
Abstract
The Eligen® technology, an oral delivery technology, is based on the design and development of proprietary delivery carriers referred to as Emisphere delivery agents. The major limitations for noninvasive delivery such as rapid metabolism, chemical instability, and minimal absorption through gastrointestinal (GI) tract are overcome by the application of these delivery agents. Most of the delivery agents consist of amino acids having a molecular weight of 250–300 Da that are structurally diverse with different physiochemical properties. They facilitate the absorption of macromolecules through a transcellular mechanism. These agents have been evaluated for their ability to enhance the delivery of a wide range of therapeutic macromolecules. This review discusses the major challenges and strategies employed to overcome these difficulties. It also focuses on the application of novel proprietary delivery agents that have enabled the oral delivery of a large number of therapeutic proteins and peptides.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Eligen®
- Macromolecules
- Absorption
- Transcellular
- Sodium N-[8-(2-hydroxybenzoyl)amino] caprylate (SNAC)
- Calcitonin
- Insulin
- Heparin
- Human growth hormone
- Vitamin B12
- Parathyroid hormone
1 Introduction
Recent advances in areas of biotechnology have resulted in the development of a large number of commercially available macromolecular drugs that have great potential for a vast range of therapeutic indications. However, the challenges of utilizing these drugs for noninvasive delivery have been daunting. Common problems that are continually faced in developing oral dosage forms include low aqueous stability , lack of permeability , rapid metabolism, intracellular trafficking, biological and chemical instability of the macromolecules, among others [1, 2]. The low permeability or absorption of biopharmaceuticals in the GI epithelium is mainly due to large molecular size and low lipophilicity [3, 4]. Over the years, research on certain formulations is ongoing on development of viable oral delivery products to overcome these issues [5–11]. On the contrary, many molecules have been limited to parental dosing because their drug properties do not facilitate oral absorption. Despite the various hurdles; oral dosing is generally considered to be the most patient friendly and convenient route of drug administration [12]. There are various companies dedicated on improving the oral delivery of existing drugs by GI absorption enhancement, which, if successful, could have the greatest impact on oral drug therapy.
Successful protein drug absorption and efficacy from the GI tract requires certain specific physicochemical properties [3, 11, 13, 14]. Moreover, it has to withstand the harsh chemical and biological milieu within the GI tract. The physiochemical properties include suitable molecular weight (typically below 500–1,000 Da), pKa (a measure of the degree of acidity or alkalinity), degree of lipophilicity (log D), as well as proper solubility [15, 16]. The hydrophilic property of these macromolecules makes it difficult for them to penetrate through the epithelial cells (via the transcellular route) because of low permeability. The absorption of large hydrophilic macromolecules is mainly limited to the paracellular pathway, which consists of aqueous pores created by the cellular tight junctions. Most available drugs are either weak acids or weak bases, and under normal conditions only the nonionized fraction (the most lipophilic) crosses biological membranes, except where active transport is involved. The new technology suggested to overcome these limitations is based on carrier molecules, of amino acids having a molecular weight of 250–300 Da that are structurally diverse with different physiochemical properties [17]. These carriers possess hydrophobic moieties that can associate with the drug molecules to create a more lipophilic drug or carrier complex, enabling transport across the epithelial membrane [18, 19]. Because of the weak association between carrier and drug, the interaction is reversible, and occurs spontaneously by simple dilution on entering the blood circulation. Studies have shown that the carriers enable the systemic absorption of the drug via transcellular absorption, a common drug absorption pathway, without compromising the integrity of the intestinal epithelium.
Numerous delivery systems for oral protein delivery have been actively developed, especially by pharmaceutical companies, in hope to make them clinically viable. Although most of the works still remain in the development stage, many of them have progressed beyond the proof-of-concept stage to the clinical trials. Some of the available technologies for oral protein delivery under development by pharmaceutical companies are listed in Table 18.1.
2 Eligen® Technology
Emisphere Technologies, Inc. is a biopharmaceutical company pioneering the oral delivery of otherwise injectable drugs and the Eligen® technology is a broad-based platform technology developed and patented by them [20]. This oral delivery technology is founded on the design and synthesis of proprietary delivery agents, known as Emisphere® delivery agents or carriers. Emisphere’s business strategy is to develop oral forms of drugs that are not currently available or have poor bioavailability in oral form, either alone or with corporate partners, by applying its proprietary Eligen® technology to those drugs or licensing its Eligen® technology to partners who, typically, apply it directly to their marketed drugs. This technology has enabled the oral delivery of proteins, peptides, and some macromolecules. At present, Emisphere maintains a library of more than 1,800 structurally diverse carriers with different physicochemical properties. Most of these delivery agents are small organic molecules with a molecular weight of 250–350 Da, and almost all of them are amino acids.
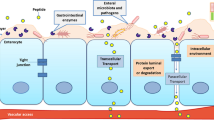
Eligen® technology is a macromolecule-delivering platform technology where a macromolecule is used as an absorption enhancer . The macromolecule interacts with the drug molecules to create a weak, noncovalent association, the drug remaining chemically unmodified. Among the existing library of absorption-enhancing compounds, sodium N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC) or salcaprozate sodium , also called sodium N-[8-(2-hydroxybenzoyl) amino] octanoate is used by Emisphere. Emisphere contends that SNAC enhances absorption by forming a noncovalent complex with the active drug that enables transcellular absorption, without altering tight junctions [19, 21, 22]. For proteins, the mechanism may involve a reversible change in protein conformation and protection against degradation, prior to absorption. Unlike the traditional penetration enhancers, Emisphere delivery agents are believed to cause minimal histological damages to the intestinal epithelium and are applicable to diverse group of drug molecules ranging in size from 500 to > 150,000 Da. The formed lipophilic drug or SNAC complex is claimed to be capable of transport across the epithelial membrane as shown in Fig. 18.1. Eligen® technology has been used to develop various types of oral formulations including solutions, tablets, and capsules. They have advanced oral formulations or prototypes of salmon calcitonin (sCT) [23–30], heparin [31–35], insulin [36–40], parathyroid hormone [18], human growth hormone [19, 21, 22, 41], Vitamin B12 [42], and cromolyn sodium. These delivery agents have been evaluated in various animal models as well as in humans for their ability to enhance the delivery of a wide array of therapeutic macromolecules that are in various stages of clinical development. To date, six oral products have undergone clinical testing with Emisphere® delivery agents. Their clinical trials have been discussed in the subsequent sections. Although the exact mechanism of absorption enhancement has not been elucidated, it has been hypothesized that the delivery agent-mediated absorption involves a sequence of advanced and novel features (Fig. 18.1) which may be generalized as follows [2, 43, 44].

