
Abstract
The pharmacodynamics of anti-cancer agents, as opposed to direct acting medications, pose a unique set of challenges. These challenges include; delayed response, inadequate number of response measurements, and limited set of biomarkers for effect or toxicity. However these challenges can be overcome and more advancements have allowed for limiting the aforementioned challenges. Pharmacodynamic assessment in early stage oncology clinical requires a study design optimized not only for the determination of clinical response and toxicity, but also for optimal measure of pharmacokinetic and pharmacodynamic measures. Pharmacodynamic models, study design, and pharmacodynamic biomarkers for anti-cancer clinical trials are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Pharmacodynamics is the mathematical relationship between a drugs concentrations and its pharmacological and clinical response. The goal of pharmacodynamic modeling is to allow prediction of an individual patient’s clinical outcome(s) based on the dosage regimen administered and that patient’s specific clinical response given the known pharmacokinetic behavior of the medication prescribed. However, the term pharmacodynamics is often misused to show the response to the drug based on a time course or a dose administered in lieu of pharmacokinetics. Optimal clinical trial design, accurate pharmacokinetic parameter estimation, and appropriate pharmacodynamic analysis are required to meet this goal.
Drug concentrations used for pharmacodynamic analysis are typically plasma or serum concentrations. However, concentrations may be measured at other available sites such as cerebrospinal fluid, ascites fluid, or other easily accessible tissues or fluids. Pharmacokinetic modeling allows prediction of plasma drug concentrations following dose administration. However, application of a pharmacodynamic model alone or in combination with a pharmacokinetic model allows determination of drug response at a specified concentration, or based on a specified measure of drug exposure at any time after the administered dose. With this knowledge, the optimal dosage regimen of the drug to be given to a specific patient or patient population can be determined and therapy can be monitored effectively.
The pharmacodynamic study of anticancer agents is beset with several pitfalls and hurdles making the analysis more difficult. These include narrow therapeutic index, delayed response to therapy, difficult to measure direct effect markers and biomarkers, difficult to extrapolate exposure parameters to the site of action, small sample size, and heterogeneous patient populations within early phase clinical trials. The therapeutic index is defined as the ratio of the concentration causing severe toxicity to the concentration at which the drug exhibits the desired pharmacological effect. The therapeutic index for cytotoxic drugs is very narrow or even nonexistent. Anticancer agents typically cause severe and potentially life-threatening toxicities prior to reaching the concentration required for complete remission or “cure.” The desired effect of “cure” based on tumor cell kill is not easily measured and is often delayed. For drugs exhibiting an immediate response, an effect can theoretically be observed simultaneously with drug concentrations. However, simultaneous measurement of cell kill based on imaging is a time-dependent process, often taking weeks to detect a change in tumor mass. The lack of a simultaneous and easily measured direct effect, such as tumor burden in many cancers, also limits pharmacodynamic modeling for classical cytotoxic agents. Although for some solid tumors CT scan is an effective measure of response, the measure of tumor shrinkage is delayed and may cause unnecessary continuation of therapy for a drug that might be under dosed or inactive in a particular patient. An alternative measure of pharmacodynamic response would be a well-validated biomarker. Although the search for reliable and easily measured biomarkers is underway, one main hurdle for identifying reliable biomarkers remains that each drug class and tumor type may possess a different “ideal” biomarker. Moreover, in cancer research, the correct exposure marker may be difficult to identify. Traditional direct response models can reliably predict clinical or pharmacological effect by modeling the measured effect versus the measured or predicted plasma concentration. However, several well-established models for anticancer agents have shown that atypical exposure measures such as time above a threshold concentration, maximum concentration (C max), or area under the concentration versus time curve ( AUC) are more applicable [1, 2]. Finally, a major drawback of reliable pharmacodynamic modeling for anticancer agents is due to lack of an ideal clinical trial design. Phase I studies for anticancer agents using healthy volunteers are neither feasible nor would this population exhibit the appropriate pharmacodynamic response. Early phase clinical trials for anticancer drugs employ a small and heterogeneous population and often a heterogeneous population for phase II investigation. Together these drawbacks have hampered the field of cancer chemotherapy pharmacodynamics and limited the usefulness of the results obtained to truly optimize therapy by predicting appropriate response based on drug pharmacokinetics. However, recent advances in clinical trial design and simulation, availability of more practical and accurate measures of tumor response, and identification of viable biomarkers are paving the way to allow optimal pharmacodynamic modeling for cancer therapy.
Novel therapies targeting tumor angiogenesis, invasion, metastases, or signal transduction present the additional challenge that they are not meant to directly kill the tumor cell but alter the biological process which can consequently enable tumor survival. Thus, current studies of anticancer agents are identifying specific pharmacological biomarkers to assess pharmacodynamics. The goal of anticancer drug pharmacodynamic modeling is to optimize the dose such that maximal benefit can be obtained with minimal toxicity. Currently drugs used to treat cancer are most often dosed on body surface area (mg/m2), weight basis (mg/kg), or as a flat-fixed dose (mg). Clearly, this is not the safest and most effective way to dose cancer therapies. Clinical investigation of anticancer agents has shown that pharmacokinetically and pharmacodynamically guided dosing are feasible strategies [3–7]. These methods would represent an improvement if they were clinically possible and cost effective. Thus, guiding the regimen of an anticancer agent based on its individual pharmacodynamic parameters would represent the most efficient means to dose these therapies [8].
2 Classical Pharmacodynamic Models
2.1 Individual Pharmacodynamic Models
Most clinical pharmacodynamic studies of anticancer therapies approach data analysis in a non-modeling fashion. One typical approach utilizes segregation of patients by response and statistical comparison of the difference in mean or median systemic exposure (C max, or AUC) values (Fig. 1). Initial evaluation of an effect can often most efficiently be conducted by investigation of pharmacokinetics within groups with the largest differences in outcomes. For instance, it may be easier to see differences in the pharmacokinetics of a potential hepatotoxin when the assessed population contains groups of patients who experience no hepatic toxicity or extreme toxicity as compared to a population of patients with all degrees of toxicity. Obviously, later studies will need to determine the feasibility of identifying patients who will subsequently have only mild toxicity compared to morbid effects.

