
Abstract
Over the last decade, the transcription factor PPARγ, previously known for its essential role in regulation of metabolic processes in adipose tissue, emerged as highly promising new target for the treatment of many neurological conditions, including ischemic and hemorrhagic stroke. Based on many cell culture and animal studies, activation of PPARγ was demonstrated to be associated with a broad range of biological effects (via genomic and non-genomic mode of action in virtually all brain cell types) which could effectively ameliorate pathogenic processes triggered by stroke, including inflammation, oxidative damage, edema, BBB preservation, and excitotoxicity, as well as help in the post-stroke recovery process by modulating the macrophage-mediated brain cleanup process. Some key aspects of PPARγ as target for stroke treatment are reviewed in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The peroxisome proliferator-activated receptors (PPARs), including α, γ, and δ/β, are encoded by separate genes and are members of the nuclear hormone receptor superfamily of ligand-activated nuclear transcription factors. PPARγ, also known as NR1C3 (nuclear receptor subfamily 1, group C, member 3), is a pleiotropic type II nuclear receptor, which was termed for its ability to induce proliferation of hepatic peroxisomes in response to xenobiotic stimuli in mice [1]. Three different PPARγ transcripts (PPARγ 1, 2, and 3), each a derivative of the PPARγ gene through differential promoter usage and alternative splicing, have been identified [2, 3]. While PPARγ 2 is the form primarily expressed in adipose tissue, PPARγ 1 has broader tissue distribution including presence in the brain [2]. As a transcription factor that regulates target gene expression through binding to the conserved DNA sequence termed peroxisome-proliferator response element (PPRE) [2, 4, 5], PPARγ was initially described in adipose tissue as a key regulator of metabolic processes [6–9]. Soon after, PPARγ was shown to be a unique therapeutic target for the treatment of metabolic disorders, e.g., diabetes (insulin resistance), obesity, hyperlipidemia, and hyperglycemia [10–12]. Among many compounds, ligands for PPARγ activation include fatty acids (especially the oxidized form) [13–15], cyclopentanone prostaglandins (e.g., 15-deoxy-Δ12,14-prostaglandin J2; -15d-PGJ2) [16], lipoxygenase products [17, 18], the nonsteroidal anti-inflammatory drugs (NSAIDs) [19, 20], and a class of clinically relevant compounds, the thiazolidinediones (TZDs) [10, 21]; of which pioglitazone and rosiglitazone are used to treat the type 2 diabetes mellitus [22–25]. In addition, PPARγ transactivation is regulated by its phosphorylation [26, 27]. Specifically, phosphorylation of PPARγ by the extracellular signal-regulated kinase (ERK1/2) and C-Jun N-terminal kinase (JNK) reduces PPARγ activity [26, 27]. Since JNK is activated by H2O2, oxygen–glucose deprivation (OGD), NMDA or ischemic stroke and acts as pro-death signal [28–32], the deleterious JNK functions may be secondary to the phosphorylation-mediated PPARγ inhibition.
Later studies on the mechanism of PPARγ action in other than fat tissue demonstrated its important role in regulation of anti-oxidative and anti-inflammatory processes [33–35]. It is primarily the anti-inflammatory properties of PPARγ ligands that ultimately brought the closer attention to PPARγ and PPARγ-activating agents to vascular diseases process [36–38]. PPARγ (and primarily PPARγ1) expression is ubiquitous regarding the type of tissues and cells it is expressed. In terms of neurological conditions, PPARγ in preclinical studies was shown to act as potential target for the treatment of ischemic stroke [39–51], intracerebral hemorrhage [52], neurotrauma [53–58], Alzheimer’s and neurodegenerative diseases [59–69], autoimmune encephalomyelitis (EAE), a model for multiple sclerosis [70–72]. In this chapter, our focus is mainly on the role of PPARγ in ischemic stroke, attempting to discuss the interactions of PPARγ with the NF-E2-related factor 2 (Nrf2) and the nuclear factor kappa B (NF-κB) signaling pathways in regulating pro- and anti-inflammatory responses in the brain.