Emisphere Eligen® oral protein delivery technology. Absorption of drug-delivery agent complex via the intestine
A noncovalent interaction occurs between the macromolecule and the delivery agent. This interaction transiently alters the physicochemical properties of the macromolecules (e.g., hydrophobicity, conformation, stability, etc.) . The complex formed has a conformation of the macromolecule that has a higher transport rate compared to its physiological conformation. This complex mimics the body’s natural biomolecular transport mechanisms and the GI absorption is facilitated. Once the complexes along with the drug are transported across the epithelial cells to the circulation, the delivery agent dissociates from the drug and the drug reestablishes its native conformations, ensuring its therapeutically active state. No histological damage to the intestinal epithelium represents a significant advantage over traditional penetration enhancers that have been reported to be associated with significant disruption of the tight junctions, change in membrane fluidity, and toxicity, among others.
2.1 Calcitonin
Calcitonin discovered by Copp and Cameron has been available as a therapeutic agent for metabolic bone disease for more than 30 years [45]. It is a naturally occurring 32-amino acid polypeptide produced by the parafollicular C cells of the thyroid. Synthetic or recombinant calcitonin has been derived from a number of different species including human, porcine, and salmon. However, sCT , believed to be 100 times more potent than human calcitonin, has been widely preferred in clinical practice [45]. It has been approved for the treatment of postmenopausal osteoporosis, Paget’s disease, bone associated pain conditions, and hypercalcemia. A unique advantage of sCT, unshared by any other antiresorptive agents, is its analgesic effect on bone pain previously demonstrated in clinical studies in patients with osteoporotic vertebral fractures and musculoskeletal disorders. sCT is commercially available as an injectable form and as a nasal spray. The short half-life of calcitonin in serum has led to several attempts to increase plasma concentrations. One very recent approach of oral calcitonin formulation is the use of the Eligen® technology. In this new formulation the carrier studied extensively is disodium salt of N-(5-chlorosalicyloyl)-8-aminocaprylic acid (5-CNAC oral calcitonin) salt [29, 46]. This carrier has been found to bind with calcitonin without changing its biological properties, thereby reducing the compound’s susceptibility to degradation. It creates an insoluble entity at low pH which later dissolves at higher pH and facilitates passive absorption by the transcellular pathway by enhancing peptide transport over nonpolar biological membrane. After passage through the intestine, the delivery agent disassociates from the peptide, and the peptide is absorbed into the hepatic vein with subsequent systemic absorption . The metabolism and disposition of 5-CNAC have been investigated and they were found safe and well tolerated [47].
The pharmacokinetic profile of 5-CNAC oral calcitonin has also been evaluated in a randomized, crossover double-blinded Phase I trial, controlled by both a placebo and a parenteral verum [24]. This study demonstrated that oral delivery of sCT is feasible with reproducible absorption and systemic biological efficacy. Eight healthy volunteers received single doses of 400, 800, and 1,200 µg of sCT orally, a placebo, and a 50 IU sCT intravenous infusion. sCT was readily and reliably absorbed from the oral formulation, with an absolute bioavailability of around 1.2 % depending on the applied dose. It also demonstrated a marked, dose-dependent drop in blood and urine C-terminal telopeptide of type I collagen, a sensitive and specific bone resorption marker, with the effects of 1,200 µg exceeding those of the other formulations. It also decreased blood calcium and phosphate, and increased the circulating levels of parathyroid hormone and, transiently, the urinary excretion of calcium. It was well tolerated, with some subjects presenting mild and transient nausea, abdominal cramps, diarrheic stools, and headaches.
The efficacy and safety of another Eligen® technology-based oral formulation to deliver sCT to the circulation was assessed on postmenopausal women. A multicentric randomized, double-blind, placebo-controlled, dose-ranging clinical trial has been carried out that included 277 healthy postmenopausal women in the age group of 55–85 [30]. The women were treated with doses of sCT in combination with 200 mg of delivery agent 5-CNAC or placebo for 3 months. Subjects received treatment with daily doses of 0.15, 0.4, 1 and 2.5 mg or with 1 mg every intermittent day. They were also given 1,000 mg calcium supplements and 400 IU vitamin D daily throughout the study. Acute changes in serum urinary C-terminal telopeptide of type I collagen (CTx), N-mid osteocalcin, bone-specific alkaline phosphatase, calcium, and parathyroid hormone measured by immunoassays, were the studied efficacy parameters. The first dose of sCT brought about dose-dependent decrease in serum CTx compared with placebo and reached lowest value within 3 h after drug intake, after which gradual increase had been noticed. The results depicted effective enteral absorption, a pronounced inhibition of bone resorption with minimal alteration of formation, and reproducibility of responses over 3 months. At month 3, the placebo-corrected changes in the predose value of serum and urinary CTx were significant only in the 1.0 mg dose group (− 18.9 % and − 20.5 %, respectively, p < 0.05). The results thus suggested that the oral formulation was well tolerated, with mild to moderate GI and skin manifestations apparent mainly in the high-dose groups. The above 3-month trial shows that the novel Eligen® technology-based oral formulation of sCT has potential to become a safe and effective treatment for postmenopausal bone loss. Future trials are, however, necessary to assess the impact of long-term administration on changes in bone mineral density (BMD) and fracture risk.
