
Abstract
Overview of Peripheral T Cell Homeostasis
The lymphocyte is a major class of white blood cell that defends the body from attack by infectious agents and confers long-term protection through immunological memory. Lymphocytes are also involved in pathological immune reactions including graft rejection, allergies, and autoimmune disorders. Their remarkable capacity to proliferate up to 5,000-fold in response to antigen must be counterbalanced by a controlled process of removing cells. This is achieved through molecular pathways of programmed cell death, which maintain selective and specific homeostasis of the numbers of lymphocytes and other immune cells. We focus principally on cell death mechanisms in T lymphocytes that control the number of T cells of a given antigen specificity represented in the finite T cell niche. Here, we discuss the central role of caspases in the regulation of the general pathways of cell-extrinsic and cell-intrinsic T cell apoptosis and programmed necrosis with an overview of their importance for human health.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
3.1 Forms of T Cell Death
Although the concept of a programmed form of cell death was envisioned as early as the 1950s, the major supporting data for this process were published in the 1970s and 1980s [1]. These seminal studies were performed in Caenorhabditis elegans as part of an effort to trace the ontogeny of the entire worm’s embryonic cell lineages. The process of programmed cell death was given the moniker “apoptosis,” which is a Greek term referring to leaves falling off trees or petals falling off flowers, by pathologists in 1972 [1]. In recognition of their contributions to understanding apoptosis pathways, Brenner, Horvitz, and Sulston received the 2002 Nobel Prize in Physiology or Medicine [2, 3]. In 1988, independent studies of cell death in mammalian cells by Vaux/Cory/Adams [4] revealed the critical role of BCL-2 (described in detail below) in blocking programmed cell death of tumor cells. These studies were extended by Korsmeyer [5] to demonstrate that BCL-2 also inhibits apoptosis of non-transformed cells. The breadth of studies on cell death has become remarkably vast, and multiple forms of programmed cell death beyond apoptosis have now been identified [6]. The focus of this chapter is on apoptosis in mature T lymphocytes with a brief discussion of a non-apoptotic form of cell death classified as programmed necrosis.
3.1.1 Apoptosis
As elegantly demonstrated in C. elegans, apoptosis is a cellular process that results from highly regulated biochemical events terminating in a controlled form of cell death. During C. elegans development, 1,090 cells are generated, and 131 of these cells invariably disappear through apoptosis. The genes controlling this process in C. elegans are ced-3, ced-4, ced-9, and egl-1 via a pathway of epistatic relationships in which EGL-1 –| CED-9 –| CED-4 → CED-3 → cell death [7]. Mammalian cells undergoing apoptosis display distinctive hallmarks including cell shrinkage, nuclear condensation, DNA fragmentation, apoptotic bodies, and membrane blebbing (Fig. 3.1) [8]. In late-stage apoptosis, the so-called eat-me signals (e.g., phosphatidylserine) appear on the cell surface and induce phagocytes to rapidly clear dying cells by engulfment [9].

Electron micrographs of healthy (a) and apoptotic (b) cells. Red asterisk: Condensed nucleus; white arrow: apoptotic bodies; blue arrow: intact membrane. Micrographs courtesy of Dr. Lixin Zheng
T lymphocytes have great potential for rapid expansion as they carry out their task of orchestrating and executing immunological responses against invading pathogens [10]. As such, apoptosis plays a critical role in homeostatic control of T cell numbers and is important in safeguarding against the autoimmune diathesis caused by rare pathogenic T cells that may expand during a response to infection and overcome peripheral tolerance mechanisms. There are two main pathways discussed in detail below that induce apoptosis: receptor-mediated/extrinsic and mitochondrial/intrinsic. The intrinsic pathway of cell death is an ancient one closely related to the cell death pathway delineated in nematodes. The extrinsic pathway, however, only exists in vertebrates and is initiated by ligation of surface “death” receptors that have major physiological roles in controlling lymphocytes. Interestingly, the emergence of the extrinsic pathway in vertebrates [11] corresponds with the evolution of lymphoid cells, which first appear in basal jawed vertebrates [12], suggesting possible coevolution of death receptor signaling for control of lymphocyte homeostasis.
3.1.2 Non-apoptotic Cell Death
The forms of cell death were originally simply categorized by the dichotomy of either traumatic, injury-induced cell death called necrosis or noninflammatory, programmed cell death via apoptosis. We now appreciate that there are multiple, individually nuanced forms of programmed cell death. Those observed in T cells include apoptosis, necroptosis [13], and autophagic cell death [14]. There are also a variety of mechanisms involving specific molecular pathways that account for pathological cell death due to infectious agents, particularly viral cytopathicity [15]. The importance of non-apoptotic forms of cell death for T cell biology is still being established, but the critical role of apoptosis is irrefutable.