Pleiotropic Effect of PPARγ Agonists in Ischemic Stroke
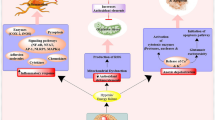
Based on the known function of gene targets, PPARγ acts as a key regulator in a broad range of processes virtually in all brain cells including neurons [45, 73], astroglia [74–76], oligodendroglia [77–79], microglia [54, 80, 81], and endothelial cells [82, 83]. Primarily through the use of various PPARγ agonists but also through the use of cell-specific PPARγ knockouts, PPARγ was demonstrated to protect brain from damages caused by ischemic [41, 42, 45, 84–87] and hemorrhagic stroke [53–55]. The beneficial effects of PPARγ activation was linked to (1) repression of pro-inflammatory mediators production (at least in part through inhibition of NF-κB either directly or by upregulation of endogenous NF-κB inhibitor, IκB [33, 34, 53, 88–95]), (2) upregulation of antioxidant enzymes including CuZn-superoxide dismutase (SOD) and catalase [41, 54], (3) inhibition of excitotoxicity [96, 97], and (4) activation of phagocytotic activities by microglia and macrophages via mechanism involving the PPARγ-target gene—scavenger receptor CD36, the molecule that assists in cleanup of damaged brain tissue, a process necessary for efficient recovery and the termination of deleterious pro-inflammatory cascade (Fig. 17.1) [54, 98–101].

PPARγ regulated pathways after stroke—role of PPARγ activators. PPARγ transcriptionally controls expression of numerous genes including the anti-oxidative enzymes, such as catalase and superoxide dismutase (SOD), as well as the transcription factor Nrf2. Nrf2 plays a key role in amplifying the expression of many anti-oxidative genes including catalase and SOD, similar to PPARγ. This anti-oxidative feature of PPARγ is critical in combating oxidative damage imposed by cerebral ischemia. Importantly, since PPARγ and Nrf2 are ubiquitously expressed, this anti-oxidative mechanism may apply to all brain cell types affected by stroke. In addition, both PPARγ and Nrf2 regulate expression of CD36, a scavenger receptor that is abundant on microglia/macrophages. CD36 plays important role in endocytosis of oxidized lipids and phagocytosis of dead (including apoptotic) cells and other cellular debris, thereof aiding in cleanup—process allowing for a faster inflammation resolution and more efficient tissue repair. Another important task of PPARγ is to inhibit NF-κB, a proinflammatory transcription factor implicated in BBB disruption and brain edema formation. Ultimately, augmented PPARγ activation improves inflammation resolution, tissue repair, and functional recovery after stroke
PPARγ and Neuroprotection
In response to the prolonged ischemia, neurons that are localized in the ischemic core die rapidly as consequence of ischemia-induced energy failure, anoxic depolarization, and excitotoxicity, which is the result of glutamate receptors overactivation, calcium overload, and a breakdown of ion homeostasis [102–109]. Using oxygen–glucose deprivation (OGD) or glutamate/NMDA toxicity (in vitro models of ischemia) to study the neuroprotective capacity of PPARγ agonists [including pioglitazone, rosiglitazone, or cyclopentanone prostaglandins (CyPG)], we and other groups demonstrated that activation of PPARγ potently reduces the neuronal death in the primary neurons [96, 97, 110], implying that PPARγ may act as pro-survival factor for neurons under the ischemic/excitotoxic stress. The anti-excitotoxic effect of PPARγ agonists was observed not only in cultured neurons but also in the animal injury model that assess the extent of brain damage caused by intracortical injection of NMDA [97]. Finally, we have established that neurons derived from animals engineered to lack PPARγ, selectively in neurons, demonstrated significantly increased susceptibility to excitotoxic damage and to OGD [84]. In agreement with the in vitro data, mice lacking PPARγ in neurons were significantly more susceptible to the ischemic damage caused by focal cerebral ischemia [84].