Example of a pharmacodynamic segregation analysis. Patients were given a potentially nephrotoxic chemotherapeutic agent by continuous infusion over 3 days. Renal toxicity was assessed as a change from baseline. Daily, steady-state concentrations were segregated into groups based on the presence (tox) or absence (nl) of postexposure nephrotoxicity. Reproduced from Ref. W. D. Figg and H. L. McLeod. Handbook of Anticancer Pharmacokinetics and Pharmacodynamics. Humana Press (edition 1), Totowa, N., 2004.)
Given the limited nature of the phase I studies and the discontinuity of some of the pharmacodynamic measures, the statistical approach may be the only practical method of pharmacodynamic assessment. Comparisons between pharmacological response value and drug concentration can be modeled using a cumulative linear logistic (logit) model. For pharmacological response parameters described best by a binary function [i.e., response (+) or no response (+)], a simple logit model or a Wilcoxon Rank-Sum analysis may be the most appropriate method to assess differences in response based on exposure. A cumulative logit model analysis might be performed to assess the association between level of clinical response and pharmacokinetic exposure parameter. The ordinal data parameters of treatment response categorized as complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD) as previously defined would be used for this analysis. Similarly a cumulative logit model might be used to assess the effect of plasma concentrations on the ordinal parameters of toxicity grade (NCI grade I through IV). If the toxicity data are better described by a binary function, a simple logit model can be used to assess the toxicity as a function of changed pharmacological parameter.
Data measures using a continuous variable may employ comparison of two or more groups using a paired t-test or analysis of variance (ANOVA) assuming the data meet the criteria for parametric testing. An alternative method places patients into discrete groups based on their degree of systemic exposure (e.g., mean or median value of exposure) and then compares any difference in the pharmacodynamic response amplitude or duration between the groups (Fig. 2).

Example of a pharmacokinetic segregation analysis. Kaplan–Meier survival analysis of patients who were segregated based on the median population systemic exposure to a prodrug (lower values = more exposure to active moiety). Reproduced from Ref. W. D. Figg and H. L. McLeod. Handbook of Anticancer Pharmacokinetics and Pharmacodynamics. Humana Press (edition 1), Totowa, N., 2004.)
Determination of an appropriate approach to model an association with systemic exposure and pharmacodynamics is often initially conceived by the outcome measures and the mechanistic basis for the effect. In situations in which one has multiple response outcomes for one patient (e.g., time-dependent change in biomarker versus changes in drug concentration), the evaluation of each patient’s data set can be done via standard two-stage approach. It is typical with oncology studies to have just one pharmacodynamic outcome for each patient (e.g., survival). In the latter setting, the approach would be to evaluate patients in a single stage approach such as grouping all the systemic exposure and survival data points into one file and evaluating at the same time.
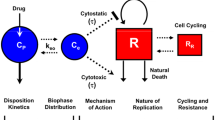
Model selection should be based on the mechanistic relationship between the drug and the pharmacodynamic measure expected. Many models used in oncology are based on receptor occupancy (Fig. 3). In these situations one expects no observable effect until a minimal exposure is achieved, followed by a nonlinear increase in effect, a pseudo-linear change in response, and a maximization of the effect, above which no significant increase in response is noted.

Examples of pharmacodynamic modeling approaches based on receptor interaction theory. Reproduced from Ref. W. D. Figg and H. L. McLeod. Handbook of Anticancer Pharmacokinetics and Pharmacodynamics. Humana Press (edition 1), Totowa, N., 2004.)
Despite the difficulties with pharmacodynamic measures for cancer therapy, the Hill equation (1) [9] and its variants have been used to describe the pharmacodynamic relationship for many oncologic drugs.
where E is the effect response, E max is the maximum effect response, C is a measure of drug exposure/concentration, EC50 is the concentration/exposure producing one-half the maximum effect, and s is the Hill constant.
These relationships can take the form of direct or indirect-response models and can relate various drug exposure parameters, such as discrete concentration at the time of the response (C), C max, time above a threshold concentration, or AUC, to the pharmacodynamic response variable. Pharmacodynamic response entails changes in circulating plasma proteins involved in tumor growth and metastases, toxicity measurements, radiologic response, or clinical response. Another feasible method employs comparisons between pharmacological response value and drug concentrations obtained from the various dose or exposure groups using a regression analysis to assess validity.
Although it may be relatively easy to identify a relationship between pharmacokinetics and pharmacodynamics, the strength of the association may not be sufficient to justify a therapeutic target without further analysis and data interpretation.
2.2 Pharmacodynamic Model Assessment
Arguably, one of the most appropriate techniques to assess a model’s utility is the evaluation of bias and precision [10]. The bias can be easily calculated as the mean prediction error (me) (2), and the precision can be measured as the root-mean-squared prediction error (rmse) (3). Determination of these parameters will enable accurate assessment of how relevant the model will be for future clinical use.
where me is the mean prediction error, rmse is the root-mean-squared prediction error, and pe is the error of the predicted pharmacodynamic parameter.
2.3 Sampling and Measurement
Pharmacokinetic studies are typically designed with rigor to ensure adequate evaluation of patient-specific parameters such as systemic exposure. Such detail is typically not applied to many pharmacodynamic endpoints; thus their accuracy is not as well controlled. The reasons for this vary from lack of attention to adequate evaluation of such variability to dealing with logistical issues. A good example is modeling of pharmacokinetic data with the pharmacodynamic endpoint of chemotherapy-induced myelosuppression. Routine clinical practice would entail weekly WBC monitoring for many drugs. However, in order to establish a good association between the nadir WBC and chemotherapy systemic exposure, one may need to monitor the WBC at least several times per week or ideally once daily.
Routine lab tests conducted by accredited clinical labs have sufficient quality control such that their data could be utilized in pharmacodynamic studies. However, tests conducted for research purposes on pharmacodynamic endpoints may not have sufficiently stringent controls or acceptable variability (e.g., <15 % (20 % at limit of quantitation) is acceptable for the analytical range of drug concentrations) in order to conduct pharmacodynamic modeling. This could be due to inexperience of the lab conducting the test or the nature of the test itself.
