are then exported to the cytosol for reutilization by the cell. This process is poorly understood.
6.2 Selective Types of Autophagy
Early studies suggested that autophagy was a nonselective process in which cytoplasmic structures were randomly sequestered into autophagosomes before being delivered to the mammalian lysosome or the plant and yeast vacuole for degradation. Now there is growing evidence that unwanted cellular structures can be selectively recognized and exclusively eliminated within cells. This is achieved through the action of specific autophagy receptors, such as p62 and Nbr1. Thus, excess or damaged organelles including mitochondria , peroxisomes, lipid droplets, endoplasmic reticulum, and ribosomes can be specifically sequestered by autophagosomes and targeted to the lysosome for degradation. Importantly, there is growing evidence that selective autophagy subtypes also have a wide range of physiological functions. In yeast, the cytosol-to-vacuole (Cvt) pathway transports hydrolases into the vacuole. In eukaryotes, autophagy plays a central role in both innate and acquired immunity [65]. In pancreatic cells, autophagy has recently been shown to specifically turn over secretory granules damaged by acute pancreatitis as a protective cellular response [40].
6.2.1 Mitophagy, the Selective Autophagic Degradation of Damaged Mitochondria
Three major pathways of mitochondrial quality control have been described so far. Two AAA protease complexes can degrade misfolded mitochondrial membrane proteins with catalytic sites facing both sides of the inner membrane. Mitochondrial proteins can also be degraded by translocation to lysosomes; vesicles budding from mitochondrial tubules sequester selected mitochondrial cargos, and deliver those mitochondrial components to the lysosome for degradation. The third pathway, known as mitophagy , involves sequestration of an entire mitochondrion within the autophagosome, followed by fusion with a lysosome.
Concomitant with the energy production through oxidative phosphorylation, mitochondria also generate ROS, which in excess cause damage through the oxidation of proteins, lipids, and DNA often inducing cell death. Therefore, the quality control of mitochondria is essential to maintain cellular homeostasis and this process appears to be achieved via autophagy . It has been postulated that mitophagy contributes to differentiation and development by participating in the intracellular remodeling that occurs, for example, during hematopoiesis and adipogenesis. In mammalian red blood cells, the expulsion of the nucleus followed by the removal of other organelles, such as mitochondria , are necessary differentiation steps. Nix/Bnip3L, an autophagy receptor whose structure resembles that of ATG32, is also an outer mitochondrial membrane protein that interacts with gamma-aminobutyric acid receptor-associated protein (GABARAP) [66, 67] and plays an important role in mitophagy during erythroid differentiation [68, 69]. Although autophagosome formation probably still occurs in Nix/Bnip3L-deficient reticulocytes, mitochondrial elimination is severely impaired. Consequently, mutant reticulocytes are exposed to increased levels of ROS and die, and Nix/Bnip3L knockout mice suffer severe anemia. Depolarization of the mitochondrial membrane potential of mutant reticulocytes by treatment with an uncoupling agent results in restoration of mitophagy [69], emphasizing the importance of Nix/Binp3L for the mitochondrial depolarization and implying that mitophagy targets uncoupled mitochondria . Hematopoietic-specific ATG7 knockout mice also exhibited severe anemia as well as lymphopenia, and the mutant erythrocytes markedly accumulated degenerated mitochondria but not other organelles [70]. The mitochondrial content is regulated during the development of T cells as well; that is, the high mitochondrial content in thymocytes is shifted to low mitochondrial contents in mature T cells. ATG5- or ATG7-deleted T cells fail to reduce their mitochondrial content resulting in increased ROS production as well as an imbalance in pro- and anti-apoptotic protein expression [71–73]. All together, this evidence demonstrates the essential role of mitophagy in hematopoiesis.
Recent studies have described the molecular mechanism by which damaged mitochondria are selectively targeted for autophagy , and have suggested that the defect is implicated in familial Parkinson’s disease (PD) [74]. PINK1, a mitochondrial kinase, and Parkin, an E3 ubiquitin ligase, have been genetically linked to both PD and a pathway that prevents progressive mitochondrial damage and dysfunction. When mitochondria are damaged and depolarized, PINK1 becomes stabilized and recruits Parkin to the damaged mitochondria. Various mitochondrial outer membrane proteins are ubiquitinated by Parkin and mitophagy is then induced. Of note, PD-related mutations in PINK1 and Parkin impair mitophagy [75–78], suggesting that there is a link between defective mitophagy and PD. How these ubiquitinated mitochondria are recognized by the autophagosome remains unknown. Although p. 62 has been implicated in the recognition of ubiquitinated mitochondria, elimination of the mitochondria occurs normally in p62-deficient cells [79, 80].
Mitochondrial function is essential for cancer cells. However, different cancer cell types undergo different bioenergy alterations, some to more glycolytic and others to more oxidative, depending in part on the developmental state of the cell undergoing neoplastic transformation. Therefore, different alterations in bioenergy metabolism or mitochondrial ROS production and redox biology can be found depending on the specific environment of the cancer cells promoting the cell survival [81]. In this context, for example, mitophagy is an important mechanism to promote cell survival by the clearance of damaged mitochondria that are potential sources of ROS [82].
6.3 Autophagy in Cancer Cell Metabolism
Both downregulated and excessive autophagy have been implicated into the pathogenesis of diverse diseases, such as certain type of neuronal degeneration, diabetes and its complications, and cancer [83]. Autophagy has also been implicated in cell death called autophagic or type II programmed cell death, which was originally described on the basis of morphological studies detecting autophagic vesicles during tissue involution [84] .
Cancer cells in general tend to undergo less autophagy than their normal counterparts, at least for some tumors [85, 86]. The Beclin1 autophagy gene is monoallelically deleted in 40–74 % of cases of human sporadic breast, ovarian, and prostate cancer [86]. Heterozygous disruption of Beclin1 increases the frequency of spontaneous malignancies and accelerates the development of virus-induced premalignant lesions [86] suggesting that defective regulation of autophagy promotes tumorigenesis. It has been proposed that autophagy suppresses carcinogenesis by a cell-autonomous mechanism involving the protection of genome integrity and stability, and a nonautonomous mechanism involving suppression of inflammation and necrosis. On the other hand, autophagy may support the survival of rapidly growing cancer cells that have outgrown their vascular supply and are exposed to an inadequate oxygen supply or metabolic stress. By contrast, excessive levels of autophagy promote cell death [87]. Accordingly, it has been proposed that autophagy plays an important role both in tumor progression and in promotion of cancer cell death [88], although the molecular mechanisms responsible for this dual action of autophagy in cancer have not been elucidated .