Reactive oxygen species (ROS) are well known to represent one of the most important components of brain injure in response to ischemia/reperfusion insult. ROS are generated by the ischemia-affected brain cells, the activated microglia, and infiltrating neutrophils that collectively impose oxidative stress to cells located in proximity to the ischemia [111–114]. To combat the oxidative stress, cells have developed a number of self-defense mechanisms including upregulation of enzymes with anti-oxidative functions. Superoxide dismutase along with catalase and glutathione peroxidase plays key roles in eliminating ROS through catalytic decomposition of superoxide or H2O2 [84, 115, 116]. Catalase is a large homotetrameric protein that is usually localized in peroxisomes (the membrane-bound organelles that house β-oxidation of very long chains of fatty acids, in which toxic peroxides are generated as side products) [117], where it acts to protect the cells from the toxic effects of H2O2 by catalyzing its decomposition. As a ubiquitous enzyme to most cells in our body including neuroglia and neurons [118], catalase expression is regulated by PPARγ and Nrf2 [115, 119]. The distribution pattern of catalase-immunopositive neurons throughout the brain inversely corresponds to increased susceptibility to damage induced by global cerebral ischemia [118], suggesting that catalase plays important role in cell survival. Overexpression of catalase in rat striatum through virus-mediated gene transfer decreases the vulnerability to ischemic stroke [120]. In response to PPARγ activation, expression of catalase rapidly increased in the ischemia-affected brain [118, 121] and in the OGD-injured neurons [122], which likely reflect an adaptive response aiming at improving the antioxidant buffering capacity under the pathological scenarios. In agreement with this notion, treatment with catalase of neurons in culture subjected to H2O2-induced injury provided a robust cytoprotection [123, 124]. Thus, catalase upregulation by PPARγ may reflect a self-protective mechanism to combat oxidative stress in stroke. It is important to point out that in addition to catalase, PPARγ regulates expression of superoxide dismutase (including in neurons), an enzyme well recognized for decades as a key player in mitigating oxidative injury and brain damage after cerebral ischemia [41, 84, 125, 126].
PPARγ-Induced CD36 Expression on Phagocytes and the Endogenous Cleanup Mechanism
After cerebral ischemia, the infarcted/dead tissue not only acts as a reservoir of various cytotoxic and pro-inflammatory molecules that harm the adjacent healthy brain tissue, but it also forms a biological and physical barrier hampering neural reorganization, repair, and ultimately, neurological recovery. Thus, in order to minimize such detrimental effects, infarcted tissue needs to be removed to facilitate recovery. Microglia and hematogenous macrophages (MMΦ) are the cells primarily responsible for such cleanup and repair processes. Successful removal of the disintegrated and apoptotic brain cells or debris (including the neutrophils that accumulate in brain in response to injury and consequently die through apoptosis) by MMΦ is also essential in achieving resolution of inflammation. While apoptotic cells appear to be considerably benign to the surrounding brain tissue, an apoptotic cells non-phagocytosed in a timely manner may undergo secondary necrosis causing spill of the intracellular toxic content, leading to the damage to the neighboring cells and causing inflammation. Several macrophage scavenger receptors that mediate cleanup process have been identified. These include not only CD36 but also CD91, SR-A, and several others [54, 127–133]. Regarding apoptotic cell efferocytosis by macrophages, the phosphatidyl serine on the sickle red blood cells, symmetric red cell ghosts [134–136], or apoptotic neutrophils was suggested to act as the recognition molecule for CD36, a class II scavenger receptor on macrophages [137–139]. Expression of CD36 on macrophages (MΦ) is transcriptionally regulated by both PPARγ [98, 140, 