3.2 Central Role of Caspases in Apoptosis
As with most tightly controlled cellular processes, the cascade of events leading to apoptosis involves activation of a series of proteins with several regulatory steps along the way. In the case of apoptosis, the cysteine-dependent, aspartate-directed protease (caspase) enzymes, which are homologous to C. elegans’ CED-3, are central mediators that function by cleaving substrate proteins after an aspartate residue. Caspases are first synthesized as zymogens that include two enzymatic subunits and an N-terminal pro-domain that, in one class of caspases, contains an 80–100-amino acid protein–protein interaction motif of the death effector domain (DED) or caspase recruitment domain (CARD) variety [16]. These domains form a hexahelical bundle of tightly coiled alpha helices known as the death fold that binds through homotypic interactions (i.e., DED–DED or CARD–CARD) to appropriate signaling complexes containing the same death fold [17]. Following processing, the two enzymatic subunits are released and the prodomain is destroyed. Crystal structures of the mature enzyme indicate that two copies each of the enzymatic subunits interact non-covalently to form a heterotetramer [18]. Caspases are involved in apoptosis and inflammation. The inflammatory roles involve processing precursor forms of cytokines and will not be further discussed. Those mediating apoptosis fall into two categories: termed “initiator” and “effector” caspases (Fig. 3.2a). Initiator caspases can be activated by cell-extrinsic signals (caspases 8 and 10) transduced via surface receptors or by cell-intrinsic pathways (caspase 9) initiated by cell stress, such as DNA damage or growth factor withdrawal. Initiator caspases contain either two tandem DED domains (caspases 8 and 10) or a CARD domain (caspase 9), whereas effector caspases (including caspases 3, 6, and 7) lack either recruitment domain. An additional initiator caspase called caspase 2 has more recently been proposed to be important in inducing apoptosis after DNA damage through a signaling complex known as the PIDDosome [19]. Once activated, initiator caspases cleave (and thereby activate) full-length pro-forms of the effector caspases. Effector caspases act by cleaving various protein substrates to inhibit or alter their function and execute the apoptotic program. Cell death substrates that are targeted for cleavage by effector caspases during apoptosis include cytoskeletal proteins (e.g., actin, plectin, ROCK1, gelsolin), nuclear lamins, and inhibitor of the caspase-activated DNase (ICAD) [20], which upon cleavage lead to cell shrinkage/blebbing, nuclear condensation, and DNA fragmentation, respectively.

Key protein mediators of extrinsic (a) and intrinsic (b) cell death. Colored boxes denote protein domains as indicated in the legends
Caspase activity is negatively regulated by proteins in the inhibitor of apoptosis (IAP) family [21], which includes XIAP, survivin, c-IAP-1, c-IAP-2, and NAIP. IAP family proteins have multiple functions including ubiquitin ligase activity and inhibition of active caspases. XIAP [22] is the best studied in this family, and it has been demonstrated to bind caspases 3 and 7 with high affinity through its BIR2 domain and caspase 9 with low affinity through its BIR3 domain [23]. This binding profile reveals the role of XIAP as a regulator of the downstream effector caspases that function in both the extrinsic and intrinsic apoptosis pathways discussed below. Once bound to a caspase, XIAP sterically hinders the enzyme active site to prevent execution of the apoptotic program [24]. IAP proteins are, in turn, inhibited by the mitochondrial proteins SMAC/DIABLO [25, 26] and OMI [27] (described further in the discussion of the intrinsic pathway below) to allow apoptosis to proceed. Regulation of caspase 8 is more complex, likely because this protease is unique in its ability to participate in non-apoptotic signaling. Importantly, full-length caspase 8 participates in transducing signals required for T cell activation [28] by augmenting NF-κB activation downstream of the TCR [29]. Moreover, basal caspase 8 activity is required to prevent T cells from undergoing autophagic cell death [14].
3.3 Extrinsic, Death Receptor-Induced Apoptosis and Necroptosis
Cell-extrinsic death pathways are initiated by ligation of death receptors expressed on the surface of lymphoid cells.
3.3.1 TNFR Superfamily Death Receptors
The most important death-inducing receptors on the surface of T cells are members of the TNF receptor (TNFR) superfamily that share structural similarities and conserved cytoplasmic domains. Their signaling depends on trimer formation that is characteristic of TNFR superfamily members and mediated by the pre-ligand-binding assembly domain (PLAD) [30]. The ligands for the DD-containing TNFR proteins are expressed as membrane-bound trimers [31] homologous to TNF-α and can be cleaved from the cell surface by metalloproteinases to generate soluble forms [32]. However, elegant mouse studies have demonstrated that TNFR ligands are most active when in cell-surface, membrane-bound trimer form [31]. The death receptors contain a death domain (DD), which forms the hexahelical death fold described above, that nucleates formation of a protein complex required to transduce the death signal. Receptors included in this family of DD-containing TNFR proteins are FAS, TNFR1, TRAIL-R1, TRAIL-R2, DR3, and DR6 [32]. DR3 and DR6 biology is relatively poorly understood, though ligands have been identified (TL1A [33] for DR3 and APP [34] for DR6). Although they both have some functions in the immune system [35–37], DR3 primarily promotes T cell activation [33], and DR6 functions primarily in the nervous system [34]. Because our current understanding attributes little role in T cell death, DR3 and DR6 will not be discussed further in this chapter.