It has been suggested that autophagy may be a cancer cell survival response to tumor-associated hypoxia. Tumor hypoxia has been used as a marker of poor prognosis [89]; however, how cancer cells become more malignant or survive with an extremely poor blood supply is poorly understood. When cancer cells are exposed to hypoxia, anaerobic glycolysis increases and provides energy for cell survival, but as the glucose supply is also insufficient because of the poor blood supply, there must be an alternative metabolic pathway that provides energy when both oxygen and glucose are depleted [90, 91]. Hypoxia in pancreatic cancer has been reported to increase its malignant potential [89]. Proliferating cancer cells require more nutrients than surrounding noncancerous cells do, though nutrition is supplied via functionally and structurally immature neo-vessels. Because autophagy-specific genes promote the survival of normal cells during nutrient starvation in all eukaryotic organisms, autophagy may react to the cancer microenvironment to favor the survival of rapidly growing cancer cells. LC3 expression in surgically resected pancreatic cancer tissue, showed activated autophagy in the peripheral area, which included the invasive border and concomitantly shows enhanced expression of carbonic anhydrase [92]. This observation suggests that autophagy may promote cell viability in hypo-vascularized cancer tissue .
It has also been proposed that autophagy is a cancer cell survival response to tumor-associated inflammation [93]. Cancer-associated inflammation results in promotion of carcinogenesis and resistance to therapy. Several phenotypic alterations observed in cancer cells are a result of inflammatory signals found within the tumor microenvironment [93]. The receptor for advanced glycation end products (RAGE) is an induced inflammatory receptor constitutively expressed on many murine and human epithelial tumor cell lines [94, 95] and the highest levels of RAGE expression were observed in murine and human pancreatic adenocarcinoma tumors. Genotoxic and/or metabolic stress lead to modest but reproducible increases in overall expression of RAGE on epithelial cell lines. RAGE expression correlates directly with the ability of both murine and human pancreatic tumor cell lines to survive cytotoxic insult. Targeted knockdown of RAGE significantly increases cell death, whereas forced overexpression promotes survival. Recently, it was reported that the enhanced sensitivity to cell death in the setting of RAGE knockdown is associated with increased apoptosis and decreased autophagy. In contrast, overexpression of RAGE is associated with enhanced autophagy, diminished apoptosis, and enhanced cancer cell viability. Knockdown of RAGE enhances mTOR phosphorylation in response to chemotherapy, thus preventing induction of a survival response. Inhibition of autophagy by means of silencing Beclin1 expression in pancreatic cancer cells enhances apoptosis and cell death [96]. These observations suggest that RAGE expression in cancer cells has a role in tumor cell response to environmental stress through the enhancement of autophagy. However, increased sensitivity to chemotherapeutic agents in RAGE-knockdown pancreatic cancer cells is dependent on ATG5 expression but independent of BECN1 expression [96]. These last findings suggested that the role of autophagy in the resistance to microenvironment insult or in the sensitivity to chemotherapeutic agent is the result of complex molecular pathways in the tumor cell .
On the other hand, repression of autophagy has been suggested as a cancer cell response to prolonged hypoxic conditions. Pancreatic cancer cell response to prolonged hypoxia may consist of inhibition of autophagic cell death. The short isoform of single-minded 2 (SIM2s) is a member of the basic helix–loop–helix family of transcriptional regulators [97] and is upregulated in pancreatic cancer. Microarray studies identified the pro-cell death gene BNIP3 as a target of SIM2s repression. Prolonged hypoxia induces cell death via an autophagic pathway involving the hypoxia-inducible factor 1 (HIF1)-mediated upregulation of BNIP3 [30, 98]. The deregulation of both SIM2s and BNIP3 is associated with poor prognostic outcomes [99]. Decreased BNIP3 levels and poor prognosis clearly correlate with elevated SIM2s expression in pancreatic cancer. The loss of BNIP3, either by hypermethylation or by transcriptional repression, is correlated with inhibition of cell death [100, 101], whereas upregulation of BNIP3 sensitizes pancreatic carcinoma cells to hypoxia-induced cell death [102]. SIM2s expression, concomitant with its repression of BNIP3, enhances tumor cell survival under prolonged hypoxic conditions. Recent data link increased SIM2s expression with enhanced cell survival during hypoxic stress concomitantly with BNIP3 repression and the attenuation of hypoxia-induced autophagic processes. Thus, the inhibition of autophagic cell death by BNIP3 repression enhances tumor cell survival under prolonged hypoxic conditions .
Decreased autophagy in some cancer cells has been related to malignant stages of the disease. Cancer cells, in general, tend to undergo less autophagy than their normal counterparts, supporting the contention that defective autophagic cell death plays a role in tumor progression. Studies of carcinogen-induced pancreatic cancer in animal models have shown that pancreatic adenocarcinoma cells have lower autophagic capacity than premalignant cells [103]. The WIPI protein family, which includes ATG18, the WIPI-1 homolog in Saccharomyces cerevisiae, was genetically identified as a gene contributing to autophagy [103]. Human WIPI-1a is a member of a highly conserved WD-repeats protein family. hWIPI-1 is linked to starvation-induced autophagy in the mammalian system. Amino acid deprivation triggers an accumulation of endogenous hWIPI-1 protein to large vesicular and cup-shaped structures, where it colocalizes with LC3. Starvation-induced hWIPI-1 formation is blocked by wortmannin, a principal inhibitor of PI-3 kinase-induced autophagosome formation [104]. Interestingly, WIPI proteins are linked pathologically to cellular transformation because all human WIPI genes are reportedly expressed aberrantly in a variety of matched human cancer samples. Strikingly, hWIPI-2 and hWIPI-4 messenger RNA (mRNA) expression is substantially decreased in 70 % of matched kidney (ten patients) and 100 % of pancreatic (seven patients) tumor samples. The majority of these samples were derived from advanced stage tumors, such as pancreatic adenocarcinomas stages I–IV. Hence, cancer-associated downregulation of hWIPI-2 and hWIPI-4 supports the possibility that decreased autophagic activity is necessary for the malignant stages of pancreatic cancer .
6.4 Otto Warburg and the “Warburg Effect”
Born in Freiburg, Germany, in 1883, Otto Warburg was one of the leading chemists of the first half of the twentieth century. The son of a very famous physicist, student of the eminent chemist Emil Fisher, and Nobel Prize laureate, Warburg devoted several years of his life to elucidate the mechanisms by which cancer cells obtain energy especially under fast-growing conditions [105].