A subsequent study was carried out to induce significant dose-dependent reductions in the biochemical marker of cartilage degradation envisaging potential chondroprotective effects [48]. This was a randomized double-blind; placebo-controlled clinical study which included 152 Danish postmenopausal women aged 55–85. The subjects received treatment with different doses of sCT coupled with Eligen® technology-based carrier molecule, or placebo for 3 months. The efficacy parameter was evaluated with the changes in the 24-h excretion of urinary CTx-I/CTx-II biomarkers of bone resorption and cartilage degradation respectively. The results depict that the 3 month treatment with oral sCT induced significant dose-dependent decreases in both urinary CTx-I and CTx-II. The maximum responses in both biomarkers were associated with treatment using 1.0 mg daily dose of sCT and it has also been noticed that women with accelerated cartilage degradation at baseline (high CTx-II) seemed to be more responsive to clinically effective dose of oral sCT. Another important finding was that women with elevated baseline urinary CTx-II were more likely to manifest with joint-related symptoms and respond with the largest decreases in the degradation product of collagen type II. So subsequently, women with high cartilage turnover are more likely to benefit from potential chondroprotective therapy. The above-mentioned study is a post hoc analysis of a completed clinical trial, investigating the efficacy and safety of sCT for the inhibition of bone turnover in postmenopausal women. However, sCT is not yet an established drug for treatment of patients with osteoarthritis. Further studies are on the anvil to assess the impact of optimal doses of sCT on cartilage mass using MRI.
A study further reports that the bioavailability and efficacy of orally administered calcitonin SMC021, is heavily influenced by meal time, amount of water used to take the tablet, and proximity to intake of a meal [29]. SMC021 is an oral formulation of sCT consisting of the peptide hormone and 5-CNAC, a unimolecular enhancer of GI peptide absorption, licensed to Novartis. The study clearly suggested that drug uptake of SMC021 is influenced by the amount of water given with the tablet. A water volume of 50 ml resulted in a two- to three-fold higher absorption of sCT in comparison with a volume of 200 ml of water. This doubling of absorption was obtained irrespective of the timing of the meal suggesting that the volume of water strongly impacts digestion and absorption. Further the biochemical marker of bone resorption demonstrated improved efficacy. These data were the first to demonstrate that water intake has an important effect on oral peptide uptake with the Eligen® technology, improving bioavailability as much as 400 %, and even more if placebo corrected. A similar study carried out was a randomized, partially blind, placebo-controlled, single dose, exploratory crossover Phase I study involving 56 healthy postmenopausal women. sCT of 0.8 mg with 50 ml of water taken 30 and 60 min prior to meal time resulted in optimal pharmacodynamic and pharmacokinetic parameters. The data suggest that this novel oral formulation may have improved absorption and reduction of bone resorption compared to that of nasal calcitonin.
Similar studies have also been reported previously with a 14-day clinical trial of twice daily oral calcitonin with 5-CNAC suggesting potentially useful reductions in biomarkers of bone resorption and cartilage degradation [26]. An abstract presented at the recent 2011 American College of Rheumatology meeting reported that an oral formulation of sCT with the Eligen® delivery system has entered Phase III clinical trials for the treatment of osteoarthritis. Oral sCT at a dose of 0.8 mg twice daily for 2 years, significantly reduced pain and stiffness, improved physical function, and slowed cartilage loss in a placebo-controlled clinical trial involving 1,169 patients with painful knee osteoarthritis. The patients had Kellgren-Lawrence grade 2 disease with mean age of trial subjects being 64 years, and mean body mass index (BMI) of 28.9 kg/m2. Sixty-eight percent of patients were women. At month 24, oral calcitonin was also superior to placebo on 24-h visual analogue scale pain scores (P00.018), patient global assessment (P00.008), and physician global assessment (P00.014). However, by month 24, oral calcitonin subjects demonstrated a 4.5 % loss in cartilage volume on MRI in both the signal and nonsignal knee; placebo subjects demonstrated a 7 % loss in both knees. The differences were statistically significant. The most common adverse events in the oral calcitonin group versus placebo were hot flushes, nausea, dyspepsia, and diarrhea. However, despite the mentioned drawbacks the potential for a compound with improved bioavailability and efficacy in an oral preparation, combined with the established safety profile of sCT, hold suitable promise in the future.
2.2 Insulin
Oral insulin is an exciting area of development in the treatment of diabetes because of its potential benefit in patient compliance , rapid insulinization of liver, adequate insulin delivery while potentially avoiding adverse effects of weight gain and hypoglycemia. Insulin consists of two polypeptide chains (A and B) of 21 amino acids and 30 amino acids, respectively. Its molecular weight in monomeric form is 6,000 Da. The mechanism of the GI absorption of insulin has been studied using an Emisphere delivery agent. The molecule appears to be absorbed throughout the GI tract following oral administration, but the best site of absorption following coadministration with an Emisphere delivery agent appears to be the colon [49, 50]. A study with insulin also revealed that Emisphere delivery agents facilitate drug transport via transcellular pathways without permealization of the plasma membrane or tight junction disruption. Another study went on to investigate the mechanism of insulin absorption across Caco-2 cell monolayers with one of these drug delivery agents, SNAC. The results showed that SNAC increases insulin permeability approximately ten fold across cell monolayers and does so without affecting mannitol permeability or disrupting cell membranes. Confocal microscopy and immunocytochemistry revealed that insulin is transported transcellularly without detectable alteration of the tight junctions between adjacent cells. SNAC also appears to play some role in protecting insulin from proteolytic degradation, potentially allowing for more intact insulin to be available at the site of absorption [40].