141] and Nrf2 [142–145]. Although CD36 has various functions, one of its primary roles is to mediate endocytosis of (oxidized) fatty acids and phagocytosis of dead/apoptotic cells [129, 137, 146–148]. Deficiency of CD36 in macrophages due to genetic deletion of PPARγ leads to delayed uptake of oxidized LDL by macrophages and aggravation of atherosclerotic lesions [149]. In CD36-KO mice, aberrant phagocytotic capacity of macrophages was proposed to explain the deficiency in remyelination in response to sciatic nerve crush injury [150]. In addition, transfection of non-phagocytic cells with CD36 renders these cells capable of ingesting apoptotic neutrophils, lymphocytes, and fibroblasts [138], further confirming the important role of CD36 in phagocytosis. As pointed above, since CD36 transcription is under control of Nrf2 and PPARγ, the upregulation of CD36 by MMΦ in response to Nrf2 and/or PPARγ activators may ensure a more efficient interaction between the MMΦ and their targets for phagocytosis. This may allow for more efficient phagocytosis-mediated clearance of dead cells/tissues from the ischemic brain. However, despite its beneficial role in the cleanup process, CD36 may have detrimental effect which is normally characterized by increased oxidative stress and pro-inflammatory responses, as adult animals deficient in CD36 suffer from the less profound damage in response to cerebral ischemia [151, 152]. The nature of these responses is not known; however, the likelihood is that upon engulfment of cellular debris including oxidized lipids, the MMΦ generate damaging levels of oxidative stress during degradation of debris in the phagolysosomes. Interestingly, CD36 knockout neonates subjected to cerebral ischemia experienced more damage (suggesting beneficial function of CD36), which was suggested to be in part due to the impaired cleanup mechanism [153]. Independent of the natural responses that were tested in experiments using CD36 knockout mice, we suggest that under conditions using pharmacologic agents to activate PPARγ, MMΦ not only express higher levels of CD36 for a more efficient phagocytosis but also produce more anti-oxidative enzymes (e.g., catalase) that are regulated by PPARγ. Recently, we provided the evidence that MMΦ in culture challenged with PPARγ or Nrf2 activators, despite expressing CD36 at much higher level and demonstrating the augmented phagocytosis, experienced less oxidative damage and showed reduced pro-inflammatory gene expression [54].
Thus, in response to PPARγ in activated microglia, the upregulation of the antioxidant enzymes (in addition to CD36) may play a protective role allowing for effective and safe phagocytosis. Consequently, cleaning the apoptotic/dislocated/damaged cells or debris will help to reestablish the nurturing environment necessary for restoring tissue structure and neurological function recovery [154, 155].
PPARγ Activation and the Interaction of PPARγ and RXR
PPARγ regulates target gene expression by binding to PPRE as heterodimers with the retinoic acid receptor (RXR). Interestingly, existing studies indicate that activation of PPARγ–RXR complex can be achieved with either PPARγ and/or by RXR ligand (e.g., 9-cis retinoic acid), indicating some level of the promiscuity in activation of PPARγ [156, 157]. Although each ligand can initiate transactivation independently, the effect of co-activation appears to be stronger [9], suggesting that the occupancy of both PPARγ and RXR ligand (e.g., 15d-PGJ2 plus 9-cis retinoic acid) is needed for the maximal receptor activity [9, 158–160]. In agreement with this notion, we found that co-treatment of cultured neurons with 15d-PGJ2 and 9-cis retinoic acid was more effective in reducing the OGD-induced damage, as compared to each ligand alone [53]. This beneficial interaction between PPARγ and RXR ligands in our neuroprotection assay is consistent with an earlier report showing that combination use of 15d-PGJ2 and 9-cis retinoic acid was superior to each drug alone in reducing behavioral dysfunction in a mouse model of experimental autoimmune encephalomyelitis [161].