3.3.1.1 FAS
The prototype for the death receptor family is the FAS receptor (also known as APO-1, CD95, or TNFRSF6), which exists as a multimer, likely a trimer, on the surface of activated T cells due to homotypic interaction of the PLAD [38] domains. Upon binding of membrane-bound, trimeric FAS ligand (FASL), higher order clusters of FAS trimers termed signaling protein oligomerization transduction structures [39] (SPOTS) are formed and permit downstream signaling. The first step in FAS receptor signaling is formation of the death-inducing signaling complex (DISC), which is formed by homotypic interaction of the DD domain of FAS with the DD domain of the adaptor molecule FADD (Fig. 3.3). Structural analysis has revealed that the stoichiometry of FAS:FADD interactions is 5–7 FAS molecules with 5 FADD molecules [40], consistent with the formation of SPOTS [39]. In addition to a DD domain, FADD also contains a DED domain, which mediates FADD–FADD interactions that stabilize the DISC and recruit the DED-containing pro-caspases 8 and 10. Once pro-caspase 8 and 10 are recruited to the DISC, the pro-caspase zymogens oligomerize and initiate a two-step proteolytic cleavage event that first removes a C-terminal p10 or p12 peptide for caspases 8 and 10, respectively, and then liberates the p18 or the p17 peptide for caspases 8 and 10, respectively, to enable formation of the active caspase heterotetramer consisting of two p10/p12 molecules and two p18/p17 molecules [41]. Active caspases 8 and 10 then cleave the effector caspases to execute the death program.

Caspase 8 and 9 activation pathways
The susceptibility of a cell to FAS-induced apoptosis is variable, leading to a categorization paradigm in which type 1 cells (e.g., T cells) die efficiently after activation of effector caspases by activated caspases 8 and 10 and type 2 cells (e.g., hepatocytes and pancreatic beta cells) are resistant to death by this pathway unless the caspase cascade is amplified. This amplification occurs when caspase 8 cleaves BID [42–44], generating a pro-apoptotic truncated BID (tBID) protein that amplifies apoptosis signaling by initiating the intrinsic pathway of cell death [45, 46]. XIAP has also been described to be a key reason why type 2 cells are more resistant to FAS death [47].
Negative regulation of FAS signaling is achieved through the activity of c-FLIP, a pro-caspase 8-like protein that is recruited to the DISC via its two tandem, N-terminal DED domains. C-FLIP is a catalytically inactive caspase 8 paralog that decreases the sensitivity of cells to FAS-induced death [48–51] by binding the DISC and inhibiting processing of initiator pro-caspases [52, 53]. Another layer of negative regulation of caspase 8 is provided by TIPE2 [54], a member of the TNF-α-induced protein-8 (TNFAIP8) family that contains a DED domain. TIPE2 functions by binding caspase 8 and inhibiting activation of the transcription factors AP-1 and NF-κB while promoting FAS-induced apoptosis. Mice deficient in TIPE2 succumb to premature death with splenomegaly and multi-organ inflammation [54]. Formation of the DISC can also be impaired by sequestration of FADD by the DED-containing PEA-15 molecule [55].
3.3.1.2 TNFR1
The cytokine TNF-α, which binds TNFR1 and TNFR2, has pleiotropic effects including both cell activation and cell death [56]. TNFR1 but not TNFR2 contains a DD domain important in recruiting adaptor molecules that signal for cell death by apoptosis or necroptosis [32]. Binding of TNF-α to TNFR1 induces consecutive formation of two different signaling complexes [57] on the cell surface (complex I) and then in the cytoplasm (complex II) [58]. Recruitment of the adaptor molecule TNF receptor-associated protein with death domain (TRADD) via its DD domain nucleates complex I and recruits the adaptor TNF receptor-associated factor-2 (TRAF2), a kinase called receptor-interacting protein-1 (RIP1), c-IAP-1, and c-IAP-2, leading to poly-ubiquitination of RIP1 by the c-IAPs and downstream activation of NF-κB and AP-1 transcription factors [32]. In a second step, the TNFR1 complex is internalized, followed by de-ubiquitination of RIP1 by A20 and CYLD (cylindromatosis), allowing for formation of a cytoplasmic DISC/complex II when TRADD and RIP1 associate with RIP3, FADD, and pro-caspase 8 [58]. Complex II signals the cell to die by either caspase 8-mediated apoptosis [59] or RIP3-mediated necroptosis [60, 61]. RIP1 but not RIP3 contains a DD domain, but both contain a RIP homotypic interaction motif (RHIM) that mediates RIP1:RIP3 interactions [62]. RIP1 and 3 are kinases in the RIP family (including RIP1, RIP2/RICK/CARDIAK, RIP3, RIP4/DIK/PKK, RIP5/SgK288, RIP6/LRRK1, and RIP7/LRRK2) whose substrates and functions are as yet largely unidentified [62].
The disparate cellular outcomes of TNFR1 ligation (i.e., activation, apoptosis, or necroptosis) are determined through complex feedback inhibition that hinges on the ubiquitination status of RIP1. Poly-ubiquitinated RIP1 acts as a prosurvival scaffold molecule that activates NF-κB through complex I, and its kinase activity is not required. NF-κB inhibits cell death through a process involving upregulation of anti-apoptotic molecules including the c-IAP ubiquitin ligases and c-FLIP, which translocates to complex II and inhibits caspase 8 processing [58]. Upon removal of ubiquitin, RIP1 functions as a pro-death kinase as a part of the pro-apoptotic DISC formed by complex II. If caspases are blocked, the RIP kinases can divert death signaling to the pro-necroptotic pathway [57, 58]. An additional regulator of TNFR1 signaling is a DD-containing adaptor called silencer of death domains (SODD), which has been found to associate with TNFR1 and DR3 [63] and negatively regulate their signaling, though the details of its action are still being defined. The importance of TNFR1 in T cell homeostasis is still an open question, as animals deficient in TNFR1 do not accumulate T cells (as would be expected if cell death mechanisms were impaired) but instead exhibit deficient host defense and inflammatory responses [64]. Overall, TNFR1 signaling in T cells appears to lead predominantly to cytokine secretion, pro-inflammatory responses, and cell survival [65] in vivo.