The activity of the absorbed insulin from the GI tract was evaluated using SNAC in combination with insulin [36]. The capsules containing insulin and SNAC, in various combinations, were administered orally, as a single dose, to 12 nondiabetic subjects and four control subjects (receiving SNAC or insulin only) in order to assess its biological effect and safety. Plasma glucose levels, insulin and C-peptide concentrations, as well as SNAC levels, were determined, at timed intervals up to 4 h. In all cases, a glucose-lowering effect was demonstrated, preceded by an increase in plasma insulin levels. The nadir of plasma glucose levels appeared after 30–50 min, following the ingestion of the mixture. The plasma insulin levels were found to parallel the blood SNAC levels. Plasma C-peptide levels were suppressed by the lowered glucose levels achieved concurrent with the increasing amount of exogenous insulin absorbed, indicating that the secretion of endogenous hormone was partially abolished. There were no biological effects regarding blood glucose levels upon administration of SNAC or insulin when given alone. No adverse effects were detected during the trial or several weeks after the trial. So it was concluded that the insulin in combination with a novel delivery agent, SNAC, given orally, is absorbed through the GI tract in a biologically active form. This was also demonstrated by a glucose-lowering effect of the mixture as well as a suppression of an endogenous insulin secretion.
The oral delivery of insulin has been investigated as a representative example in a clinical trial with 10 fasted healthy volunteers following oral administration of insulin in combination with the delivery agent [38]. The results indicate that insulin was rapidly absorbed into the systemic circulation and peak plasma concentration occurred within 25 min. The corresponding maximum reductions in both plasma glucose and C-peptide (a marker of endogenous insulin production) concentrations occurred within 1 h. The results were clinically significant because insulin alone or the delivery agent alone dosed orally did not affect plasma levels of insulin or glucose. In another recent clinical study in patients with type II diabetes, a capsule preparation of insulin containing 10 mg of insulin and 200 mg of the delivery agent was evaluated. The data demonstrated that oral administration of this unformulated insulin, when administered 30 min prior to the standardized meal, reduced postprandial excursion, produced a marked increase in systemic insulin levels, and a concomitant reduction in C-peptide . In addition, plasma insulin concentrations peaked faster using Emisphere’s oral unformulated dosage as compared to fast-acting injectable insulin (30 min for oral versus approximately 45 min for injectable formulations) [37].
Emisphere’s oral insulin uses a proprietary permeation enhancer which helps in the absorption of insulin. The pharmacokinetics studies indicated a rapid absorption time of around 20 min from the time of administration and the plasma insulin levels return to baseline within 2 h. Based on the rapidity of absorption, it was proposed that the absorption is from the upper GI tract. In another 2-week clinical trial on patients, well controlled under dietary conditions, Emisphere’s oral insulin was shown to improve both glycemic control and insulin sensitivity. Emisphere also reported completion of a placebo-controlled four treatment arm; 90 days Phase II study in 2004 [51]. The dose of insulin was fixed for the entire duration of the trial, with insulin dose ranging from 20 mg per day to 40 mg day. However, the highest studied dose showed statistically significant reduction in HbA1c over placebo, that too in patients with baseline HbA1c of 8 % and above. HbA1c decrease of 0.74 % from baseline was observed in patients on highest dose of oral insulin, while no change (0.00 %) was observed in patients on placebo (n = 17, p = 0.03). Emisphere’s oral insulin product demonstrated a good safety profile as there were no significant differences in hypoglycemic events, serious adverse events or insulin antibody formation in comparison to placebo (additional data reported as press release). In this study, only the highest dose showed a clinically meaningful drop in HbA1c after 3 months of therapy. The high dose increases the cost of therapy and points to researchers having to pay significant attention to ensure the commercial viability of such an oral insulin drug in the marketplace.
2.3 Heparin
Heparin an anionic pentasaccharide is one of the most important anticoagulant drugs in current clinical use. Heparin is composed of glucosamine and L-iduronic acid or D-glucuronic acid in chains of variable length, having a molecular weight range of 5,000–30,000 [52]. It is widely used for the prevention and treatment of deep venous thrombosis, pulmonary embolism in patients undergoing orthopedic surgery and for patients with renal failure. Its hydrophilic nature, anionic structure due to presence of SO3− groups and large molecular weight prevents absorption through the GI tract.
SNAC is an acetylated amino acid molecule that has been shown to facilitate the GI absorption of codelivered heparin. SNAC-mediated GI absorption of heparin occurs in a passive transcellular process without causing apparent damage to the intestinal epithelium. The pathway of oral absorption of heparin was evaluated using fluorescence microscopy to follow the transport of heparin across Caco-2 cell monolayers [32]. The localization of fluorescently labeled heparin was determined using epifluorescence and confocal microscopy. DNA dyes were used to determine the effect of SNAC on the plasma membrane integrity. F-actin was labeled with fluorescent phalloidin to investigate the stability of perijunctional actin rings in the presence of SNAC. Heparin was detected in the cytoplasm only after incubation of the cells with heparin and SNAC. No DNA staining was observed in cells incubated with a DNA dye in the presence of SNAC concentrations at which heparin transport occurred. In addition, no signs of actin redistribution or perijunctional ring disbandment were observed during the transport of heparin. The results indicate that SNAC enables heparin transport across Caco-2 monolayers via the transcellular pathway. Heparin transport in the presence of SNAC is selective and does not involve permeabilization of the plasma membrane or tight junction disruption.