By 1920, measuring lactate production and oxygen consumption on rat liver carcinoma tissue, Otto Warburg and colleagues proposed that cancer cells display some very relevant differences when compared with normal tissues with regard to their glucose metabolism by favoring glycolysis despite oxygen availability. Warburg hypothesized that cancer growth is caused by the fact that tumor cells mainly generate energy (in the form of ATP) by the non-oxidative breakdown of glucose. This view contrasts with the observation that normal cells produce ATP through oxidative phosphorylation obtaining “fuel” by the oxidative breakdown of glucose [105].
The ATP yield from glycolysis under anaerobic conditions (2 ATPs per molecule of glucose) is much smaller than the yield from the complete oxidation of glucose to CO2 under aerobic conditions (30 or 32 ATPs per molecule of glucose) [106]. About 15 times more glucose is consumed anaerobically in contrast to the aerobic pathway to yield the same amount of ATP. As a consequence, glucose uptake proceeds about ten times faster in most solid tumors than in normal tissues [107]. Tumor cells commonly experience hypoxia (limited O2 supply), and as a result, cancer cells depend on anaerobic glycolysis for their ATP production.
This phenomenon of preferred aerobic glycolysis was denominated the “Warburg effect” resulting in increased lactate production, even in the presence of adequate oxygen partial pressures. It was suggested that this cellular behavior relies on a respiratory impairment, characterized by a mitochondrial dysfunction, which results in a switch to glycolysis . It was proposed that the high glycolytic rate might also result from a decreased number of mitochondria in tumor cells [108].
This effect, first described in cancer tissues, was further identified in many other rapidly dividing normal cells [109]. Several mechanisms have been proposed to explain the Warburg effect in cancer tissues. These mechanisms may be involved in transcriptional and posttranslational related metabolic changes. Transcriptional upregulation of glycolytic enzymes was extensively studied for decades. Some well-characterized transcriptional regulators have been associated with the molecular basis of the Warburg effect . The HIF1 transcription factor increases the glycolytic enzymes and glucose transmembrane transport, and upregulates pyruvate dehydrogenase kinases (which results into a reduction of the pyruvate flux to the tricarboxylic acid cycle) [110]. The degradation of this transcriptional regulator involves some mediators, like the Von Hippel–Lindau tumor suppressor ubiquitin ligase, which seems to be consistently altered in some cancer cells [110]. In these tumors, even in normoxic conditions, HIF1 seems to increase the glycolytic rate, to elevate lactate production and to activate the PI3K/AKT/mTOR pathway [110].
In cancer cells, the reduced expression of the tumor suppressor protein, p53, might be also linked to the Warburg effect . In fact, p53 reduces the glycolysis rate by increasing the activity of fructose-2,6-bisphosphatase , a mechanism also involved in the regulatory pathways of apoptosis [111]. It also seems to increase the oxidative phosphorylation process. Other transcriptional regulators might be linked to the Warburg effect, such as the alpha estrogen-related receptor (of potential relevance in breast cancer); in the same direction, increased expression of oncogenes such as MYC also seems to be associated with an increased glycolytic rate and might be involved in the pathophysiology of the metabolic modifications found in tumors [112]. Besides, glycolytic enzymes and glucose transmembrane transport are activated by MYC overexpression.
As mentioned before, the posttranslational regulation of the Warburg effect was also under scrutiny. As a relevant example, activation of the PI3K/AKT downstream pathway leads to an increased glucose influx and the phosphorylation of hexokinase and phosphofructokinase-2 with a concomitant upregulation of the glycolytic pathway [110]. Several posttranslational modifications of the M2 isoform of pyruvate kinase result in a change in its activity, modulating the glycolytic pathway in several tissue types. The K305 acetylation of this M2 isoform reduces its enzymatic activity and increases the enzyme degradation via CMA [110]. By oxidation, acetylation, phosphorylation, etc., the posttranscriptional modification of the M2 isoform of the pyruvate kinase influences glycolysis in various models and experimental conditions.
Tumor overexpression of endogenous microRNA (miRNA) was recently linked with metabolic regulation of cancer cells and the “Warburg effect” [110]. Although attractive, the biological impact of this association remains to be clarified.
All of these mechanisms, heterogeneous by nature, were proposed as possible explanations of this phenomenon in cancer cells. Often these mechanisms were extrapolated from isolated cancer cell experiments in vitro not including other components of the neoplastic tissue. Other cellular components apart from the cancer cells, vascular growth rates, and oxygen partial pressure in different tumor segments, as well as differential concentrations of distinct transcriptional factors across the tumor volume were not considered in many of these experiments. Nevertheless, some findings collected from the first decade of the twenty-first century changed our form of understanding of this metabolic behavior in cancer tissues. These findings will be summarized in the following section.
6.5 The “Reverse Warburg Effect”
6.5.1 The Concept
The Warburg effect was thought to occur only in cancer cells until recently. In 2008, Vincent et al. [113] showed that human skin keloid fibroblasts display similar bioenergetic mechanisms as cancer cells in generating ATP mainly from glycolysis. This observation may be explained by the similarity in the hypoxic microenvironment in solid tumors and keloids [113]. In line with this previous study, Lisanti et al. proposed in 2010, a new hypothesis for understanding the Warburg effect in tumors [114]. They suggested that epithelial cancer cells induce the Warburg effect in neighboring stromal fibroblasts.
As a first step, these cancer-associated fibroblasts undergo myofibroblastic differentiation and secrete lactate and pyruvate through the glycolytic pathway. This process is induced, as previously stated, by cancer cells by a mechanism involving oxidative stress with overproduction of reactive oxygen species, loss of Caveolin-1 , mitophagy and/or mitochondrial dysfunction, and increased production of NO [115].
In a second step, epithelial cancer cells take up the energy-rich metabolites that enter in the tricarboxylic acid (TCA) pathway and, in consequence, ATP is generated by oxidative phosphorylation. These cells expand their mitochondrial mass to satisfy an increased metabolic demand, upregulate enzymes involved in antioxidant defense to cope with oxidative stress, and increase the tumor aggressive behavior [116].
This interesting hypothesis was based in studies performed in a co-culture system mimicking tumor–stroma co-evolution, where stromal fibroblasts (human telomerase reverse transcriptase (hTERT)-BJ1 cells) interact with human breast cancer cells (MCF-7) [117]. The conclusions are consistent with Warburg’s original view, but one must take into consideration that the phenomenon is occurring in the tumor stroma.
For a deeper analysis of the phenomenon, a division into two general steps can be made as follows.
6.5.2 Step 1: Cancer-Associated Fibroblasts Undergo Aerobic Glycolysis to Produce Energy-Rich Nutrients
Epithelial cancer cells firstly induce the Warburg effect in adjacent cancer-associated fibroblasts through the downregulation of Caveolin-1 [114]. The loss of Cav-1 expression may be sufficient to induce this constitutive fibroblastic phenotype, although this mechanism needs further investigation. The absence of stromal Cav-1 is associated with a high rate of tumor recurrence, metastasis, and poor clinical outcome [118].