Interaction of PPARγ and Nrf2 and NF-κB
The pro-survival role of PPARγ includes the non-genomic inhibition of deleterious pro-inflammatory transcription factor, nuclear factor kappa B, NF-κB. In the ischemia-injured brain, the delayed cell death is in part triggered by the overproduction of pro-inflammatory molecules including pro-inflammatory cytokines (such as tumor necrosis factor alpha, TNF-α or interleukin-1 beta, IL-1β), adhesion molecules (such as intercellular adhesion molecule 1, ICAM-1 or vascular cell adhesion molecule, VCAM), matrix metalloproteinases (including MMP9) or the pro-oxidative inducible form of nitric oxide synthase (iNOS) capable of generating large quantities of nitric oxide, that in presence of superoxide generated by NADPH oxidase is converted to a highly cytotoxic peroxynitrites [109, 162–165]. Once perpetuated by ischemia, these potentially deleterious factors act in concert to damage blood–brain barrier (BBB) and cause edema and/or hemorrhage [166–168]. Interestingly, the expression of all these factors is tightly regulated by NF-κB. The activation of PPARγ can antagonize these harmful effects through inhibition of NF-κB [33, 34], which may be achieved by at least three independent mechanisms (Fig. 17.1) [33, 34, 53, 88–95]. First, PPARγ may directly bind to the NF-κB subunits, p50 and p65, resulting in NF-κB inactivation [169]; second, PPARγ may indirectly inhibit NF-κB by sequestering the common transcription co-activators such as SRC-1 [170] and p300/CBP (CREB-binding protein) [88–90]; and third, PPARγ may upregulate the production of inhibitor kappa B (IκB) [91, 93–95], the protein that directly inhibit NF-κB activation. Inhibition of NF-κB by PPARγ agonists may reduce generation of pro-inflammatory mediators involved in the secondary brain damage.
Nrf2 is a ubiquitous pleiotropic transcription factor and a key genomic homeostatic regulator of intracellular stress [171]. By combining with Mif family proteins, Nrf2 forms heterodimeric complexes capable of transactivating the antioxidant response elements (ARE) within the regulatory region of many cytoprotective target genes including catalase, superoxide dismutase, glutathione-S-transferase, thioredoxin, NQO1, and many other proteins with important role in neutralization of oxidative stress and detoxification [172]. In most cells, Nrf2 is present at low concentrations due to continuous Nrf2 degradation through the proteasome pathway [173, 174]. Nrf2 contributes to cytoprotection and amelioration of tissue damage through reducing the oxidative stress in many pathogenic conditions including cerebral ischemia [175–181], neurodegenerative diseases [182], and mitochondrial metabolic stress [183]. The growing body of evidence suggests that PPARγ may play important role in regulation of Nrf2 and thus Nrf2 target genes (Fig. 17.1). The interaction between PPARγ and Nrf2 may involve several layers of interaction. Most importantly, PPARγ was demonstrated to regulate Nrf2 gene expression and Nrf2-regulated genes containing putative PPREs [184]. Interestingly, it appears that Nrf2 also regulates PPARγ and PPARγ-regulated genes containing the ARE [185]. Next, PPRE and ARE coexist in the same genes, such as CD36 and catalase, suggesting an interactive function of Nrf2 and PPARγ in expression of these genes. Finally, an interaction between PPARγ and Nrf2 may be through NF-κB inhibition. Since NF-κB activation requires the presence of oxidative stress [186], the effect of Nrf2 in ameliorating oxidative stress was proposed to inhibit NF-κB [187]. As different mechanisms are used by Nrf2 and PPARγ in inhibiting NF-κB, it is likely that the mutual effect may lead to a synergistic role [188–190].
Adverse Effects of PPARγ Agonists
There is a small number of observations reporting the dose-dependent neurotoxic effects of the endogenous PPARγ ligand 15d-PGJ2 in cerebellar granule cells [191], primary cortical neurons [192], and spinal cord motor neurons [193]. The mechanism that underlies this neurotoxicity is unclear and some reports indicate that these harmful actions are probably not directly linked to PPARγ [191]. In our studies using mouse and rat neurons in culture, we have not observed neurotoxicity using PPARγ activating ligands to date. In fact, all the tested PPARγ agonists including 15d-PGJ2, 15d-PGD2, ciglitazone, rosiglitazone, and pioglitazone demonstrated potent cytoprotective effects in models of OGD and excitotoxicity [45, 50, 97]. The only instance showing toxicity was when the doses of the agonists were higher than these needed for the cytoprotection. Unlike synthetic TZDs that display rather significant levels of PPARγ specificity, prostaglandin D2 derivatives, including 15d-PGJ2, have a limited selectivity toward PPARγ and many of their biological activities are independent of PPARγ [92, 194–198]. However, the clinical use of PPARγ ligands, and primarily rosiglitazone, was associated with hemodilution, peripheral edema, increase in body weight, as well as cardiomyopathies and heart failure [46, 199–201]. Again, these are the known side effects of long-term use of these medications and as such should not necessarily influence the safety of patients subjected to short-term treatment. The study evaluating the safety of pioglitazone in patients with hemorrhagic stroke is currently ongoing [52].