3.3.1.3 TRAIL-R1 and TRAIL-R2
The third major death-promoting TNFR superfamily signaling pathway is induced by a ligand called TNF-related apoptosis-inducing ligand (TRAIL) [66]. TRAIL has five distinct receptors with two, TRAIL-R1 (DR4) and TRAIL-R2 (DR5), containing a functional DD domain and three acting as “decoy” receptors that impair TRAIL-induced signaling by sequestering DR4 and DR5 chains via the PLAD domain [67]. The three “decoy” receptors include the membrane-associated proteins TRAIL-R3/DcR1 and TRAIL-R4/DcR2 and the soluble protein osteoprotegerin/TNFRSF11B [68]. Upon binding of trimeric TRAIL to DR4 or DR5, higher order clusters are formed, resulting in DISC assembly [69]. Similar to the FAS DISC, FADD, pro-caspases 8 and 10, and sometimes c-FLIP are recruited to the DD domain of DR4 or DR5. Unlike the other death receptors, DR4 and DR5 require O-glycosylation in order to form the higher order clusters necessary for DISC formation [69]. Moreover, polyubiquitination of pro-caspase 8 after formation of the TRAIL DISC has been found to lead to aggregation of pro-caspase 8 and augmented processing/activation to facilitate cell death [70]. The major roles of DR4 and DR5 signaling seem to be primarily in controlling inflammation and tumor susceptibility [71], but TRAIL can also induce cell death of CD8 T cells that failed to receive cytokine signals from CD4 T cells (i.e., CD4 help) during priming [72].
3.3.2 Restimulation-Induced Cell Death
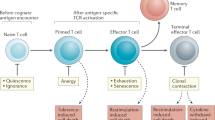
A major physiological process that employs death receptor-induced apoptosis in T cells is restimulation-induced cell death (RICD). This death mechanism is also called activation-induced cell death (AICD), but we like to distinguish the process of activation from death induction since the latter requires restimulation. RICD functions as a negative feedback mechanism to prevent overexpansion of T cells during an immune response and occurs when T cells that are activated and cycling in the presence of lymphokines (e.g., IL-2) undergo apoptosis in response to a subsequent TCR stimulus [10, 73] (Fig. 3.4). This form of propriocidal regulation of the T cell pool is initiated by signaling through the TCR [74] and depends largely on extrinsic death signaling through FAS [75] and intrinsic death signaling through BIM [76] (discussed below). In appropriate mouse strains with gene deficiencies in FAS (lpr mice) [77] or FASL (gld mice) [75], severe lymphoproliferation and autoimmunity occur in vivo with defective RICD observed in vitro. Although FAS and FASL are broadly expressed on activated T cells, FAS-dependent RICD is only initiated in cells that receive a TCR stimulus and not in bystander cells of different specificities [74]. The TCR-induced signal that makes a cell competent to die has not been clearly defined, though new protein synthesis is not required [74, 78]. Further studies examining the susceptibility of activated T cells to RICD have identified an important role for cell cycling, as only cells in late G1 or S phase were found to die by RICD [79]. As discussed in the final section of this chapter, FAS-mediated RICD is a critical process for healthy T cell homeostasis, and a deficiency in any of the required signaling molecules results in severe accumulation of lymphocytes and associated pathologies discussed further below.

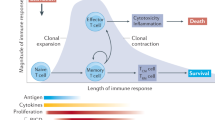
T cell homeostasis in health and disease. Shading indicates antigen load from infectious agent. Adapted from Snow et al. Immunol Rev. 2010 Jul;236:68–82
3.4 Intrinsic, Mitochondrion-Dependent Apoptosis
In addition to cell-extrinsic death signals propagated by cell surface receptors, a cell-intrinsic pathway of apoptosis has also been well defined. The intrinsic pathway of cell death is triggered by cell stress (e.g., DNA damage or growth factor deprivation) and is dependent on signals propagated from the mitochondria. Mitochondria are organelles within the cell whose major function is the efficient generation of ATP through oxidative phosphorylation in cell metabolism. However, mitochondria also play a critical role in cell death, mainly through release of proteins that induce apoptosis by caspase activation. This release occurs when the outer mitochondrial membrane integrity is disrupted by pore formation, a process tightly controlled by members of the BCL-2 family.