Cav-1 is a structural component of caveolae. These structures are flask-shaped invaginations of the plasma membrane occupying up to 30 % of the cell surface and represent a predominant location of endothelial nitric oxide synthase (eNOS). Among other functions, Cav-1 can regulate eNOS activity and NO release [119]. NO plays an important signaling role in vascular function and a regulatory role in mitochondrial function [120, 121]. However, if overproduced, mitochondrial dysfunction accompanied by increased production of ROS may develop. Oxidative stress is inseparably linked to mitochondrial dysfunction and mitochondrial turnover is dependent on autophagy [122].
ROS is a term that actually groups a range of oxygen-derived molecules formed by the incomplete reduction of O2 during oxidative metabolism that have both specific mechanisms of production and intracellular targets [123]. The biologically important species of this group are superoxide anion (O2•−) and hydrogen peroxide (H2O2), as they are formed by controlled mechanisms, and H2O2 is a signaling molecule. A major endogenous source of both O2•− and H2O2 is the mitochondrial electron-transport chain, where continuous electron leakage occurs during aerobic respiration [124]. In addition, low levels of these two species are produced by the membrane-localized nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. The low-level steady state of these species in mitochondria (10−8 M H2O2 and 10−10 M O2•−) [125] is accomplished by a group of antioxidants species that includes compounds of nonenzymatic (as glutathione) and enzymatic nature (as superoxide dismutase or catalase). Oxidative stress is an imbalance between oxidants and antioxidants in favor of the oxidants, potentially leading to damage [123].
It is imperative to briefly discuss the importance of the H2O2steady-state concentrations related to biological effects. At concentrations lower than 0.7 µM, H2O2 mainly acts as a signaling molecule redox-regulating several physiological processes. Apoptosis may be triggered at the 1–3 µM range, and necrosis may develop at concentrations higher than 3 µM [126, 127]. This brief analysis indicates the relevance of taking into account steady-state levels when addressing fundamental questions on biological effects of H2O2 and the existence of a cellular fine regulation of the subcellular concentrations of this species.
Not surprisingly, mitochondria and ROS are emerging as important players in autophagy . The cross talk between autophagy, redox signaling, and mitochondrial dysfunction is not well understood. Recently, it was suggested that chronic expression of RCAN1-1 L (stress-inducible protein) induces mitochondrial autophagy and metabolic shift from oxidative phosphorylation to glycolysis [128]. Moreover, mitophagy may also be important in attenuating apoptosis or necrosis, by clearance of damaged mitochondria [122].
The occurrence of mitophagy (including a decrease in mitochondrial mass) is not only favored by the occurrence of oxidative stress, but by the activation of HIF1 (a key factor involved and activated in hypoxic conditions) as well [115]. The overproduction of ROS is sufficient to induce HIF1 through its stabilization under normoxic conditions [129]. In this scenario, the cancer-associated fibroblasts are obliged to produce ATP through aerobic glycolysis with an increased glucose consumption rate (due to a low-energy yield, as previously explained) and the concomitant production of a high amount of lactate and pyruvate (energy-rich nutrients).
6.5.3 Step 2: Cancer Cells Uptake Nutrients and Produce ATP by Oxidative Phosphorylation
Stromal metabolism produces high-energy nutrients (for example, lactate) and recycled chemical building blocks (as nucleotides, amino acids, and fatty acids obtained through the process of mitophagy) that are taken up by cancer cells to power their own growth [117]. Lactate is taken up by a monocarboxylate transporter situated in the cancer cell membrane, converted into acetyl-coenzyme A (CoA), which subsequently condenses with oxaloacetate to initiate the TCA cycle for energy production [130]. NADH is formed as a product of these cyclic reactions and channeled into the mitochondrial respiratory chain where it is oxidized to NAD+, a process coupled to the synthesis of ATP [131]. This results in a unilateral and net energy transfer from the catabolic tumor stroma to the anabolic cancer cells [116]. As a consequence, cancer cells synthetize their own ATP and increase mitochondrial mass through biogenesis.
In normal mammalian cells, mitochondria have an average half-life of 4–20 days, depending on the organ and age. The respiratory chain is located in the mitochondrial inner membrane and consists of four complexes: Complex I (NADH dehydrogenase), Complex II (Succinate dehydrogenase), Complex III (ubiquinone-cytochrome c oxidoreductase), and Complex IV (cytochrome oxidase). These complexes may interact to form multicomplexes with defined stoichiometry [132]. Due to the reducing power of NADH (utilized to reduce O2 to water), an electrochemical gradient produced by the respiratory chain is used by ATP synthase (sometimes called Complex V) as a driving force to phosphorylate adenosine diphosphate (ADP) to ATP. This important bioenergetic process occurs in organelles that are not static. The increase in mitochondrial size, number and mass, a process known as mitochondrial biogenesis, is triggered by a variety of stimuli and involves a complex network connecting different regulatory pathways that are tightly coordinated [133, 134].
Therefore, it is obvious that the increased production of additional energy in the form of ATP occurring in cancer cells will be sensed, and adaptive changes in mitochondrial content will be triggered culminating in increased and coordinated biogenesis of new mitochondria. For example, it has been shown that the number of mitochondria in co-cultured MCF7 cells is greatly increased, as compared to homotypic cultures of MCF7 cells [115, 135].
It is important to point out that, in addition to increasing mitochondrial mass, cancer cells escape oxidative mitochondrial damage by upregulating enzymes involved in antioxidant pathways, including catalase and peroxiredoxin-1 [115]. The pathways and regulation of the “reverse Warburg effect” need further analysis, opening and interesting area of investigation. Understanding the mechanisms involved in each of the steps will lead to the development of new therapeutic strategies for cancer prevention .
A scheme of the two steps described above is shown in Fig. 6.4.

Schematic representation of the reverse Warburg effect. STEP 1: Cancer-associated fibroblasts (stroma) undergo fibroblastic differentiation triggered by oxidative stress generated by cancer cells. The occurrence of mitophagy obliges differentiated fibroblasts to produce ATP and energy-rich nutrients by aerobic glycolysis. STEP 2: Cancer cells take up ATP and nutrients, increasing their proliferative capacity and aggressive behavior
6.6 Drugs, Warburg Effect, and Reverse Warburg Effect
Several lines of evidence suggest that both inhibitors and activators of autophagy may have utility in the treatment of patients with chemotherapy-resistant cancers, since strong overactivation as well as strong inhibition of autophagy induces death in highly aggressive cancer cells, such as pancreatic cancer cells, and sensitizes them to hypoxia starvation [136]. Such autophagy activating (e.g., rapamycin derivatives, sirolimus and temsirolimus or sulforaphane—a naturally occurring dietary substance enriched in broccoli) or inhibiting drugs (e.g., antibiotic monensin, antimalarial drug chloroquine) are available and generally are well tolerated by patients.