The accurate assessment of effect is essential to the success of a pharmacodynamic model. This is best accomplished if the measure of effect is made at the effect site. These measures can safely be performed for many diseases. For example, a pharmacodynamic study of a proton pump inhibitor may include direct measurement of gastric pH by the placement of a nasogastric tube for sample collection of gastric fluids at appropriate time points. However, the assessment of antitumor effect of an antitumor agent in patients with solid tumors may require repeated tumor biopsy; the risk of tumor biopsy often outweighs the benefit. Although bone marrow biopsy is more easily performed than a solid tumor biopsy, serial samples are typically not obtained. To perform a robust pharmacodynamic study, repeated measurements would be necessary, which is not an option in patients with either solid malignancies or leukemias. Thus the clinical pharmacologist must rely on surrogate measurements for pharmacological response in patient plasma or circulating white blood cells. Changes in transcription or protein expression can be measured in peripheral lymphocytes, but the question is whether changes in normal circulating cells reflect the pharmacology at the tumor site. Preclinical correlative studies are required to validate these types of studies. In tumor xenograft models, the pharmacologist can measure changes in proteins and/or messenger RNA at the tumor site and in the circulating lymphocytes and plasma concentration determination simultaneously. If properly controlled, these preclinical studies can identify the most appropriate surrogate biomarker(s) for clinical pharmacodynamic studies.
2.4 Clinical Trial Design for Early Phase Pharmacodynamic Evaluation
Human pharmacodynamic modeling is most commonly conducted during clinical phase I trials. The reasons for this are twofold: (1) extensive pharmacokinetic data is available, and (2) the largest range of doses is explored in phase I. The doses administered in phase I studies typically vary from ones not exerting any measurable biological effect to those producing intolerable toxicity, thus providing a broad range of dose or systemic exposure versus response profile. If a relationship exists, it should be evident with such a strategy; however this is not an optimal approach to validate associations between systemic exposure and outcome. This is due to the fact that since there is almost always a direct correlation between dose and systemic exposure, associations of the latter term with pharmacodynamic measures may simply be a reflection of the dose–effect relationship. The optimal setting for evaluation of a correlation between systemic exposure and a pharmacodynamic parameter would be in situations where the dose is fixed for all patients (or normalized to body size) such as phase II and III trials. Despite this issue, pharmacokinetic–pharmacodynamic associations are important in many contemporary phase I studies since biomarkers may be used as the sole determinant of dose selection for phase II studies. Later phase studies will typically evaluate relationships between targets demonstrating usefulness on earlier studies with tumor response and/or survival.
The typical phase I trial for first-in-human anticancer agents employs a dose escalation using a modified Fibonacci scheme using a 3 + 3 enrolment strategy. Thus, at lower dose levels, it is likely that only three patients per dose level will be enrolled, leaving a small sample set from which to assess the potential pharmacodynamic response. Often times the sampling scheme is established based on the pharmacokinetic parameters determined in rodents from preclinical toxicology and pharmacology studies. Assumptions are often made that the drug will behave in a similar manner. However it is infrequent that an allometric approach is used to scale up the dose selection and sampling scheme based on scaled clearance and volume of distribution. Allometric scaling could be used to predict the human clearance, volume of distribution, and thus drug exposure (Fig. 4) [11, 12]. By using a typical scale up using only one species, one could predict human clearance and volume of distribution as

Example of allometric scaling of benzoylphenylurea clearance. Where a allometric coefficient; W weight, CL clearance, and b allometric exponent (Unpublished data, Bauer KS 2002)
where Y is the human PK parameter of interest, a is the allometric scaling coefficient (log a is the y-intercept), W is the body weight (average human body weight), and b is the allometric exponent (b is the slope). a and b are derived by log–log regression of the known species PK parameter versus species weight (Fig. 4).
Studies have shown that the starting dose in humans can be determined based on a single species, assuming a b of 0.67 using the FDA guidance “Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers” [13] using the following approach:
where HED is the human equivalent dose, NOAEL is the no observed adverse effect dose in the most sensitive species, W a is the average weight of the most sensitive species, W h is the human weight, and (1–b) is the allometric slope factor (1–0.67), or 0.33.
Predicted pharmacokinetic parameters (e.g., clearance and volume) and a more rigorous sampling scheme could be employed to accurately define the pharmacokinetic parameters in human. Likewise, information obtained from preclinical pharmacological and biomarker studies could be used to predict an optimal sampling scheme for the identified markers of pharmacodynamic effect. Even by using these strategies, one cannot always obtain a reasonable prediction for human PK as demonstrated in the first-in-human studies with UCN-01 [14]. Thus, it is recommended that first-in-human studies use a real-time pharmacokinetic evaluation plan such that the drug concentrations and PK parameters can be determined following each dose escalation and allow for protocol update to most accurately study the pharmacokinetic and pharmacodynamic of anticancer agents in this setting. An alternate approach to human pharmacokinetic parameter determination uses a micro-dosing technique expected to have no toxicologic effect on the subject while allowing for determination of pharmacokinetic parameters in humans prior to initiating the phase I trial. This type of clinical assessment is now termed a phase 0 clinical trial (see Phase 0 Trials in Oncology Chapter).
3 Population PK/PD Models
3.1 Population Models
Later phase evaluation of pharmacokinetic–pharmacodynamic effects may employ sparse pharmacokinetic sampling approaches. These could entail utilization of Bayesian algorithms or traditional population-based models such as those employed by NONMEM. There are several contemporary large studies that have utilized this strategy to identify potentially important physiologic markers which are associated with inter-patient variability in pharmacokinetics. The typical approach is to follow up this type of analysis with a study which then validates the relationships using traditional full assessments of pharmacokinetics and pharmacodynamics. For more information on population models, see the Pharmacometrics: The Science and Application in Oncology Drug Development Chapter.
4 Classical Pharmacodynamic Measures
If the ultimate goal of pharmacodynamic study is to optimize the drug regimen, then the primary markers of effect representing this endpoint for anticancer agents would be absolute tumor burden. However, this is very difficult, if not impossible. Using categorical measures of toxicity and response, establishing easily obtainable continuous measures for toxicity and identifying surrogate markers and biomarkers for effect are the only measurements that can be performed. The type of data obtained from the effect measures may be continuous, scalar, categorical, or discrete/binomial variables. The type of variable plays a major role in the choice of pharmacodynamic model that can be used. Thus, for any anticancer agent being studied, the investigator must determine the best choice of endpoint to be measured based on sampling feasibility, site of sample acquisition, availability of resources for measurement determination, cost, and, ultimately, how well it relates with actual clinical response.