3.4.1 BCL-2 Family Proteins
The BCL-2 family of proteins is a key regulator of mitochondrion-dependent apoptosis, and the family’s founding member, BCL-2, is the mammalian homolog of C. elegans’ CED-9. There are approximately 25 genes in the BCL-2 family, and each contains one or more BCL-2 homology (BH) domains consisting of BH1 through BH4 (Fig. 3.2b). The family is broadly divided into three groups: anti-apoptotic (e.g., BCL-2, BCL-xL, BCL-w, MCL-1, BFL-1), multi-domain pro-apoptotic (e.g., BAX, BAK, BOK), and BH3-only pro-apoptotic (e.g., BIM, BID, BAD, PUMA, NOXA, BMF, BIK, HRK, BLK) [80]. BAX and BAK (and, in certain cell types, BOK) are pore-forming proteins and are termed “activators,” while BH3-only proteins are generally considered “sensitizers” since they function to sense various apoptotic stimuli (e.g., growth factor withdrawal sensed by BIM and DNA damage sensed by PUMA). The BH3-only proteins are homologous to C. elegans’ EGL-1 and promote apoptosis when triggered by appropriate stimuli by binding anti-apoptotic BCL-2 family members and sequestering them to prevent their association with the multi-domain “activators” of apoptosis. Protein–protein interactions within this family are mediated primarily by the BH3 domain [81]. Regulation of mitochondrion-dependent, intrinsic cell death relies heavily on shuttling of BCL-2 family members between different compartments in the cell. Several of the BCL-2 family members contain a carboxy-terminal transmembrane domain that anchors them into the mitochondrial outer membrane (MOM); however, presence of this transmembrane domain does not dictate constitutive localization to mitochondria. In the case of the major pore-forming, pro-apoptotic proteins, BAK remains inserted in the MOM, while BAX is prevented from accumulating in the MOM by the action of BCL-xL and possibly other anti-apoptotic BCL-2 proteins [81]. The pro-apoptotic, BH3-only protein BID, on the other hand, promotes insertion of BAX in the MOM once it is cleaved by caspase 8 to form tBID [81]. Once BAX and BAK oligomerize, a pore (sometimes called the mitochondrial apoptosis-induced channel, MAC) is formed in the MOM, leading to mitochondrial outer membrane permeabilization (MOMP) and release of mitochondrial proteins that trigger downstream intrinsic apoptotic signaling.
3.4.2 Role of Mitochondrial Intermembrane Proteins
Upon MOMP, soluble mitochondrial proteins including cytochrome c, second mitochondria-derived activator of caspase (SMAC, also called DIABLO), OMI, and apoptosis-inducing factor (AIF) are released. Cytochrome c is a soluble protein normally sequestered in the intermembrane space of mitochondria and functions as an essential component of the electron transport chain. Upon release into the cytoplasm, cytochrome c binds to inositol (1,4,5) triphosphate receptor (IP3R) present on the membrane of the endoplasmic reticulum (ER) and initiates the release of ER calcium stores [82]. The resulting waves of increased cytosolic calcium induce all mitochondria to release cytochrome c as a feed-forward mechanism of amplifying apoptotic signaling [82]. Studies of the BH4 domain of BCL-2 have recently revealed its capacity to bind the IP3R on ER, blocking release of calcium stores and inhibiting pro-apoptotic calcium signaling [83]. Once cytochrome c enters the cytoplasm, it nucleates the formation of a structure termed the apoptosome (described in detail below).
SMAC, OMI, and AIF are also pro-apoptotic mitochondrial intermembrane proteins that are released upon MOMP. SMAC and OMI promote apoptosis by inhibiting the IAP proteins discussed in the caspase section above. SMAC inhibits IAP proteins by physical interaction via its amino-terminal region [84], whereas OMI irreversibly cleaves IAP proteins [85]. AIF mediates caspase-independent death when liberated from mitochondria by calpain-mediated cleavage [86]. Once in the cytoplasm, its nuclear localization signal directs AIF to the nucleus where it condenses chromatin and fragments DNA [87].
3.4.3 APAF-1 Apoptosome
Upon its release from the mitochondria, cytochrome c binds a soluble protein called apoptotic protease-activating factor 1 (APAF-1). APAF-1, which is the mammalian homolog of C. elegans’ CED-4 [88], is present in the cytoplasm and forms a heptameric [89], wheel-shaped signaling platform [90] upon binding cytochrome c (Fig. 3.3). APAF-1 is able to form this soluble receptor because it is a tripartite protein containing an N-terminal CARD domain, a nucleotide-binding and oligomerization domain (NOD) with ATPase activity, and C-terminal WD40 repeats [88]. Under steady-state, non-apoptotic conditions, APAF-1 is present in the cytoplasm as an autoinhibited monomer bound to dATP. Once cytochrome c binds the WD40 repeats of APAF-1, the NOD ATPase hydrolyzes dATP to dADP to allow for an initial conformational change [91]. However, this is not sufficient for apoptosome assembly, and studies have shown that the dADP must be exchanged for dATP to oligomerize APAF-1 [92]. Once oligomerized, the CARD domain of APAF-1 becomes accessible and enables recruitment of pro-caspase 9 through CARD:CARD interactions. As described for the extrinsic pathway above, oligomerization of the pro-caspase zymogen enables proteolytic processing that produces active caspase 9 [93]. The effector caspases 3, 6, and/or 7 are subsequently activated through caspase 9-mediated cleavage to proteolyze cell death substrates.