Metformin , a well-known antidiabetic agent, is one of the most studied agents in this area. This drug has been proposed as a potentially multi-faceted agent for cancer prevention. Meformin acts as an indirect activator of AMPK and is able to reduce mitochondrial respiratory chain Complex I activity. These have been proposed as mechanisms for reducing hepatic glucose output in patients with type 2 diabetes. In p53-deficient cancer cells, metformin treatment was associated with increased cell death. In normal cells, metformin treatment is followed by an increase in glycolytic rates as an alternative ATP-producing mechanism. In fact, one very rare but still possible adverse event of metformin is lactic acidosis. P53-deficient cells seem to experience problems in switching their metabolic pattern, which is followed by an enhanced cell death rate. By reducing the activity of the respiratory chain Complex I, metformin diminishes ROS generation in mitochondria [137]. The role of ROS in tumorigenesis and on cancer growth has been widely recognized. Metformin , as well as thrombospondin and endostatin, exhibits a mild to moderate antiangiogenic effect. This effect on angiogenesis may be the basis for its potential actions on cancer cells and/or its stroma [137].
As mentioned before, metformin activates the ATM/LKB1/AMPK axis. The tumor suppressor LKB1, well characterized in the pathophysiology of melanoma, pancreatic, and lung cancer, might participate in the mechanism of action of metformin. Part of the preventative effects of metformin might be mediated by this suppressing factor. By activating AMPK, metformin may inhibit the mTOR pathway; this effect has been proposed as an explanation for the potential antineoplastic effects of metformin in breast and renal tumors [138]. Many of the mentioned mechanisms may explain the effects of metformin on the Warburg effect . Metformin has been suggested to reduce glycolysis and to increase mitochondrial respiration in tumors, associated with growth arrest [138]. It has been proposed that pyruvate kinase expression in the fibroblasts of tumor stroma is linked to cancer growth. ROS produced by cancer cells promotes oxidative stress in fibroblasts, which results in activation of HIF1 and nuclear factor kappa B (NFkB). NFkB increases proinflammatory cytokines and HIF1 alpha promotes autophagy and anaerobic glycolysis. Pyruvate kinase activity results in an increase in ketones and lactate. These nutrients are transferred to cancer cells and used for mitochondrial oxidative metabolism. Conversely, metformin reduces the mitochondrial chain activity by inhibiting Complex I. In this manner, metformin may alter some of the mechanisms involved into the reverse Warburg effect [139]. Metformin may also affect cell reprogramming by modifying the lipogenic enzymes acetyl-CoA carboxylase and fatty acid synthase [140]. These changes may also affect the metabolic behavior of both stroma and tumor cells. As mentioned before, the clinical impact of these modifications is still uncertain.
Other drugs exhibit a potential for the modification of the Warburg effect and autophagy rates. Mild autophagy induction such as hypoxia or starvation seems to protect the cells, but rapamycin or sulforaphane lead to elimination [136]. By contrast, an excessive autophagy rate may induce cell death. Inhibition of autophagy by nonensin or 3-methyladenine is able to eliminate highly aggressive pancreatic adenocarcinoma cells [136], as these drugs may totally block continuous recycling of cellular components necessary for new synthesis and survival.
In advanced cancer, autophagy may be necessary for the maintenance of the tumor and multiple clinical trials are underway to test this as a therapeutic approach in patients using hydroxychloroquine (HCQ) [141, 142]. Standard cancer chemotherapies may affect autophagy in different ways. Gemcitabine monotherapy or its combination with other agents has become the standard chemotherapy for the treatment of advanced pancreatic cancer. Gemcitabine is a relatively effective chemotherapeutic agent acting by competition with deoxycytidine triphosphate (dCTP) for the incorporation into DNA causing chain termination; on the other hand, gemcitabine serves as an inhibitory alternative substrate for ribonucleotide reductase and leads to a reduction of deoxynucleotide pools [143, 144]. This molecule inhibits cells that are insensitive to classic anticancer drugs, including other nucleoside analogs with similar structures. Although gemcitabine seems to exert its toxicity at least in part by activation of apoptosis [143], it was recently suggested that gemcitabine also induces autophagy in pancreatic cancer cells [45]. It has been proposed that the early induction of autophagy with gemcitabine may be mediated by an increased expression of VMP1 [145]. Capecitabine, a pyrimidine analog, induces apoptosis in several cancer lines and is of modest efficacy in locally advanced PDAC when associated with limited field radiotherapy [144]. By displaying a Src kinase modulatory effect, capecitabine has been proposed to modulate autophagy [146]. The results in this area are still contradictory. Irinotecan is a topoisomerase I inhibitor which prevents DNA from unwinding. In a phase III trial, the combination of 5-fluouracil, leucovorin, oxaliplatin, and irinotecan resulted in better responses, progression free survival, and overall survival when compared with the standard single drug therapy with gemcitabine for metastatic PDAC [147]. In small cell prostatic carcinoma, irinotecan promoted an increase in autophagy of treated tumors as indicated by an increase in LC3B expression [148]. Even though authors of this research state that the role of autophagy is complex, there is evidence that autophagy supports both promotion and suppression of cancer growth. In general, as mentioned before, a considerable amount of caution should be exercised for the interpretation of the consequences of cancer chemotherapy on autophagy. Other chemotherapeutic agents like the glycoside oleandrin, some platinum compounds, the multikinase inhibitor sorafenib, and some histone deacetylase inhibitors have demonstrated effects on the autophagy rate in pancreatic carcinoma cell lines [149]. As proposed, autophagy may be involved in carcinogenesis, tumor progression, and dissemination, as well as may be associated at least in part with the actions of some chemotherapy for PDAC. All these modifications may alter both the Warburg and reverse Warburg effects. Nevertheless, the real contribution of these metabolic changes on tumor cell survival and clinical prognosis remains unclear.
6.7 Perspectives
The dysregulation of autophagic function has been implicated in a growing list of disease processes and has underscored the selective or substrate-specific versions of the pathway. In terms of cancer biology, autophagy has been viewed as having dual roles in both tumor suppression and progression, and the activation of autophagy selective forms can be used as a potential therapeutic approach for the treatment of specific cancers [150]. Autophagy is a major contributor to maintain cellular homeostasis and metabolism, and continued studies are required to identify key molecules regulating autophagy and a better understanding of the process at a molecular level.