SNAC was evaluated with escalating oral heparin doses in a randomized, double-blind, controlled clinical study for safety, tolerability, and effects on indexes of anticoagulation [35]. Investigations, both in vitro [33] and in vivo [34], revealed that the n-acylated nonalpha amino acid SNAC has no pharmacological activity. When dosed with 10.5 g SNAC/20,000 IU heparin, an increase in concentration of activated partial thromboplastin time (aPTT) , and tissue factor pathway inhibitor (TFPI) concentrations were detected. For the entire group, 30,000 IU SNAC and heparin elevated TFPI from 74.967.6 to 254.2612.3 mg/ml (P, 0.001) 1 h after dosing (P, 0.001). Similar changes occurred in antifactor IIa and antifactor Xa . aPTT rose from 2860.5 to 42.266.3 s 2 h after dosing (P, 0.01). No significant changes in vital signs, physical examination, ECGs, or clinical laboratory values were observed. Neither 30,000 IU heparin alone nor 10.5 g SNAC alone altered the haemostatic parameters. Emesis was associated with 10.5 g SNAC. A taste-masked preparation of SNAC 2.25 g was administered orally with heparin 30,000–150,000 IU. Both aPTT and antifactor Xa increased with escalating doses of heparin. This preparation was well tolerated. These results established the feasibility of oral delivery of anticoagulant doses of heparin in humans and were believed to have broader implications for the absorption of macromolecules. Phase II clinical studies on hip replacement patients have also been promising with heparin/SNAC being comparable to subcutaneous heparin for the prevention of deep venous thrombosis [31].
2.4 Recombinant Human Growth Hormone (rhGH)
Human growth hormone is a protein drug (22 kDa) and has been used by patients with growth failure due to inadequate secretion of endogenous growth hormone, Turner syndrome, chronic renal insufficiency in children, and as replacement therapy for adults. The possibility of using the Emisphere delivery agent to deliver rhGH orally was first tested in rodents. A series of N-acetylated, nonalpha, aromatic amino acids were prepared and shown to promote the absorption of (rhGH) from the GI tract. Seventy compounds in this family were tested in vivo in rats [41]. Of the compounds tested, 4-[4-[(2-hydroxybenzoyl) amino] phenyl butyric acid was identified as a preclinical candidate and was used to demonstrate the oral delivery of rhGH in primates. A significant positive correlation was found between the relative log k′ of the delivery agents, as determined by HPLC on an immobilized artificial membrane (IAM) column, and serum rhGH concentrations following oral or colonic dosing in rats. Structure-activity relationships have also been developed on the basis of electronic effects and hydrogen-bonding characteristics of the aromatic amide substituent. Subsequently, the macromolecule was delivered in cynomolgus monkeys (n = 4). A mean peak serum concentration of 55 ng/ml rhGH was obtained following administration of a single oral dose of rhGH in combination with the delivery agent. Oral administration of either rhGH or delivery agent alone to these monkeys did not result in measurable circulating levels of rhGH [53].
Studies have been conducted to investigate the mechanisms of GI absorption of rhGH in the presence of an Emisphere delivery agent. The results of these mechanistic studies suggest that the oral delivery of rhGH is dependent on both the dose of the delivery agent and rhGH, and that P-glycoprotein may be involved in the hGH absorption mechanism in the presence of these delivery agents. An early phase clinical trial has been conducted in collaboration with Eli Lilly and Company to evaluate an oral formulation of rhGH in combination with an Emisphere delivery agent [54]. In another small proof of concept study, 8 GH deficient patients were given oral rhGH [19]. Novartis investigated pharmacokinetics as well as the pharmacodynamic properties of the orally delivered rhGH. The study showed that growth hormone peaks were recorded in all patients at some time points, although with considerable variability and minor endogenous growth hormone interference. An increase in IGF-I was seen in some patients, leading to a statistically significant increase in mean serum IGF-I at day 7 compared with end of wash-out. Phase I data indicated that rhGH can be absorbed when given to growth hormone-deficient (GHD) patients in a prototype oral formulation using Emisphere’s Eligen® delivery technology.
2.5 Vitamin B12
Vitamin B12 is important for the normal functioning of the brain and nervous system and for the formation of blood. Cyanocobalamin is the stable and most widely used form of B12. Present work has been dedicated to improve available cyanocobalamin (B12) formulations directed toward achieving repletion of active B12 in B12-deficient individuals. Low levels of B12 can be the result of a lack of the vitamin in the diet, but are most likely to occur because of deficiencies in an individual’s ability to absorb B12 through the natural intricate mechanism. Conditions resulting in reduced stomach acidity (such as long-term use of proton pump inhibitors or age related stomach atrophy) or GI disturbances (such as bariatric surgery, Crohn’s disease, or celiac disease) can lead to B12 deficiency. In a study [42] completed in 2011, B12 deficient patients were given either a typical B12 injection regimen for 12 weeks (5 injections of 1,000 μg over the first 15 days and then one each at 21, 30, 60, and 90 days) or 1,000 μg of Eligen® B12 as a daily pill. All individuals in the study achieved rapid repletion of active B12 within 15 days (the first time point in the study) whether on injection or the oral tablet. Furthermore, all participants in the study had B12 levels that continued to be at normal levels till the end of the study. This performance placed the Eligen® B12 formulation on par with the standard regimen of frequent B12 injections, without the extra cost and inconvenience of drug injections.