The typically used categorical measures for toxicity are CR, PR, SD, and PD. An additional parameter of minor response (MR) may also be used on occasion. Clinical toxicity is a scalar variable with a value of 1–5 based on a predetermined grading scheme such as the NCI common toxicity criteria. Measures of toxicity are often a simple measure, while surrogate biomarkers must take into account the mechanism of action of the agent. As previously mentioned, one of the challenges in the study of pharmacodynamics of cancer therapy is the delayed measurable effect of decreased tumor burden (e.g., tumor shrinkage) or measurable toxicity (e.g., decreased blood cell counts). For classical cytotoxic chemotherapies, several relatively noninvasive easily measured indices have been established (Table 1). However, molecularly targeted agents, which act on specific signal transduction pathways or cellular and molecular processes, require more complex measures to determine the early effects of the drug in the clinical setting. Since many of these new drugs are not directly cytotoxic and target aberrant cellular processes and pathways, they may not exhibit the hematologic toxicity seen with the classical cancer chemotherapies. However, they are not without side effects; nonetheless, identifying an easily obtainable marker for effect may present more of a challenge (Table 2). For agents targeting tumor angiogenesis, markers of angiogenic signaling might be measured in the plasma, urine, or in biopsy tissues if available. For agents targeting specific signaling pathways, a downstream protein or event might be measured in lymphocytes, skin, or tumor tissue. One approach for monitoring agents that are designed to stimulate apoptosis is a radiolabeled annexin V product. Annexin V has been shown to bind to phosphatidylserine, an intracellular membrane-associated protein. During apoptosis phosphatidylserine is expressed on the external cell membrane. By labeling annexin V with a radio-imaging agent such as 99Tc, the rate of apoptosis can be measured [35]. This approach may allow quick, noninvasive, and direct measure of tumor response in patients with a number of solid tumors. This technology could lead to the ideal pharmacodynamic marker, a means to directly determine tumor burden in real time. In addition to the mechanism of action of the agent providing the means for determining a biomarker, the disease itself may also produce a specific biomarker for tumor burden. In the cases of prostate cancer and ovarian cancer, prostate-specific antigen (PSA) and CA-125 are commonly used as clinical biomarkers for disease progression and can also be used as pharmacodynamic measures for drug response.
Once the optimal endpoint for the agent has been selected, practical issues involving the clinical setting and sampling issues are then addressed to appropriately fit the model to the data. The endpoints for pharmacodynamic assessments are typically determined by what is suspected to be the drug’s primary target for effect. Broad classifications include receptor binding, alteration of enzymes/proteins, membrane interactions, or transport blockade. Target identification will allow for speculation as to the immediacy and duration of response. For example, if a drug is known to stimulate an alpha adrenergic receptor, an immediate effect may be anticipated, but one which may display altered response over continuous exposure time. Tachyphylaxis and hypersensitivity reactions are good examples of time-dependent pharmacodynamic effects. Perhaps the most difficult to evaluate are chronic pharmacodynamic effects of cancer therapy. Examples of such chronic effects include delayed growth effects, impaired learning, cardiac toxicity, and secondary malignancies.
It is important to realize that pathophysiologic factors may also influence the pharmacodynamic effect in an individual patient. For instance, a person who has been previously exposed to multiple cycles of myelosuppressive chemotherapy is likely to experience a greater effect from subsequent myelosuppressive agents due to depleted bone marrow reserves.
5 Pharmacodynamic Biomarkers
5.1 Oncology Biomarkers
Complexity of treatment decision-making in malignancies is in part due to intra-patient variability in response to chemotherapy and in part due to the complex and heterogeneous biology of malignancy. Without the ability to identify patients who may or may not benefit from a certain therapy, there is a high possibility of exposing the nonresponders to significant toxicity. For example, more than 50 % of patients undergoing chemotherapy for advanced and metastatic colorectal cancer do not show significant evidence of benefit. Identifying a measurable indicator (e.g., appropriate biomarker), which acts as a surrogate marker to chemotherapy response variability, allows us to decide between implementing an immediate treatment and deferring therapy and to select the appropriate chemotherapy agent(s) as well as modifying the regimen if needed [36–38]. Individualized anticancer therapy combined with biomarker-based monitoring for the efficacy of the regimen holds enormous potential for optimizing anticancer therapy and reducing cancer-related mortality and morbidity [39].
Biomarkers include an array of different modalities such as, but not limited to, physical symptoms, mutated DNAs and RNAs, processes such as cell death or proliferation, and serum or tissue concentrations of molecules or secreted proteins [36]. Although a biomarker can be a physical or physiological parameter, the term is now typically refers to molecular biomarkers [37].
Biomarkers are defined as molecules that are objectively measured and evaluated to be used as surrogate indicator of normal or disease processes as well as pharmacological responses to therapy [40]. Biomarkers can be prognostic, predictive, or surrogate. A prognostic biomarker identifies patients with differing risks of a specific outcome, while a predictive biomarker can be used to predict the response to a given therapy [41, 42]. A surrogate biomarker can be used “to substitute as an intermediate for a clinically meaningful endpoint” [42]. Predictive and surrogate biomarkers are more important in direct treatment decision-making strategies [42].
Proteins as biomarkers, measured in serum and/or tissue, can be used as indicators of the existence, progression, or recurrence of cancer. In addition to early detection of cancer, measurement of panels of protein biomarkers holds promise for personalized cancer therapy, treatment monitoring and optimization, and identifying response to therapy or progression of the disease [39, 42]. A single protein biomarker can be cancer and non-cancer specific, such as elevated prostate-specific antigen (PSA) in prostate cancer, benign prostatic hypertrophy (BPH), or prostatitis, or overexpressed in several disease states, such as interleukin 6 (IL-6), in oral, prostate, lung, multiple myeloma, and renal cell cancers [39, 43]. Therefore, a single biomarker might not offer adequate predictive value, and panels of proteins may be necessary when reliable cancer detection and monitoring is required [39]. However, detection of panels of biomarkers is complicated not only because both normal and elevated serum levels of biomarkers need to be accurately measured but also because their concentrations may vary widely in the serum [39].