Recently, two interesting approaches have been employed to identify new autophagy regulators: small molecule screening [151, 152] and studies on structural information of Atg proteins. These screens allowed the identification of compounds that can induce autophagy and promote long-lived protein degradation. Interestingly, some compounds are FDA-approved drugs for the treatment of human diseases [151]. The search for new autophagy regulators is a good way to explore the mechanism of autophagy and identify new molecules that may be useful for the treatment of human diseases.
Future research on the mechanism and regulation of selective autophagy and the physiological importance of this pathway in human disease may reveal new therapeutic strategies. Some pharmacological interventions may modify the Warburg and the reverse Warburg effects. Several mechanisms for such actions were reported, but in general, the clinical relevance of these findings is still being clarified. Potential pharmacological interventions modulating autophagy and the Warburg and reverse Warburg effects are shown in Fig. 6.5.

Pharmacological interventions that may modulate autophagy and the Warburg and reverse Warburg effects
References
Cuervo AM (2004) Autophagy: in sickness and in health. Trends Cell Biol 14:70–77
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Yang Z, Klionsky DJ (2009) An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 335:1–32
Hara T, Nakamura K, Matsui M et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441:885–889
Qu X, Zou Z, Sun Q et al (2007) Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128:931–946
Pattingre S, Levine B (2006) Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res 66:2885–2888
Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306:990–995
Klionsky DJ (2005) Autophagy. Curr Biol 15:282–283
Klionsky DL, Emr SD (2000) Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721
Mizushima N (2005) The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ 12:1535–1541
Klionsky DJ, Cregg JM, Dunn WA et al (2003) A unified nomenclature for yeast autophagy-related genes. Dev Cell 5:539–545
Liang XH, Jackson S, Seaman M et al (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676
Pattingre S, Tassa A, Qu X et al (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Liang C, Feng P, Ku B et al (2006) Autophagic and tumour suppressor activity of a novel Beclin 1-binding protein UVRAG. Nat Cell Biol 8:688–699
Kihara A, Noda T, Ishihara N et al (2001) Two distinct Vps34 phosphatidylinositol 3-kinase complexes complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces. J Cell Biol 152:519–530
He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93
Vaccaro MI, Ropolo A, Grasso D et al (2008) A novel mammalian trans-membrane protein reveals an alternative initiation pathway for autophagy. Autophagy 4:388–390
Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6:764–776
Yang Z, Klionsky DJ (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 22:124–131
Høyer-Hansen M, Jäättelä M (2007) AMP-activated protein kinase: a universal regulator of autophagy? Autophagy 3:381–383
Zheng M, Wang YH, Wu XN et al (2011) Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat Cell Biol 13:263–272
Mortimore GE, Pösö AR (1987) Intracellular protein catabolism and its control during nutrient deprivation and supply. Ann Rev Nutr 7:539–564
Nobukuni T, Joaquin M, Roccio M et al (2005) Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA 102:14238–14243
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124:471–484
Sancak Y, Bar-Peled L, Zoncu R et al (2010) Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141:290–303
Gwinn DM, Shackelford DB, Egan DF et al (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30:214–226
Levine B, Sinha S, Kroemer G (2008) Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 4:600–606
Botti J, Djavaheri-Mergny M, Pilatte Y et al (2006) Autophagy signaling and the cogwheels of cancer. Autophagy 2:67–73
Green DR, Wang R (2010) Calcium and energy: making the cake and eating it too? Cell 142:200–202
Tracy K, Dibling BC, Spike BT et al (2007) BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 27:6229–6242
Sherr CJ (2006) Autophagy by ARF: a short story. Mol Cell 22:436–437
Crighton D, Wilkinson S, Ryan KM (2007) DRAM links autophagy to p53 and programmed cell death. Autophagy 3:72–74
Xia HG, Zhang L, Chen G et al (2010) Control of basal autophagy by calpain1 mediated cleavage of ATG5. Autophagy 6:61–66
Mills KR, Reginato M, Debnath J et al (2004) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci USA 101:3438–3443
Pyo JO, Jang MH, Kwon YK et al (2005) Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 280:20722–20729
Sarkar S, Rubinsztein DC (2006) Inositol and IP3 levels regulate autophagy: biology and therapeutic speculations. Autophagy 2:132–134
Sarkar S, Perlstein EO, Imarisio S et al (2007) Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol 3:331–338
Mizushima N (2010) The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 22:132
Ropolo A, Grasso D, Pardo R et al (2007) The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem 282:37124–37133
Grasso D, Ropolo A, Lo Ré A et al (2011) Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem 286:8308–8324
Lo RAE, Fernández-Barrena MG, Almada LL et al (2012) Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J Biol Chem 287:25325–25334
Molejon MI, Ropolo A, ReAL et al (2013) The VMP1-Beclin 1 interaction regulates autophagy induction. Sci Rep 3:1055
Tian Y, Li Z, Hu W et al (2010) C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141:1042–1055
Calvo-Garrido J, Escalante R (2010) Autophagy dysfunction and ubiquitin-positive protein aggregates in Dictyostelium cells lacking Vmp1. Autophagy 6:100–109
Pardo R, Lo RA, Archange C et al (2010) Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology 10:19–26
Mariño G, López-Otín C (2004) Autophagy: molecular mechanisms, physiological functions and relevance in human pathology. Cell Mol Life Sci 61:1439–1454
Kuma A, Mizushima N, Ishihara N et al (2002) Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 277:18619–18625
Mizushima N, Noda T, Yoshimori T et al (1998) A protein conjugation system essential for autophagy. Nature 395:395–398
Kim J, Dalton VM, Eggerton KP et al (1999) Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol Biol Cell 10:1337–1351
Yuan W, Stromhaug PE, Dunn WA Jr (1999) Glucose-induced autophagy of peroxisomes in Pichia pastoris requires a unique E1-like protein. Mol Biol Cell 10:1353–1366
Tanida I, Tanida-Miyake E, Ueno T et al (2001) The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem 276:1701–1706
Shintani T, Mizushima N, Ogawa Y et al (1999) Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J 18:5234–5241
Mizushima N, Noda T, Ohsumi Y (1999) Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 18:3888–3896
Ichimura Y, Kirisako T, Takao T et al (2000) A ubiquitin-like system mediates protein lipidation. Nature 408:488–492