Another study [55] compared the efficacy and safety profile of a new proprietary oral vitamin B12 formulation (oral B12) with intramuscular (IM) vitamin B12 (TM B12) in restoring normal serum B12 concentrations in patients with low cobalamin levels. Patients were recruited from five centers and randomly assigned to receive oral B12 1,000 µg, taken daily for 90 days, or IM B12 1,000 µg, given on study days 1, 3, 7, 10, 14, 21, 30, 60, and 90. The patients were aged between 18 and 60 years and had GI abnormalities or were on a restricted diet. The primary efficacy outcome compared the proportion of patients in each treatment arm in whom cobalamin levels were normalized (≥ 350 ng/ml) following 60 days of treatment. Secondary objectives included comparing the efficacy of the two formulations after 90 days of treatment, assessing time to normalization of B12 levels, and evaluating the changes in the levels of biomarkers methylmalonic acid (MMA) and homocysteine (HC). The effect on holotranscobalamin II (active B12) levels was assessed as an exploratory end point and correlated to serum cobalamin levels in both treatment groups. Blood samples were collected at baseline (day 1) and on days 15, 31, 61, and 91. Fifty patients were recruited. Forty-eight patients (96.0 %) completed the study (22 patients [91.7 %] in the oral B12 group and 26 patients [100 %] in the IM B12 group). All patients (100 %) in both treatment groups and in both populations had a cobalamin level ≥ 350 pg/ml on day 61 and maintained it on day 91. The difference between the IM and oral treatment groups did not reach the planned level of statistical significance (p < 0.05) for mean percent change from baseline (PCFB) in serum cobalamin levels on day 61 and day 91. The difference between the IM and oral treatment groups did not reach the planned level of statistical significance for mean PCFB in serum MMA levels on day 61. There was a statistical difference between the IM and oral treatment groups for mean PCFB in serum MMA levels on day 91 (p = 0.033), with lower values in the oral B12 group. The difference between the IM and oral treatment groups did not reach the planned level of statistical significance for mean PCFB in plasma HC levels on day 61 and day 91. All patients in each treatment group achieved normalization of serum cobalamin levels by day 15. All patients in both treatment groups and in both populations had plasma holotranscobalamin levels ≥ 40 pmol/L on day 61 and on day 91. No statistical analysis was planned or performed for safety end points, which were reported only descriptively. Most observed adverse effects were considered mild or moderate in intensity. Adverse effects that were considered severe in intensity were also considered to be not related to the studied drug by the investigator. The treatment regime in this selected study population consisted of individuals with low cobalamin levels who received oral B12 (1,000 mu g/d) or IM B12 (1,000 mu g in nine injections over 3 months) for a total of 3 months. Both the oral and IM formulations were effective in restoring normal levels of serum cobalamin in all patients studied (100 %). Both formulations used in this study were well tolerated at the dose studied.
2.6 Parathyroid Hormone (PTH)
Parathyroid hormone (PTH) , the only drug known to stimulate bone formation, is a peptide therapeutic indicated in the treatment of osteoporosis [18, 56]. It is an 84-amino acid protein and is used to regulate calcium homeostasis. Unfortunately, PTH is only effective when dosed by injection because it has no oral bioavailability . PTH is produced by the parathyroid glands to regulate the amount of calcium and phosphorus in the body. When used therapeutically, it increases bone density and bone strength to help prevent fractures. It is approved to treat osteoporosis, a disease associated with a gradual thinning and weakening of the bones that occurs most frequently in women after menopause. Untreated postmenopausal osteoporosis can lead to chronic back pain, disabling fractures, and lost mobility.
In July 2008, Emisphere announced that its partner, Novartis Pharma AG, launched a Phase I study in postmenopausal women to determine the safety and tolerability of an oral formulation PTH1–34, a combination of human PTH1–34 and the absorption enhancer 5-CNAC using Eligen® technology, for the treatment of postmenopausal osteoporosis. The study was designed to assess the bioavailability profile of increasing doses of PTH1–34 combined with different amounts of 5-CNAC administered orally. On October 19, 2009, Novartis reported results of this study which showed potentially relevant therapeutic exposure and safety profiles similar to those of the currently available injectable dosage form. These were presented at the 73rd Annual Scientific Meeting of the American College of Rheumatology in Philadelphia, PA, USA.
In April 2010, Novartis initiated a second Phase I trial for an oral PTH1–34 for the treatment of postmenopausal osteoporosis. The study was a partially blinded, placebo-controlled, active comparator study to explore the safety, tolerability, pharmacokinetics and pharmacodynamics in postmenopausal women after daily doses of PTH1–34. The study was divided into two parts (A and B) and enrolled approximately 120 women. In Part A ascending doses of oral PTH1–34 were tested for safety, tolerability, and pharmacokinetics and compared to Forsteo®. In Part B, in addition to safety and tolerability of oral PTH, pharmacodynamic responses were measured by bone biomarker levels and bone mineral density and compared to Forsteo®. On June 17, 2011, Novartis informed the results of its recently completed study for an oral PTH1–34 using Emisphere’s Eligen® technology in postmenopausal women with osteoporosis or osteopenia. Novartis stated that although the study confirmed that oral PTH1–34 was both safe and well tolerated, several clinical endpoints were not met. Based on the data analyzed, Novartis has terminated the study and anticipates no further work on oral formulation of PTH1–34.
Another study demonstrated that a single dose of the novel oral PTHPTH1–34, which utilizes Eligen® technology and absorption-enhancer carrier molecule 5-CNAC, achieved potentially therapeutically relevant exposure and safety profiles to those of the currently available injectable formulation in healthy postmenopausal women. These results were from a single-center, partially blinded, incomplete crossover study conducted by Emisphere’s partner Novartis Pharma AG and were presented on October 19, 2009, in a poster session at the 73rd Annual Scientific Meeting of the American College of Rheumatology in Philadelphia.
This Phase I single-center partially blinded incomplete crossover study that was designed to assess the exposure and safety of orally administered doses of PTH1–34 and different amounts of the absorption enhancer 5-CNAC was conducted in 32 healthy postmenopausal women. The subjects were randomized to receive a single dose of placebo, 20 µg of subcutaneously injected parathyroid hormone PTH1–34 (Forteo®) , or one of several orally administered doses of PTH1–34 formulated with either 100 or 200 mg of Emisphere’s absorption-enhancer 5-CNAC. While all doses of oral PTH1–34 were rapidly absorbed and showed appreciable blood concentrations in a dose-dependent manner, the 2.5 and 5 mg doses of oral PTH1–34 containing 200 mg 5-CNAC achieved exposure levels closest to those of 20 µg injectable PTH1–34, with a comparable incidence of adverse events. Ionized calcium remained within normal limits in all treatment groups. There were no serious adverse events in the study. Nine participants withdrew from the study. Of these, five (one on placebo, one on Forteo® and three on either 2.5 or 5 mg PTH1–34) withdrew because of symptomatic hypotension. Three patients on either 2.5 or 5 mg PTH1–34 withdrew because of delayed vomiting. One patient on 2.5 mg PTH1-34 (100 mg 5-CNAC) withdrew because of symptomatic, but unconfirmed hypercalcemia.