3.4.4 Cytokine Withdrawal-Induced Death
For T cell biology, the most appreciated inducer of intrinsic apoptosis is cytokine withdrawal. At the conclusion of an immune response to infection, the expanded T cell populations must contract to maintain homeostasis of T cell numbers. This vital contraction is mediated largely by cytokine withdrawal-induced death (CWID) [10, 94]. CWID is primarily understood in the context of IL-2 deprivation but can also occur when other common gamma chain cytokines (e.g., IL-4, IL-7) are abruptly removed from the environment [95]. The pro-apoptotic, BH3-only protein BIM is a key mediator of CWID [96, 97] whose expression is tightly controlled by signals downstream of cytokine receptors. When key cytokines are withdrawn, T cells sense this as growth factor deprivation, resulting in upregulation and stabilization of bim mRNA as well as reduced turnover of the BIM protein [98, 99]. Moreover, the pro-apoptotic protein PUMA is also upregulated upon cytokine withdrawal and functions synergistically with BIM, though it can mediate BIM-independent CWID under certain conditions [100]. Finally, degradation of the anti-apoptotic MCL-1 protein also contributes to cell death induced by withdrawal of growth cytokines [101]. These events collectively result in a shift in the balance between anti- and pro-apoptotic BCL-2 family members, tilting in favor of cell death through MOMP and apoptosome formation. In patients with defective CWID, abnormally high numbers of lymphocytes accumulate as a result of impaired T cell contraction (Fig. 3.4) [102].
3.5 Diseases of Failed Lymphocyte Apoptosis
The importance of homeostatic control of lymphocyte cell numbers in health and disease has become evident through identification and characterization of patients with failed lymphocyte apoptosis (Fig. 3.4).
3.5.1 Autoimmune Lymphoproliferative Syndrome
Autoimmune lymphoproliferative syndrome (ALPS) is a clinical condition in which RICD fails to occur due to mutations in genes critical for homeostatic control of T lymphocytes through FAS receptor signaling. An enigmatic population of mature double-negative (CD4−CD8−) T (DNT) cells accumulates in ALPS patients and is used as a diagnostic indicator [103], though the nature of the DNT cells is still far from completely understood. Massive accumulation of T cells in the lymph nodes and spleen occurs due to disruption of the canonical FAS pathway [104] and results in autoimmune cytopenias. Mutations in FAS, FASL, and caspase 10 are responsible for the large majority of ALPS disease cases [103]. Mutations in the FAS receptor have been found that can be classified as homozygous, heterozygous, or somatic, where heterozygous, dominant-interfering mutations are most common followed by heterozygous somatic (acquired instead of inherited) mutations [103]. Notably, in somatic ALPS, mutations in the FAS gene are found only in the DNT cells, emphasizing their connection with disease pathogenesis. Dominant interference of FAS signaling by the presence of mutated FAS within a pool of wild-type FAS is caused by poisoning of the trimeric receptor complex with a signaling-incompetent chain [38]. The small subset of patients with an ALPS-type presentation who have been screened for but lack mutations in known disease genes are categorized as ALPS-unknown patients. There are also family members of affected ALPS patients who carry a mutation in the disease-causing gene and have in vitro defects in RICD but do not display clinical manifestations of ALPS. These individuals are referred to as healthy mutation-positive relatives (HMPR), and their resistance to disease highlights the importance of genetic modifiers and background genes in disease penetration [105].
Other unique diseases related to ALPS have also been discovered. Patients with mutated caspase 8 have a distinct disease termed caspase eight deficiency state (CEDS), which is characterized by immunodeficiency, lymphoaccumulation, and autoimmunity [28] due to the role of caspase 8 in non-apoptotic signaling pathways. Another subcategory of ALPS-like disease, RAS-associated ALPS-like disease (RALD), is caused by somatic, activating mutations in NRAS [102] or KRAS [106, 107] in hematopoietic cells. RALD patients exhibit defective CWID [102] and RICD [76] due to reduced levels of the pro-apoptotic BIM protein caused by exuberant protein degradation and mRNA instability caused by RAS signaling. Interestingly, the same genes mutated in RALD are also mutated in a subpopulation of pediatric patients with the severe myelodysplastic/myeloproliferative disease juvenile myelomonocytic leukemia (JMML) and a subpopulation (approximately 35 %) of adult patients with chronic myelomonocytic leukemia (CMML) [108]. However, the expanded monocyte population in these cancers is monoclonal, while in RALD the accumulating cells carrying the mutated NRAS or KRAS are of multiple different hematopoietic lineages and, therefore, likely occurred in an early progenitor cell that has not yet undergone malignant transformation [108].
3.5.2 X-Linked Lymphoproliferative Disease-1
A more rare lymphoproliferative disease of failed lymphocyte apoptosis is X-linked lymphoproliferative disease-1 (XLP1), which is caused by deficiency of the SH2-containing adapter protein (SH2D1A/SAP) [109]. XLP1 patients are immunodeficient and exhibit spontaneous lymphoproliferation and fulminant, often fatal, infectious mononucleosis after infection by the B cell-tropic Epstein–Barr virus (EBV). EBV infection in XLP1 patients is also often associated with secondary hemophagocytic lymphohistiocytosis (HLH) in which a pro-inflammatory state induces macrophage expansion and phagocytosis of other blood cells. The SAP adaptor binds to SLAM family members as well as TCR signaling components [110], making it an important player in the adhesion and signaling events required for robust T cell activation [111]. These defects have recently been found to be the cause for lethal CD8 T cell accumulation in response to EBV infection since T cells from SAP-deficient XLP1 patients fail to form robust T cell:B cell conjugates or transduce a strong enough TCR signal to pass the threshold required for RICD [112].