A comprehensive biomarker pipeline includes six essential process components including candidate discovery, qualification, verification, research assay optimization, biomarker validation, and commercialization [37]. In summary, a widely used coherent and comprehensive process for novel cancer protein biomarker’s discovery and validation has three phases: discovery, verification, and validation [44].
5.2 Biomarker Identification
Pharmacodynamic studies investigate the molecular, biochemical, and physiological effects of the drug on the organism. Such investigations enable identifying how the drug binds and modulates its target, initiates the mechanism of action, and produces a therapeutic or secondary effect. Therefore, pharmacodynamic studies can help to determine and quantify these biological effects which, consequently, help to determine the optimal biological dose in vivo. Since a therapeutic agent can also affect the downstream signaling pathways, identifying reliable biomarkers would permit prediction of the individual response of each patient. A reliable biomarker needs to be a marker of both sensitivity and resistance to a therapeutic compound. Biomarkers are identified during preclinical in vitro or in vivo studies. Thousands of potential biomarkers can be generated by utilizing high-throughput techniques and powerful discovery and screening technologies, such as DNA microarrays and proteomic profiling of various biological sources such as tissue, proximal biological fluids, cell culture supernatants, and serum [39, 44, 45]. Several proteomic technologies have allowed significant progress in cancer biology including matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS), surface-enhanced laser desorption/ionization (SELDI), and material-enhanced laser desorption/ionization (MELDI). These are relatively simple and high-throughput techniques that analyze with high sensitivity and specificity intact proteins expressed in complex biological mixtures, such as serum, urine, and tissues [46]. The generated list of candidate proteins can then be shortened to about 50–100 molecules by applying a set of predefined criteria, involving semiquantitative assessments, multiple bioinformatic analysis, and literature search. These candidates are then moved along to verification phases, during which their discriminatory potential for differentiating cases from controls in order to diagnose cancer, predict prognosis, stratify therapy, or detect recurrence is assessed through quantitative analysis in about 50 to several hundred patients. Unfortunately, the majority of these potential candidates are rejected either because they cannot discriminate between cases and controls or because they are outperformed by other clinical biomarkers currently in use [44].
5.3 Biomarker Validation
A clinically relevant biomarker needs to be directly useful in modifying treatment algorithm. Although a host of different biomarkers have been verified, only a few hold actual clinical validity. Some of these biomarkers are discussed below. In non-small cell lung cancer (NSCLC), perfusion CT can be considered a reliable biomarker because it provides direct quantification of vascular function and consequently provides assessment of perfusion and angiogenesis in lung cancer [47]. EGFR gene copy number can also be assessed as a potential biomarker associated with survival in patients with advanced NSCLC receiving single-agent treatment with EGFR tyrosine kinase inhibitors (TKIs) [40]. Some of the prognostic and predictive markers in colorectal cancer (CRC) include carcinoembryonic antigen (CEA), lactic dehydrogenase (LDH), guanylyl cyclase C (GCC), high-frequency microsatellite instability (MSI-H), loss of heterozygosity (LOH), and thymidylate synthase (TS) [48].
Circulating tumor cells (CTCs) are considered as a biomarker to assess the severity of cancer. CTCs, “defined as nucleated cells lacking CD45 and expressing cytokeratin,” are isolated tumor cells disseminated from the tumor, e.g., breast cancer, which can be identified and measured in the peripheral blood. While CTC level does not correlate with radiographic measurable disease burden, it correlates with the extent of metastatic bone involvement and can reflect the outcome [49].
A high association has been established between expression level of class III b-tubulin and disease progression in patients with metastatic breast cancer who are receiving epirubicin/paclitaxel [50].
The only protein biomarker currently recommended by the American Cancer Society as an early cancer screening tool is PSA. The danger zone for PSA serum concentration is 4–10 ng/mL, a level indicating the possibility of early stage prostate cancer, while normal levels are typically 0.5–2 ng/mL. Late stage prostate cancer is characterized by values of 10–1,000 ng/mL [39]. However, several novel biomarkers have been investigated in castration-resistant prostate cancer including LDH, hemoglobin, Ras/Raf mutations, tubulin mutations, androgen receptor splice variants, CTCs, c-met/HGF activity, androgen synthesis precursor levels, DNA repair defects, Myc amplifications, bone turnover markers such as high urine N-telopeptide and trap-5b, p 16 levels, and ki-67 [42, 51, 52].
GCC, a brush border membrane receptor found exclusively on the epithelial cells of the small intestine and colon, is overexpressed in intestinal cancer, as well as CRC metastatic to the liver and lymph nodes. Quantitative GCC mRNA analysis in lymph nodes by RT-PCR can identify micrometastases with a sensitivity of 93 % and a specificity of 97 % in patients with resected colorectal cancer [41].
Presence of high-frequency microsatellite instability (MSI-H), a phenotype characterized by impaired DNA mismatch repair, is associated with improved survival for patients with MSI-H tumors [41].
CA 19-9 is the most sensitive and specific biomarker for pancreatic cancer. It does not hold any value for diagnosis; however, higher preoperative CA 19-9 levels correlate with lower resectability, more advanced stage, and lower survival [41].
5.4 Validated Cancer Chemotherapy Biomarkers
Upon identification and verification, candidate biomarkers must be evaluated and validated in order to determine their real utility [45]. Usually 2–5 proteins are moved forward into the final phases of clinical validation, in which they are tested by means of established quantitative assays with high analytical sensitivity and specificity in a large cohort of clinically relevant samples (several hundred to thousands of samples per group), collected either retrospectively or prospectively [44]. Ideally, a cancer biomarker’s “repeatability, robustness, and accuracy (sensitivity, specificity)” must be validated through analytical measures while considering the “performance characteristics of the test itself, storage conditions, stability, inter- and intra-patient variability (signal to noise), and internal and external validity” in several phase three clinical trials [42]. A fully validated cancer biomarker must have high clinical specificity and sensitivity (e.g., >90 %) to avoid false positives and false negatives which are crucial parameters in avoiding misdiagnosis [39]. A validated biomarker allows confirmation of the pharmacological and biological mechanism of action in patients, contributes to optimal biological dose selection, identifies the best schedule of administration, or minimizes the secondary effects [45]. Furthermore, by permitting prediction of efficacy and safety of a specific compound, biomarkers provide a platform to optimize pharmacological development, to increase the confidence in each step of the validation, and to contribute to decision-making strategies in the final approval of a drug [45]. Immunohistochemistry techniques are important in biomarker validation in cancer tissues [39]. Serial tumor samples or indirect biological samples, such as peripheral blood or mucous or skin biopsies, can be analyzed by PD biomarker analysis involving Western blot or ELISA-based assays to determine the biological effect and to validate their equivalence [53].