PPARγ Agonists and Clinical Trials
Two of the thiazolidinediones (TZDs), pioglitazone and rosiglitazone, are currently approved by the FDA for treatment of type 2 diabetes mellitus. These insulin-sensitizing PPARγ agonists are unique among all the glucose-lowering agents as they act independent of secretion of insulin from pancreas (TZDs do not change blood insulin levels, rather make cells more sensitive to its effect) [22, 202]. The glucose-lowering effect of TZDs is of clinical importance since hyperglycemia during ischemia/reperfusion may worsens the brain damage and neurological outcome, including by increasing incidence of hemorrhage in patients subjected to thrombolysis with rt-PA [203–206]. A first case-matched controlled study reporting improved functional recovery in stroke patients with type 2 diabetes receiving pioglitazone or rosiglitazone (vs. control type 2 diabetes patients not receiving TZDs) yields a promising outlook [207]. Subsequently, PROACTIVE (PROspective pioglitAzone Clinical Trial In macroVascular Events; NCT00174993), a randomized, double-blinded, placebo-controlled study looked at the impact of pioglitazone on total mortality and macrovascular morbidity in 5,238 patients with diabetes and macrovascular disease. This secondary prevention study showed safety and a macrovascular benefit with pioglitazone in terms of major adverse cardiovascular events including all-cause mortality, nonfatal myocardial infarction, acute coronary syndrome, cardiac intervention (including coronary artery bypass graft or percutaneous coronary intervention), and stroke [208–210]. The higher beneficial rates were observed in patients with prior stroke compared with those without prior stroke [211, 212]. A meta-analysis of 19 randomized clinical trials with pioglitazone revealed a statistical difference regarding the favorable outcome including mortality, nonfatal MI, and stroke when using pioglitazone [201]. However, a recent study suggests that use of rosiglitazone may impose 1.4-fold increase in risk of acute MI and death from cardiovascular diseases compared with non-TZDs therapies [213]. As compared to pioglitazone, rosiglitazone significantly increased the risk of stroke, heart failure, and death in elderly patients [214]. In contrast, from the stroke prevention point, pioglitazone has shown significant protection from both micro- and macrovascular cardiovascular events and plaque progression [215–217].
References
Dreyer C, Keller H, Mahfoudi A, Laudet V, Krey G, Wahli W (1993) Positive regulation of the peroxisomal beta-oxidation pathway by fatty acids through activation of peroxisome proliferator-activated receptors (PPAR). Biol Cell 77:67–76
Elbrecht A, Chen Y, Cullinan CA, Hayes N, Leibowitz M, Moller DE et al (1996) Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun 224:431–437
Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R et al (1997) The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem 272:18779–18789
Greene ME, Blumberg B, McBride OW, Yi HF, Kronquist K, Kwan K et al (1995) Isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Expr 4:281–299
Adams M, Montague CT, Prins JB, Holder JC, Smith SA, Sanders L et al (1997) Activators of peroxisome proliferator-activated receptor gamma have depot-specific effects on human preadipocyte differentiation. J Clin Invest 100:3149–3153
Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K et al (1995) The nuclear receptor superfamily: the second decade. Cell 83:835–839
Lemberger T, Desvergne B, Wahli W (1996) Peroxisome proliferator-activated receptors: a nuclear receptor signaling pathway in lipid physiology. Annu Rev Cell Dev Biol 12:335–363
Berger J, Wagner JA (2002) Physiological and therapeutic roles of peroxisome proliferator-activated receptors. Diabetes Technol Ther 4:163–174