3 Conclusions and Future Perspectives
Oral administration of drugs is regarded as the most preferred route of administration, because of the convenience to large number of patient population and its cost effectiveness. However, macromolecular drugs cannot be administered orally because of the inherent properties of these drugs. Emisphere’s Eligen® technology makes it possible to orally deliver a therapeutic molecule without altering its chemical form or biological integrity. Eligen® delivery agents, or “carriers,” such as the absorption-enhancer 5-CNAC that facilitate or enable the transport of therapeutic molecules across the mucous membranes of the GI tract, to reach the tissues of the body where they can exert their intended pharmacological effect. Eligen® technology has been shown to enhance oral delivery of many different therapeutic molecules. This technology works especially well with water-soluble drugs, both positively and negatively charged. Enhanced oral delivery has been demonstrated in the clinic with large molecular weight drugs (such as unfractionated heparin and growth hormone), medium size biomolecules (such as peptides like calcitonin and insulin, as well as low-molecular weight heparin) and small molecules (such as cromolyn and cyanocobalamin) . For drugs with low aqueous solubility, Eligen® technology has been less successful but in certain cases it can enhance oral bioavailability . The commercial success of these products certainly will depend on its increased stability, bioavailability and tolerability or high patient compliance . We can expect that more products based on Eligen® technology will be available to the patients in near future, particularly an oral insulin product which the 347 million diabetic population is expecting for the last two decades.
References
Donovan MD, Flynn GL, Amidon GL. Absorption of polyethylene glycols 600 through 2000: the molecular weight dependence of gastrointestinal and nasal absorption. Pharm Res. 1990;7(8):863–8.
Aungst BJ. Absorption enhancers: applications and advances. AAPS J. 2012;14(1):10–8.
Woodley JF. Enzymatic barriers for GI peptide and protein delivery. Crit Rev Ther Drug Carrier Syst. 1994;11(2–3):61–95.
Lipka E, Crison J, Amidon GL. Transmembrane transport of peptide type compounds: prospects for oral delivery. J Control Release. 1996;39(2–3):121–9.
Yun Y, Cho YW, Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65(6):822–32.
He C, et al. Size-dependent absorption mechanism of polymeric nanoparticles for oral delivery of protein drugs. Biomaterials. 2012;33(33):8569–78.
Griffin BT, O’Driscoll CM. Opportunities and challenges for oral delivery of hydrophobic versus hydrophilic peptide and protein-like drugs using lipid-based technologies. Ther Deliv. 2011;2(12):1633–53.
Rekha MR, Sharma CP. Oral delivery of therapeutic protein/peptide for diabetes—future perspectives. Int J Pharm. 2013;440(1):48–62.
Muller G. Oral delivery of protein drugs: driver for personalized medicine. Curr Issues Mol Biol. 2011;13(1):13–24.
Werle M, Makhlof A, Takeuchi H. Oral protein delivery: a patent review of academic and industrial approaches. Recent Pat Drug Deliv Formul. 2009;3(2):94–104.
Shaji J, Patole V. Protein and peptide drug delivery: oral approaches. Indian J Pharm Sci. 2008;70(3):269–77.
Patel VF, Liu F, Brown MB. Advances in oral transmucosal drug delivery. J Control Release. 2011;153(2):106–16.
Malik DK, et al. Recent advances in protein and peptide drug delivery systems. Curr Drug Deliv. 2007;4(2):141–51.
Park K, Kwon IC, Park K. Oral protein delivery: current status and future prospect. React Funct Polym. 2011;71(3):280–7.
Fasano A. Novel approaches for oral delivery of macromolecules. J Pharm Sci. 1998;87(11):1351–6.
Goldberg M, Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov. 2003;2(4):289–95.
Henry CM. Special delivery. Chem Eng News. 2000;78(38):49–65.
Leone-Bay A, et al. Oral delivery of biologically active parathyroid hormone. Pharm Res. 2001;18(7):964–70.
Wu SJ, Robinson JR. Transport of human growth hormone across Caco-2 cells with novel delivery agents: evidence for P-glycoprotein involvement. J Control Release. 1999;62(1–2):171–7.
Leone-Bay A, et al. Compounds and compositions for delivering active agents, U.S. Patent, Editor. 2003, Emisphere Technologies Inc.: US.
Mlynek GM, Calvo LJ, Robinson JR. Carrier-enhanced human growth hormone absorption across isolated rabbit intestinal tissue. Int J Pharm. 2000;197(1–2):13–21.
Wu SJ, Robinson JR. Transcellular and lipophilic complex-enhanced intestinal absorption of human growth hormone. Pharm Res. 1999;16(8):1266–72.
Bagger YZ, et al. Oral salmon calcitonin induced suppression of urinary collagen type II degradation in postmenopausal women: a new potential treatment of osteoarthritis. Bone. 2005;37(3):425–30.
Buclin T, et al. Bioavailability and biological efficacy of a new oral formulation of salmon calcitonin in healthy volunteers. J Bone Miner Res. 2002;17(8):1478–85.
Hamdy RC, Daley DN. Oral calcitonin. Int J Womens Health. 2012;4:471–9.
Karsdal MA, et al. The effect of oral salmon calcitonin delivered with 5-CNAC on bone and cartilage degradation in osteoarthritic patients: a 14-day randomized study. Osteoarthr Cartilage. 2010;18(2):150–9.
Karsdal MA, et al. The effects of oral calcitonin on bone collagen maturation: implications for bone turnover and quality. Osteoporos Int. 2008;19(9):1355–61.