Burman C, Ktistakis NT (2010) Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett 584:1302–1312
Mari M, Griffith J, Rieter E et al (2010) An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol 190:1005–1022
Hayashi-Nishino M, Fujita N, Noda T et al (2009) A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11:1433–1437
Hailey DW, Rambold AS, Satpute-Krishnan P et al (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141:656–667
Ravikumar B, Moreau K, Jahreiss L et al (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12:747–757
Tooze SA, Yoshimori T (2010) The origin of the autophagosomal membrane. Nat Cell Biol 12:831–835
Di Bartolomeo SC, Nazio F et al (2010) The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol 191:155–168
Liang C, Lee JS, Inn KS et al (2008) Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10:776–787
Matsunaga K, Saitoh T, Tabata K et al (2009) Two Beclin 1-binding proteins, Atg14 L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396
Molejon MI, Ropolo A, Vaccaro MI (2013) VMP1 is a new player in the regulation of the autophagy-specific phosphatidylinositol 3-kinase complex activation. Autophagy 2013 Apr 4;9(6) [Epub ahead of print]
Reggiori F, Komatsu M, Finley K et al (2012) Selective types of autophagy. Int J Cell Biol 2012:156272
Schwarten M, Mohrluder J, Ma P et al (2009) Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5:690–698
Novak I, Kirkin V, McEwan DG et al (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11:45–51
Schweers RL, Zhang J, Randall MS et al (2007) Nix is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104:19500–19505
Sandoval H, Thiagarajan P, Dasgupta SK et al (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454:232–235
Mortensen M, Ferguson DJ, Edelmann M et al (2010) Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA 107:832–837
Stephenson LM, Miller BC, Ng A et al (2009) Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T-lymphocytes. Autophagy 5:625–635
Hubbard VM, Valdor R, Patel B et al (2010) Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol 185:7349–7357
Jia W, He YW (2011) Temporal regulation of intracellular organelle homeostasis in T-lymphocytes by autophagy. J Immunol 186:5313–5322
Abeliovich A (2010) Parkinson’s disease: mitochondrial damage control. Nature 463:744–745
Narendra DP, Jin SM, Tanaka A et al (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biology 8 Article ID e1000298
Jin SM, Lazarou M, Wang C et al (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191:933–942
Deas E, Plun-Favreau H, Gandhi S et al (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20:867–879
Shi G, Lee JR, Grimes DA et al (2011) Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum Mol Genet 20:1966–1974
Okatsu K, Saisho K, Shimanuki M et al (2010) P62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15:887–900
Narendra DP, Kane LA, Hauser DN et al (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–1106
Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12:685–698
Mathew R, White E (2011) Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev 21:113–119
Mizushima N, Levine B, Cuervo AM et al (2008) Autophagy fights disease through cellular self-digestion. Nature 451:1069–1075
Høyer-Hansen M, Jäättelä M (2008) Autophagy: an emerging target for cancer therapy. Autophagy 4:574–580
Toth S, Nagy K, Palfia Z, Rez G (2002) Cellular autophagic capacity changes during azaserine-induced tumour progression in the rat pancreas: Up-regulation in all premalignant stages and down-regulation with loss of cycloheximide sensitivity of segregation along with malignant transformation. Cell Tissue Res 309:409416
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the Beclin 1 autophagy gene. J Clin Invest 112:1809–1820
Levine B (2007) Cell biology: autophagy and cancer. Nature 446:745–747
Mathew R, Karantza-Wadsworth V, White E (2007) Role of autophagy in cancer. Nat Rev Cancer 7:961–967
Buchler P, Reber HA, Lavey RS et al (2004) Tumor hypoxia correlates with metastatic tumor growth of pancreatic cancer in an orthotopic murine model. J Surg Res 120:295–303
Izuishi K, Kato K, Ogura T et al (2000) Remarkable tolerance of tumor cells to nutrient deprivation: possible new biochemical target for cancer therapy. Cancer Res 60:6201–6207
Esumi H, Izuishi K, Kato K et al (2002) Hypoxia and nitric oxide treatment confer tolerance to glucose starvation in a 5’-AMP-activated protein kinase-dependent manner. J Biol Chem 277:32791–32798
Fujii S, Mitsunaga S, Yamazaki M et al (2008) Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci 99:1813–1819
DeNardo DG, Johansson M, Coussens LM (2008) Inflaming gastrointestinal oncogenic programming. Cancer Cell 14:7–9
Abe R, Yamagishi S (2008) AGE-RAGE system and carcinogenesis. Curr Pharm Des 14:940–945
Arumugam T, Simeone DM, Van GK, Logsdon CD (2005) S100P promotes pancreatic cancer growth, survival, and invasion. Clin Cancer Res 11:5356–5364
Kang R, Tang D, Schapiro NE et al (2009) The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ 16:1–11
Kewley RJ, Whitelaw ML, Chapman-Smith A (2004) The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol 36:189–204
Azad MB, Chen Y, Henson ES et al (2008) Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy 4:195–204
Burton TR, Gibson SB (2009) The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ 16:515–523
Okami J, Simeone DM, Logsdon CD (2004) Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res 64:5338–5346
Mahon PC, Baril P, Bhakta V et al (2007) S100A4 contributes to the suppression of BNIP3 expression, chemoresistance, and inhibition of apoptosis in pancreatic cancer. Cancer Res 67:6786–6795
Abe T, Toyota M, Suzuki H et al (2005) Upregulation of BNIP3 by 5-aza-2’-deoxycytidine sensitizes pancreatic cancer cells to hypoxia-mediated cell death. J Gastroenterol 40:504–510
Guan J, Stromhaug PE, George MD et al (2001) Cvt18/Gsa12 is required for cytoplasm-to-vacuole transport, pexophagy, and autophagy in Saccharomyces cerevisiae and Pichia pastoris. Mol Biol Cell 12:3821–3838
Proikas-Cezanne T, Waddell S, Gaugel A et al (2004) WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene 23:9314–9325
Warburg O (1956) On the origin of cancer cells. Science 123:309–314
Nelson D, Cox D (2008) Lehninger principles of biochemistry (chapter 14). WH Freeman and Co, New York
Bartrons R, Caro J (2007) Hypoxia, glucose metabolism and the Warburg’s effect. J Bioenerg Biomembr 39:223–229
Gogvadze V, Zhivo tovskyB, Orrenius S (2010) The Warburg effect and mitochondrial stability in cancer cells. Mol Asp Med 31:60–74
Vincent M (2011) Cancer: a de-repression of a default survival program common to all cells? Bioessays 34:72–82
Bensinger SJ, Christofk HR (2012) New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol 23(4):352–361
Bensaad K, Tsuruta A, Selak MA et al (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126:107–120