Plasma provides a source for many biomarkers such as CTCs, proteins, and metabolites; however, it might have low sensitivity and measurements can be compromised by variability in sample collection as well as handling and storage. Normal tissue surrogates, such as hair follicles, can also be considered; however, they may not exhibit the targeted pathway expressed in tumor cells [45]. Noninvasive imaging endpoints offer great advantage in this regard for assessing PD markers of drug activity. And different imaging techniques can be used to measure blood volume, blood flow, and several semiquantitative and quantitative kinetic hemodynamic parameters. Such techniques include CT, MRI, PET, single-photon emission computed tomography (SPECT), ultrasound, and near-infrared optical imaging [55].
Functional imaging techniques can also be used in measuring biological effect(s). These techniques are based on the noninvasive monitoring of target lesions by procedures such as CT, PET, or MRI [45]. For example, to monitor pathophysiologic changes in tumor vascular structure and functionality in response to antiangiogenic agents, molecular and functional imaging techniques, such as dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI), PET, and dynamic contrast-enhanced computed tomography (DCE-CT), have been developed [53]. Additionally, noninvasive imaging techniques can be used to monitor metabolism, proliferation, and cellularity of the tissue and, therefore, can be used as a method for metabolic imaging of pharmacodynamic biomarkers [54]. Moreover, perfusion computed tomography may provide prognostic and predictive pharmacodynamic information by measuring tumor perfusion and neoangiogenesis after administration of antiangiogenic agents. Currently the primary biomarker validation research has focused on early detection and prognosis of cancer; many of these biomarkers, as reasonable effect parameters, can be used for pharmacodynamic modeling to guide dosing and predict response to anticancer therapies.
6 Model Implementation
6.1 Model Implementation and Assessment
Attention to methods employed is vital in order to ensure that the parameters selected to evaluate relationships between drug exposure and pharmacodynamic response are adequately described. Typical drug exposure parameters utilized in oncology studies include total systemic exposure(AUC), maximal drug concentration, minimal concentration (for multidose), and concentration above a target value. Preclinical data (both animal and cell cultures) can often be useful to assist in determination of the optimal parameter. Most sampling schemes will be adequately constructed to allow estimation of AUC. However, accurate determination of time above a target concentration or other exposure parameters mandate a degree of attention to the concentration versus curve profile and the sampling strategy with an emphasis on duration of sampling to assess the terminal portion of the curve.
A variety of pharmacokinetic programs are used to conduct pharmacodynamic modeling. Many programs (e.g., WinNonlin, Pharsight, Inc.; ADAPT II, BMSR, UCSF) have embedded the standard receptor-based equations in their model libraries. Pharmacodynamic modeling is often conducted based on changes from a baseline value of a measurable biomarker. Measurable physiologic biomarkers can be affected by diurnal changes, day-to-day variability, as well as a variety of other factors including diet and coadministration of other medications. It is good practice to obtain at least two baseline values for pharmacodynamic evaluations that involve continuous data of an endogenous biomarker. Initial model selection also involves determination whether the model will be based on a direct effect or an inverse (inhibitory) relationship. The process of identifying the most appropriate model involves a statistical approach similar to that used for pharmacokinetic model identification (see Pharmacokinetic Modelling Chapter for more details).
While it is relatively easy to identify relationships between pharmacokinetics and pharmacodynamics, the strength of the association is often not sufficient to justify utilization of the data for establishment of a therapeutic target.
6.2 Data Interpretation
Establishment of a relationship between pharmacokinetics and pharmacodynamics is typically used to justify investigation of therapeutic drug monitoring in the clinical setting. Simply establishing the link does not mean that individualization of doses can be applied easily and accurately. However, the data may be utilized to determine the optimal schedule of administration and provide clues toward the likely biological mechanism of action. For example, if the time of plasma exposure experienced above a potentially cytotoxic concentration relates to response better than the maximal observed concentration, it would suggest a cycle-dependent mechanism and a prolonged exposure regimen may be preferred for future studies based on these data. Thus, the pharmacodynamic assessment of anticancer agents will rely heavily on effect marker determination, data analysis, and appropriate clinical trial design.
7 Conclusion
Pharmacodynamic modeling of anticancer agents presents a unique set of challenges from measurement to model building. However, diligence has proven that appropriate models can be produced to accurately predict the pharmacological effects of these drugs at given drug exposures and times. The real remaining challenge is the development of simple and cost-effective means to use these models to individualize cancer therapies to achieve maximum benefit and minimal toxicity.