Karsdal MA, et al. Investigation of the diurnal variation in bone resorption for optimal drug delivery and efficacy in osteoporosis with oral calcitonin. BMC Clin Pharmacol. 2008;8:12.
Karsdal MA, et al. Optimizing bioavailability of oral administration of small peptides through pharmacokinetic and pharmacodynamic parameters: the effect of water and timing of meal intake on oral delivery of Salmon calcitonin. BMC Clin Pharmacol. 2008;8:5.
Tanko LB, et al. Safety and efficacy of a novel salmon calcitonin (sCT) technology-based oral formulation in healthy postmenopausal women: acute and 3-month effects on biomarkers of bone turnover. J Bone Miner Res. 2004;19(9):1531–8.
Baughman RA, et al. Oral delivery of anticoagulant doses of heparin. A randomized, double-blind, controlled study in humans. Circulation. 1998;98(16):1610–5.
Brayden D, et al. Heparin absorption across the intestine: effects of sodium N-[8-(2-hydroxybenzoyl)amino]caprylate in rat in situ intestinal instillations and in Caco-2 monolayers. Pharm Res. 1997;14(12):1772–9.
Gonze MD, et al. Orally administered heparin for preventing deep venous thrombosis. Am J Surg. 1998;176(2):176–8.
Malkov D, et al. Pathway of oral absorption of heparin with sodium N-[8-(2-hydroxybenzoyl)amino] caprylate. Pharm Res. 2002;19(8):1180–4.
Rivera TM, et al. Oral delivery of heparin in combination with sodium N-[8-(2-hydroxybenzoyl)amino]caprylate: pharmacological considerations. Pharm Res. 1997;14(12):1830–4.
Kidron M, et al. A novel per-oral insulin formulation: proof of concept study in non-diabetic subjects. Diabet Med. 2004;21(4):354–7.
Abbas R, et al. Oral insulin: pharmacokinetics and pharmacodynamics of human insulin following oral administration of an insulin/delivery agent capsule in healthy volunteers. Diabetes. 2002;51:A48.
Hoffman A, Qadri B. Eligen insulin—a system for the oral delivery of insulin for diabetes. IDrugs. 2008;11(6):433–41.
Kapitza C, et al. Oral insulin: a comparison with subcutaneous regular human insulin in patients with type 2 diabetes. Diabetes Care. 2010;33(6):1288–90.
Malkov D, et al. Oral delivery of insulin with the eligen technology: mechanistic studies. Curr Drug Deliv. 2005;2(2):191–7.
Leone-Bay A, et al. 4-[4-[(2-Hydroxybenzoyl)amino]phenyl]butyric acid as a novel oral delivery agent for recombinant human growth hormone. J Med Chem. 1996;39(13):2571–8.
Castelli MC, et al. SNAC co-formulation produces significant enhancement of oral vitamin B12 bioavailability in rats. FASEB J. 2008;22:795.
Milstein SJ, et al. Partially unfolded proteins efficiently penetrate cell membranes–implications for oral drug delivery. J Control Release. 1998;53(1–3):259–67.
Alani AW, Robinson JR. Mechanistic understanding of oral drug absorption enhancement of cromolyn sodium by an amino acid derivative. Pharm Res. 2008;25(1):48–54.
Azria M, Copp DH, Zanelli JM. 25 years of salmon calcitonin: from synthesis to therapeutic use. Calcif Tissue Int. 1995;57(6):405–8.
Lee YH, Sinko PJ. Oral delivery of salmon calcitonin. Adv Drug Deliv Rev. 2000;42(3):225–38.
Gschwind HP, et al. Metabolism and disposition of the oral absorption enhancer 14C-radiolabeled 8-(N-2-hydroxy-5-chlorobenzoyl)-amino-caprylic acid (5-CNAC) in healthy postmenopausal women and supplementary investigations in vitro. Eur J Pharm Sci. 2012;47(1):44–55.
Bagger YZ, et al. Oral salmon calcitonin induced suppression of urinary collagen type II degradation in postmenopausal women: A new potential treatment of osteoarthritis. Bone. 2005;37(3):425–30.
Dinh S, Liu P, Wong V. Gastric absorption of oral insulin in rats, in Annual Meeting of American Association of Pharmaceutical Scientists. Toronto: American Association of Pharmaceutical Scientists; 2002.
Malkov D, Wang H-Z, Angelo R. Mechanism of oral absorption of insulin with Emisphere drug delivery agents, in The 29th Annual Meeting of Controlled Release Society. Seoul: Controlled Release Society; 2002.
Arbit E, et al. Oral insulin as first-line therapy in type 2 diabetes: a randomized-controlled pilot study. Diabetologia. 2004;47:A5.
Hirsh J, et al. Guide to anticoagulant therapy: Heparin a statement for healthcare professionals from the American Heart Association. Circulation. 2001;103(24):2994–3018.
Leone-Bay A, Paton DR, Weidner JJ. The development of delivery agents that facilitate the oral absorption of macromolecular drugs. Med Res Rev. 2000;20(2):169–86.
Leone-Bay A, et al. Oral delivery of rhGH: preliminary mechanistic considerations. Drug News Perspect. 1996;9(10):586–91.
Castelli MC, et al. Comparing the efficacy and tolerability of a new daily oral vitamin B-12 formulation and intermittent intramuscular vitamin B-12 in normalizing low cobalamin levels: a randomized, open-label, parallel-group study. Clin Ther. 2011;33(3):358–71.
Molina E, et al. The effect of parathyroid hormone (hPTH 1–34) on oxytocin-induced contractions of pregnant human myometrium “in vitro”. FASEB J. 1996;10(3):3800.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Victor, S., Paul, W., Sharma, C. (2014). Eligen® Technology for Oral Delivery of Proteins and Peptides. In: das Neves, J., Sarmento, B. (eds) Mucosal Delivery of Biopharmaceuticals. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-9524-6_18
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9524-6_18
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-9523-9
Online ISBN: 978-1-4614-9524-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)