Nilsson LM, Forshell TZ, Rimpi S et al (2012) Mouse genetics suggests cell-context dependency for Myc-regulated metabolic enzymes during tumorigenesis. PLoS Genet 8:e1002573. doi: 10.1371
Vincent AS, Phan TT, Mukhopadhyay A et al (2008) Human skin keloid fibroblasts display bioenergetics of cancer cells. J Invest Dermatol 128:702–709
Pavlides S, Whitaker-Menezes D, Castello-Cross R et al (2009) The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8:3984–4001
Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D et al (2010) Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implication for breast cancer and DICS therapy with autophagy inhibitors. Cell Cycle 9:2423–2433
Lisanti MP, Martinez-Outschoorn UE, Chiavarina B et al (2010) Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging roles for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol Ther 10:537–542
Bonuccelli G, Tsirigos A, Whitaker-Menezes D et al (2010) Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 9:3506–3514
Witkiewicz AK, Dasgrupta A, Sotgia F et al (2009) An absence of stromal Caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breasts cancers. Am J Pathol 174:2023–2034
Goligorsky MS, Li H, Brodski S et al (2001) Relationship between caveolae and eNOS: everything in proximity and the proximity of everything. Am J Physiol Renal Physiol 283:1–10
Ignarro LJ, Buga GM, Wood KS et al (1987) Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci USA 84:9265–9269
Boveris A, Costa LE, Poderoso JJ et al (2000) Regulation of mitochondrial respiration by oxygen and nitric oxide. Ann N Y Acad Sci 899:121–135
Lee J, Giordano S, Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signaling. Biochem J 441:523–540
Sies H (1997) Oxidative stress: oxidants and antioxidants. Exp Physiol 82:291–295
Chance B, Sies H, Boveris A (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59:527–605
Boveris A, Cadenas E (1997) Cellular sources and steady-state levels of reactive oxygen species. In: Biadasz Clerch L, Massaro, DJ (eds) Oxygen, Gene Expression and Cellular Function. Marcel Dekker, New York, pp. 1–25
Antunes F, Cadenas E (2001) Cellular titration of apoptosis with steady-state concentrations of H2O2: submicromolar levels of H2O2 induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic Biol Med 9:1008–1018
Antunes F, Cadenas E, Brunk U (2001) Apoptosis induced by exposure to a low steady-state concentration of H2O2 is a consequence of lysosomal rupture. Biochem J 356:549–555
Ermak G, Sojitra S, Yin F et al (2012) Chronic expression of RCAN1-1 L protein induces mitochondrial autophagy and metabolic shift from oxidative phosphorylation to glycolysis in neuronal cells. J Biol Chem 287:14088–14098
BelAiba RS, Djordjevic T, Bonello S et al (2004) Redox-sensitive regulation of the HIF pathway under non-hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem 385:249–257
Yao J, Hamilton RT, Cadenas E (2010) Decline in mitochondrial bioenergetics and shift to ketogenic profile in brain during reproductive senescence. Biochim. Biophys Acta 1800: 1121–1126
Tzagaloff A (1982) Mitochondria. Plenum Press, London
Vonck J, Schafer E (2009) Supramolecular organization of protein complexes in the mitochondrial inner membrane. Biochim Biophys Acta 1793:117–124
Nisoli E, Clementi E, Moncada S et al (2004) Mitochondrial biogenesis as a signaling framework. Biochem Pharmacol 67:1–15
Scarpulla RC (2008) Transcriptional paradigs in mammalian mitochondrial biogenesis and function. Physiol Rev 88:611–638
Sotgia F, Whitaker-Menezes D, Martinez-Outschoon UE et al (2012) Mitochondrial metabolism in cancer metastasis. Cell Cycle 11:1445–1454
Rausch V, Liu L, Apel A, Rettig T, Gladkich J, Labsch S et al (2012) Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J Pathol 227:325–335
Smith-Vikos T (2012) A report of the James Watson lecture at Yale University. Yale J Biol Med 85:417–419
Del Barco SVazquez-Martin, Cufi S et al (2011) Metformin: multi-faceted protection against cancer. Oncotarget 2:896–917
Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE et al (2011) Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther 12:1101–1113
Vazquez-Martin A, Corominas-Faja B, Cufi S et al (2013) The mitochondrial H(+)-ATP synthase and the lipogenic switch: new core components of metabolic reprogramming in induced pluripotent stem (iPS) cells. Cell Cycle 12:207–218
Amaravadi RK, Lippincott-Schwartz J, Yin XM et al (2011) Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 17:654–666
Mancias JD, Kimmelman AC (2011) Targeting autophagy addiction in cancer. Oncotarget 2:1302–1306
Ewald B, Sampath D, Plankett W (2008) Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene 27:6522–6237
Jackson AS, Jain P, Watkins CR et al (2010) Efficacy and tolerability of limited field radiotherapy with concurrent capecitabine in local advanced pancreatic cancer. Clin Oncol (R Coll Radiol) 22:570–577
Vivanco I, Sawyers C (2002) The phosphatidylinositol 3-kinase Akt pathway in human cancer. Nat Rev Cancer 2:489–501
Sheith R, Walsh N, Clynes M et al (2010) Challenges of drug resistance on the management of pancreatic cancer. Expert Rev Anticancer Ther 10:1647–1661
Conroy T, Desseigne F, Ychoy M et al (2010) Randomized phase III trial comparing FOLFIRINOZ (F: 5FU/leucovorine [LV], irinotecan [I] and oxaliplatin [O]) versus gemcitabine (G) as first-line treatment for metastatic pancreatic adenocarcinoma (MPA): prepanned interim analysis results of the PRODIGE 4/ACOORD 11 trial. J Clin Oncol 28 (May 20 supplement): 4010
Tung W, Wang Y, Cout PW et al (2011) Use of irinotecan for treatment of small cell carcinoma of prostate. Prostate 71:675–681
Ropolo A, Bagnes CI, Molejon M et al (2012) Chemotherapy and autophagy-mediated cell death in pancreatic cancer cells. Pancreatology 12:1–7
Hughson LR, Poon VI, Spowart JE et al (2012) Implications of therapy-induced selective autophagy on tumor metabolism and survival. Int J Cell Biol 2012:872091
Zhang L, Yu J, Pan H et al (2007) Small molecule regulators of autophagy identified by an image-based high-throughput screen. Proc Natl Acad Sci USA 104:19023–19028
Farkas T, Høyer-Hansen M, Jäättelä M (2009) Identification of novel autophagy regulators by a luciferase-based assay for the kinetics of autophagic flux. Autophagy 5:1018–1025
Acknowledgments
Our work is supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT), the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), and the University of Buenos Aires.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Vaccaro, M., Gonzalez, C., Alvarez, S., Ropolo, A. (2014). Modulating Autophagy and the “Reverse Warburg Effect”. In: Kanner, S. (eds) Tumor Metabolome Targeting and Drug Development. Cancer Drug Discovery and Development. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9545-1_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9545-1_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9544-4
Online ISBN: 978-1-4614-9545-1
eBook Packages: MedicineMedicine (R0)