References
Evans WE, Relling MV, Rodman JH, Crom WR (1989) Anticancer therapy as a pediatric pharmacodynamic paradigm. Dev Pharmacol Ther 13(2–4):85–95
Gianni L, Kearns CM, Giani A, Capri G, Viganó L, Lacatelli A, Bonadonna G, Egorin MJ (1995) Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol 13(1):180–90
Figg WD, Stevens JA, Cooper MR (1994) Adaptive control with feedback of suramin using intermittent infusions. J Clin Oncol 12(7):1523–4
Lowis SP, Price L, Pearson AD, Newell DR, Cole M (1998) A study of the feasibility and accuracy of pharmacokinetically guided etoposide dosing in children. Br J Cancer 77(12):2318–23
Ghazal-Aswad S, Tilby MJ, Lind M, Baily N, Sinha DP, Calvert AH, Newell DR (1999) Pharmacokinetically guided dose escalation of carboplatin in epithelial ovarian cancer: effect on drug-plasma AUC and peripheral blood drug-DNA adduct levels. Ann Oncol 10(3):329–34
Bauer KS, Figg WD, Hamilton JM, Jones EC, Premkumar A, Steinberg SM, Dyer V, Linehan WM, Pluda JM, Reed E (1999) A pharmacokinetically guided Phase II study of carboxyamidotriazole in androgen-independent prostate cancer. Clin Cancer Res 5(9):2324–9
Ratain MJ, Mick R, Janisch L, Berezin F, Schilsky RL, Vogelzang NJ, Kut M (1996) Individualized dosing of amonafide based on a pharmacodynamic model incorporating acetylator phenotype and gender. Pharmacogenetics 6(1):93–101
Burke GA, Estlin EJ, Lowis SP (1999) The role of pharmacokinetic and pharmacodynamic studies in the planning of protocols for the treatment of childhood cancer. Cancer Treat Rev 25(1):13–27
Wagner JG (1968) Kinetics of pharmacologic response I. Proposed relationships between response and drug concentration in the intact animal and man. J Theoret Biol 20:173–201
Sheiner LB, Beal SL (1981) Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 4:503–12
Boxenbaum H, Fertig JB (1984) Scaling of antipyrine intrinsic clearance of unbound drug in 15 mammalian species. Eur J Drug Metab Pharmacokinet 9(2):177–83
Mahmood I (1999) Allometric issues in drug development. J Pharm Sci 88(11):1101–6
Food and Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. July 2005. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078932.pdf, 07/06/05
Sausville EA, Lush RD, Headlee D, Smith AC, Figg WD, Arbuck SG, Senderowicz AM, Fuse E, Tanii H, Kuwabara T, Kobayashi S (1998) Clinical pharmacology of UCN-01: initial observations and comparison to preclinical models. Cancer Chemother Pharmacol 42(Suppl):S54–9
Egorin MJ, Van Echo DA, Tipping SJ, Olman EA, Whitacre MY, Thompson BW, Aisner J (1984) Pharmacokinetics and dosage reduction of cis-diammine(1,1-cyclobutanedicarboxylato) platinum in patients with impaired renal function. Cancer Res 44(11):5432–8
Newell DR, Siddik ZH, Gumbrell LA, Boxall FE, Gore ME, Smith IE, Calvert AH (1987) Plasma free platinum pharmacokinetics in patients treated with high dose carboplatin. Eur J Cancer Clin Oncol 23(9):1399–405
Wittes RE, Brescia F, Young CW, Magill GB, Golbey RB, Krakoff IH (1975) Combination chemotherapy with cis-diamminedichloroplatinum (II) and bleomycin in tumors of the head and neck. Oncology 32(5–6):202
Bruno R, Hille D, Riva A, Vivier N, ten Bokkel Huinnink WW, van Oosterom AT, Kaye SB, Verweij J, Fossella FV, Valero V, Rigas JR, Seidman AD, Chevallier B, Fumoleau P, Burris HA, Ravdin PM, Sheiner LB (1998) Population pharmacokinetics/pharmacodynamics of docetaxel in phase II studies in patients with cancer. J Clin Oncol 16(1):187–96
Minami H, Ohtsu T, Fujii H, Igarashi T, Itoh K, Uchiyama-Kokubu N, Aizawa T, Watanabe T, Uda Y, Tanigawara Y, Sasaki Y (2001) Phase I study of intravenous PSC-833 and doxorubicin: reversal of multidrug resistance. Jpn J Cancer Res 92(2):220–30
Ratain MJ, Schilsky RL, Choi KE, Guarnieri C, Grimmer D, Vogelzang NJ, Senekjian E, Liebner MA (1989) Adaptive control of etoposide administration: impact of interpatient pharmacodynamic variability. Clin Pharmacol Ther 45(3):226–33
Vokes EE, Mick R, Kies MS, Dolan ME, Malone D, Athanasiadis I, Haraf DJ, Kozloff M, Weichselbaum RR, Ratain MJ (1996) Pharmacodynamics of fluorouracil-based induction chemotherapy in advanced head and neck cancer. J Clin Oncol 14(5):1663–71
Kerbusch T, de Kraker J, Keizer HJ, van Putten JW, Groen HJ, Jansen RL, Schellens JH, Beijnen JH (2001) Clinical pharmacokinetics and pharmacodynamics of ifosfamide and its metabolites. Clin Pharmacokinet 40(1):41–62
Slichenmyer WJ, Rowinsky EK, Grochow LB, Kaufmann SH, Donehower RC (1994) Camptothecin analogues: studies from the Johns Hopkins Oncology Center. Chemother Pharmacol 34(Suppl):S53–7
Oliff A, Bleyer WA, Poplack DG (1979) Methotrexate-induced oral mucositis and salivary methotrexate concentrations. Cancer Chemother Pharmacol 2(3):225–6
Gianni L, Kearns CM, Giani A, Capri G, Vigano L, Lacatelli A, Bonadonna G, Egorin MJ (1995) Nonlinear pharmacokinetics and metabolism of paclitaxel and its pharmacokinetic/pharmacodynamic relationships in humans. J Clin Oncol 13(1):180–90
Stewart CF, Baker SD, Heideman RL, Jones D, Crom WR, Pratt CB (1994) Clinical pharmacodynamics of continuous infusion topotecan in children: systemic exposure predicts hematologic toxicity. J Clin Oncol 12(9):1946–54
Cianfrocca M, Cooley TP, Lee JY, Rudek MA, Scadden DT, Ratner L, Pluda JM, Figg WD, Krown SE, Dezube BJ (2002) Matrix metalloproteinase inhibitor COL-3 in the treatment of AIDS-related Kaposi’s sarcoma: a phase I AIDS malignancy consortium study. J Clin Oncol 20(1):153–9
Stopeck A, Sheldon M, Vahedian M, Cropp G, Gosalia R, Hannah A (2002) Results of a Phase I dose-escalating study of the antiangiogenic agent, SU5416, in patients with advanced malignancies. Clin Cancer Res 8(9):2798–805
Chow S, Patel H, Hedley DW (2001) Measurement of MAP kinase activation by flow cytometry using phospho-specific antibodies to MEK and ERK: potential for pharmacodynamic monitoring of signal transduction inhibitors. Cytometry 46(2):72–8