3.6 Concluding Remarks
Programmed cell death is a critical regulator of T cell homeostasis that is required to prevent pathological accumulation of lymphocytes. The study of genetic immunological diseases has yielded fascinating insight into the basic biology of programmed cell death. Moreover, the study of human pathways has revealed clear differences and additional complexity compared with C. elegans or even other mammals. The continued exploration of lymphocyte homeostasis through the lens of human genetics promises to yield additional medically important insights.
References
Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2:545–50.
Check E. Worm cast in starring role for Nobel prize. Nature. 2002;419:548–9.
Marx J. Nobel prize in physiology or medicine. Tiny worm takes a star turn. Science. 2002;298(526).
Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–2.
McDonnell TJ, et al. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88.
Snow AL, Lenardo MJ. Programmed cell death. In: Paul WE, editor. Fundamental immunology. Philadelphia, PA: Lippincott Williams & Wilkins; 2012. pp 718–40.
Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–29.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Cohen JJ, Duke RC, Fadok VA, Sellins KS. Apoptosis and programmed cell death in immunity. Annu Rev Immunol. 1992;10:267–93.
Lenardo M, et al. Mature T lymphocyte apoptosis–immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–53.
Yuan S, et al. Characterization of the extrinsic apoptotic pathway in the basal chordate amphioxus. Sci Signal. 2010;3:ra66.
Bajoghli B, et al. Evolution of genetic networks underlying the emergence of thymopoiesis in vertebrates. Cell. 2009;138:186–97.
Han J, Zhong CQ, Zhang DW. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat Immunol. 2011;12:1143–9.
Yu L, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2.
Freundt EC, et al. The open reading frame 3a protein of severe acute respiratory syndrome-associated coronavirus promotes membrane rearrangement and cell death. J Virol. 2010;84:1097–109.
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–41.
Kersse K, Verspurten J, Vanden Berghe T, Vandenabeele P. The death-fold superfamily of homotypic interaction motifs. Trends Biochem Sci. 2011;36:541–52.
Lavrik I, et al. The active caspase-8 heterotetramer is formed at the CD95 DISC. Cell Death Differ. 2003;10:144–5.
Janssens S, Tinel A. The PIDDosome. DNA-damage-induced apoptosis and beyond. Cell Death Differ. 2012;19:13–20.
Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ. 2007;14:641–50.
Deveraux QL, et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–23.
Deveraux QL, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 1997;388:300–4.
Deveraux QL, et al. Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 1999;18:5242–51.
Kaufmann T, Strasser A, Jost PJ. Fas death receptor signalling: roles of Bid and XIAP. Cell Death Differ. 2012;19:42–50.
Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42.
Verhagen AM, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53.
Suzuki Y, et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–21.
Chun HJ, et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–9.
Su H, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–8.
Chan FK, et al. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–4.
Schneider P, et al. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med. 1998;187:1205–13.
Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23:1625–37.
Migone TS, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–92.
Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–9.
Chinnaiyan AM, et al. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science. 1996;274:990–2.
Liu J, et al. Enhanced CD4+ T cell proliferation and Th2 cytokine production in DR6-deficient mice. Immunity. 2001;15:23–34.
Zhao H, et al. Impaired c-Jun amino terminal kinase activity and T cell differentiation in death receptor 6-deficient mice. J Exp Med. 2001;194:1441–8.
Siegel RM, et al. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–7.
Siegel RM, et al. SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol. 2004;167:735–44.
Wang L, et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol. 2010;17:1324–9.
Krammer PH, Arnold R, Lavrik IN. Life and death in peripheral T cells. Nat Rev Immunol. 2007;7:532–42.
Kaufmann T, et al. The BH3-only protein bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–33.
McKenzie MD, et al. Proapoptotic BH3-only protein Bid is essential for death receptor-induced apoptosis of pancreatic beta-cells. Diabetes. 2008;57:1284–92.
Yin XM, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–91.
Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90.
Jost PJ, et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature. 2009;460:1035–9.
Algeciras-Schimnich A, Griffith TS, Lynch DH, Paya CV. Cell cycle-dependent regulation of FLIP levels and susceptibility to Fas-mediated apoptosis. J Immunol. 1999;162:5205–11.
Bentele M, et al. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J Cell Biol. 2004;166:839–51.
Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity. 1998;8:615–23.
Schmitz I, et al. Resistance of short term activated T cells to CD95-mediated apoptosis correlates with de novo protein synthesis of c-FLIPshort. J Immunol. 2004;172:2194–200.
Irmler M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–5.
Ozturk S, Schleich K, Lavrik IN. Cellular FLICE-like inhibitory proteins (c-FLIPs): fine-tuners of life and death decisions. Exp Cell Res. 2012;318:1324–31.
Sun H, et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133:415–26.
Condorelli G, et al. PED/PEA-15: an anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene. 1999;18:4409–15.
Zheng L, et al. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995;377:348–51.
Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308.
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90.
Schneider-Brachert W, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–28.
Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23.
He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11.
Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7:243–9.
Jiang Y, Woronicz JD, Liu W, Goeddel DV. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science. 1999;283:543–6.
Peschon JJ, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160:943–52.
Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65.
Pan G, et al. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8.
Clancy L, et al. Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc Natl Acad Sci U S A. 2005;102:18099–104.
Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene. 2010;29:4752–65.
Wagner KW, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med. 2007;13:1070–7.
Jin Z, et al. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137:721–35.
Finnberg N, Klein-Szanto AJ, El-Deiry WS. TRAIL-R deficiency in mice promotes susceptibility to chronic inflammation and tumorigenesis. J Clin Invest. 2008;118:111–23.
Janssen EM, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434:88–93.
Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 1991;353:858–61.
Hornung F, Zheng L, Lenardo MJ. Maintenance of clonotype specificity in CD95/Apo-1/Fas-mediated apoptosis of mature T lymphocytes. J Immunol. 1997;159:3816–22.
Takahashi T, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–76.
Snow AL, et al. Critical role for BIM in T cell receptor restimulation-induced death. Biol Direct. 2008;3:34.
Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7.
Wong B, Arron J, Choi Y. T cell receptor signals enhance susceptibility to Fas-mediated apoptosis. J Exp Med. 1997;186:1939–44.
Boehme SA, Lenardo MJ. Propriocidal apoptosis of mature T lymphocytes occurs at S phase of the cell cycle. Eur J Immunol. 1993;23:1552–60.
Garcia-Saez AJ. The secrets of the Bcl-2 family. Cell Death Differ. 2012;19:1733–40.
Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101.
Boehning D, et al. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–61.
Rong YP, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A. 2009;106:14397–402.
Wu G, et al. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–12.
Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003;17:1487–96.
Polster BM, Basanez G, Etxebarria A, Hardwick JM, Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–54.
Marchetti P, et al. The novel retinoid 6-[3-(1-adamantyl)-4-hydroxyphenyl]-2-naphtalene carboxylic acid can trigger apoptosis through a mitochondrial pathway independent of the nucleus. Cancer Res. 1999;59:6257–66.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13.
Yu X, et al. A structure of the human apoptosome at 12.8 A resolution provides insights into this cell death platform. Structure. 2005;13:1725–35.
Acehan D, et al. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–32.
Reubold TF, Wohlgemuth S, Eschenburg S. Crystal structure of full-length Apaf-1: how the death signal is relayed in the mitochondrial pathway of apoptosis. Structure. 2011;19:1074–83.
Reubold TF, Wohlgemuth S, Eschenburg S. A new model for the transition of APAF-1 from inactive monomer to caspase-activating apoptosome. J Biol Chem. 2009;284:32717–24.
Pop C, Timmer J, Sperandio S, Salvesen GS. The apoptosome activates caspase-9 by dimerization. Mol Cell. 2006;22:269–75.
Razvi ES, Jiang Z, Woda BA, Welsh RM. Lymphocyte apoptosis during the silencing of the immune response to acute viral infections in normal, lpr, and Bcl-2-transgenic mice. Am J Pathol. 1995;147:79–91.
Strasser A. The role of BH3-only proteins in the immune system. Nat Rev Immunol. 2005;5:189–200.
Hildeman DA, et al. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16:759–67.
Pellegrini M, Belz G, Bouillet P, Strasser A. Shutdown of an acute T cell immune response to viral infection is mediated by the proapoptotic Bcl-2 homology 3-only protein Bim. Proc Natl Acad Sci U S A. 2003;100:14175–80.
Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ. Activation of the ERK1/2 signaling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003;278:18811–6.
Matsui H, Asou H, Inaba T. Cytokines direct the regulation of Bim mRNA stability by heat-shock cognate protein 70. Mol Cell. 2007;25:99–112.
Bauer A, et al. The NF-kappaB regulator Bcl-3 and the BH3-only proteins Bim and Puma control the death of activated T cells. Proc Natl Acad Sci U S A. 2006;103:10979–84.
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell. 2006;21:749–60.
Oliveira JB, et al. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 2007;104:8953–8.
Oliveira JB, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116:e35–40.
Fisher GH, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–46.
Jackson CE, et al. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64:1002–14.
Niemela JE, et al. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117:2883–6.
Takagi M, et al. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2011;117:2887–90.
Shannon K, Li Q. Oncogenic Ras scales the ALPS. Blood. 2011;117:2747–8.
Pachlopnik Schmid J, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 2011;117:1522–9.
Proust R, Bertoglio J, Gesbert F. The Adaptor Protein SAP Directly Associates with CD3zeta Chain and Regulates T Cell Receptor Signaling. PLoS One. 2012;7:e43200.
Nichols KE, Ma CS, Cannons JL, Schwartzberg PL, Tangye SG. Molecular and cellular pathogenesis of X-linked lymphoproliferative disease. Immunol Rev. 2005;203:180–99.
Snow AL, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest. 2009;119:2976–89.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Lucas, C.L., Lenardo, M.J. (2014). Molecular Basis of Cell Death Programs in Mature T Cell Homeostasis. In: Wu, H. (eds) Cell Death. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9302-0_3
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9302-0_3
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9301-3
Online ISBN: 978-1-4614-9302-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)