Lièvre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, André T, Bibeau F, Diebold MD, Rougier P, Ducreux M, Tomasic G, Emile JF, Penault-Llorca F, Laurent-Puig P (2008) KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 26(3):374–9
Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, Fidias P, Stubbs H, Engelman JA, Sequist LV, Tan W, Gandhi L, Mino-Kenudson M, Wei GC, Shreeve SM, Ratain MJ, Settleman J, Christensen JG, Haber DA, Wilner K, Salgia R, Shapiro GI, Clark JW, Iafrate AJ (2010) Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 363(18):1693–703, Erratum in: N Engl J Med. 2011 10;364(6):588
Baselga J, Rischin D, Ranson M, Calvert H, Raymond E, Kieback DG, Kaye SB, Gianni L, Harris A, Bjork T, Averbuch SD, Feyereislova A, Swaisland H, Rojo F, Albanell J (2002) Phase I safety, pharmacokinetic, and pharmacodynamic trial of ZD1839, a selective oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with five selected solid tumor types. J Clin Oncol 20(21):4292–302
Karp JE, Lancet JE, Kaufmann SH, End DW, Wright JJ, Bol K, Horak I, Tidwell ML, Liesveld J, Kottke TJ, Ange D, Buddharaju L, Gojo I, Highsmith WE, Belly RT, Hohl RJ, Rybak ME, Thibault A, Rosenblatt J (2001) Clinical and biologic activity of the farnesyltransferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: a phase 1 clinical-laboratory correlative trial. Blood 97(11):3361–9
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–16
Belhocine T, Steinmetz N, Hustinx R, Bartsch P, Jerusalem G, Seidel L, Rigo P, Green A (2002) Increased uptake of the apoptosis-imaging agent (99m)Tc recombinant human Annexin V in human tumors after one course of chemotherapy as a predictor of tumor response and patient prognosis. Clin Cancer Res 8(9):2766–74
Watson RG, McLeod HL (2011) Pharmacogenomic contribution to drug response. Cancer J 17(2):80–8
Rifai N, Gillette MA, Carr SA (2006) Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol 24(8):971–83
Bjartell A, Montironi R, Berney DM, Egevad L (2011) Tumour markers in prostate cancer II: diagnostic and prognostic cellular biomarkers. Acta Oncol 50(Suppl 1):76–84
Rusling JF, Kumar CV, Gutkind JS, Patel V (2010) Measurement of biomarker proteins for point-of-care early detection and monitoring of cancer. Analyst 135(10):2496–511
Manne U, Srivastava RG, Srivastava S (2005) Recent advances in biomarkers for cancer diagnosis and treatment. Drug Discov Today 10(14):965–76
Denlinger CS, Cohen SJ (2007) Progress in the development of prognostic and predictive markers for gastrointestinal malignancies. Curr Treat Options Oncol 8(5):339–51
Armstrong AJ, Eisenberger MA, Halabi S, Oudard S, Nanus DM, Petrylak DP, Sartor AO, Scher HI (2012) Biomarkers in the management and treatment of men with metastatic castration-resistant prostate cancer. Eur Urol 61(3):549–59
Vickers AJ, Ulmert D, Serio AM, Björk T, Scardino PT, Eastham JA, Berglund G, Lilja H (2007) The predictive value of prostate cancer biomarkers depends on age and time to diagnosis: towards a biologically-based screening strategy. Int J Cancer 121(10):2212–7
Makawita S, Diamandis EP (2010) The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: current strategies for candidate verification. Clin Chem 56(2):212–22
Rojo F, Dalmases A, Corominas JM, Albanell J (2007) Pharmacodynamics: biological activity of targeted therapies in clinical trials. Clin Transl Oncol 9(10):634–44
Bakry R, Rainer M, Huck CW, Bonn GK (2011) Protein profiling for cancer biomarker discovery using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and infrared imaging: a review. Anal Chim Acta 690(1):26–34
Quan G, Du X, Huo T, Li X, Wei Z, Cui H, Chang X, Cheng Y, Ye X, Cheng H (2010) Targeted molecular imaging of antigen OC183B2 in ovarian cancers using MR molecular probes. Acad Radiol 17(12):1468–76
Dahabreh IJ, Linardou H, Kosmidis P, Bafaloukos D, Murray S (2011) EGFR gene copy number as a predictive biomarker for patients receiving tyrosine kinase inhibitor treatment: a systematic review and meta-analysis in non-small-cell lung cancer. Ann Oncol 22(3):545–52
Swaby RF, Cristofanilli M (2011) Circulating tumor cells in breast cancer: a tool whose time has come of age. BMC Med 9:43
Paradiso A, Mangia A, Chiriatti A, Tommasi S, Zito A, Latorre A, Schittulli F, Lorusso V (2005) Biomarkers predictive for clinical efficacy of taxol-based chemotherapy in advanced breast cancer. Ann Oncol 16(Suppl 4):iv14–19
Chakravarti A, DeSilvio M, Zhang M, Grignon D, Rosenthal S, Asbell SO, Hanks G, Sandler HM, Khor LY, Pollack A, Shipley W (2007) Radiation Therapy Oncology Group. Prognostic value of p16 in locally advanced prostate cancer: a study based on Radiation Therapy Oncology Group Protocol 9202. J Clin Oncol 25(21):3082–9
Augustin H, Freibauer C, Bayer L, Lunglmayr G, Tschurlovich F, Kuber W, Pummer K (2006) Molecular markers and their prognostic impact in patients with advanced prostate cancer undergoing intermittent androgen suppression. Prostate Cancer Prostatic Dis 9(3):279–83
Yang X, Knopp MV (2011) Quantifying tumor vascular heterogeneity with dynamic contrast-enhanced magnetic resonance imaging: a review. J Biomed Biotechnol 2011:732848
Beloueche-Babari M, Chung YL, Al-Saffar NM, Falck-Miniotis M, Leach MO (2010) Metabolic assessment of the action of targeted cancer therapeutics using magnetic resonance spectroscopy. Br J Cancer 102(1):1–7
Miller JC, Sorensen AG (2005) Imaging biomarkers predictive of disease/therapy outcome: ischemic stroke and drug development. Prog Drug Res 62:319–56
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Bauer, K.S., Tavakkoli, F. (2014). Pharmacodynamic Modeling. In: Rudek, M., Chau, C., Figg, W., McLeod, H. (eds) Handbook of Anticancer Pharmacokinetics and Pharmacodynamics. Cancer Drug Discovery and Development. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9135-4_12
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9135-4_12
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9134-7
Online ISBN: 978-1-4614-9135-4
eBook Packages: MedicineMedicine (R0)