Berger J, Moller DE (2002) The mechanisms of action of PPARs. Annu Rev Med 53:409–435
Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA (1995) An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 270:12953–12956
Moller DE, Berger JP (2003) Role of PPARs in the regulation of obesity-related insulin sensitivity and inflammation. Int J Obes Relat Metab Disord 27(Suppl 3):S17–S21
Li Y, Zhang J, Schopfer FJ, Martynowski D, Garcia-Barrio MT, Kovach A et al (2008) Molecular recognition of nitrated fatty acids by PPAR gamma. Nat Struct Mol Biol 15:865–867
Yamamoto K, Itoh T, Abe D, Shimizu M, Kanda T, Koyama T et al (2005) Identification of putative metabolites of docosahexaenoic acid as potent PPARgamma agonists and antidiabetic agents. Bioorg Med Chem Lett 15:517–522
Itoh T, Murota I, Yoshikai K, Yamada S, Yamamoto K (2006) Synthesis of docosahexaenoic acid derivatives designed as novel PPARgamma agonists and antidiabetic agents. Bioorg Med Chem 14:98–108
Li H, Ruan XZ, Powis SH, Fernando R, Mon WY, Wheeler DC et al (2005) EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-gamma-dependent mechanism. Kidney Int 67:867–874
Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM (1995) A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83:813–819
Yuan H, Li MY, Ma LT, Hsin MK, Mok TS, Underwood MJ et al (2010) 15-Lipoxygenases and its metabolites 15(S)-HETE and 13(S)-HODE in the development of non-small cell lung cancer. Thorax 65:321–326
Cimen I, Astarci E, Banerjee S (2011) 15-Lipoxygenase-1 exerts its tumor suppressive role by inhibiting nuclear factor-kappa B via activation of PPAR gamma. J Cell Biochem 112:2490–2501
Jaradat MS, Wongsud B, Phornchirasilp S, Rangwala SM, Shams G, Sutton M et al (2001) Activation of peroxisome proliferator-activated receptor isoforms and inhibition of prostaglandin H(2) synthases by ibuprofen, naproxen, and indomethacin. Biochem Pharmacol 62:1587–1595
Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F et al (2003) Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci 23:9796–9804
Lambe KG, Tugwood JD (1996) A human peroxisome-proliferator-activated receptor-gamma is activated by inducers of adipogenesis, including thiazolidinedione drugs. Eur J Biochem 239:1–7
Grossman SL, Lessem J (1997) Mechanisms and clinical effects of thiazolidinediones. Expert Opin Investig Drugs 6:1025–1040
Nattrass M, Bailey CJ (1999) New agents for type 2 diabetes. Baillieres Best Pract Res Clin Endocrinol Metab 13:309–329
Gillies PS, Dunn CJ (2000) Pioglitazone. Drugs 60:333–343, discussion 344–335
Goldstein BJ (2000) Rosiglitazone. Int J Clin Pract 54:333–337
Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK (1997) Transcriptional activation by peroxisome proliferator-activated receptor gamma is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J Biol Chem 272:5128–5132
Camp HS, Tafuri SR, Leff T (1999) C-jun n-terminal kinase phosphorylates peroxisome proliferator-activated receptor-gamma1 and negatively regulates its transcriptional activity. Endocrinology 140:392–397
Mielke K, Damm A, Yang DD, Herdegen T (2000) Selective expression of JNK isoforms and stress-specific JNK activity in different neural cell lines. Brain Res Mol Brain Res 75:128–137
Barbin G, Roisin MP, Zalc B (2001) Tumor necrosis factor alpha activates the phosphorylation of ERK, SAPK/JNK, and p38 kinase in primary cultures of neurons. Neurochem Res 26:107–112
Borsello T, Clarke PG, Hirt L, Vercelli A, Repici M, Schorderet DF et al (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med 9:1180–1186
Borsello T, Croquelois K, Hornung JP, Clarke PG (2003) N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur J Neurosci 18:473–485
Wang Q, Wang X, Studzinski GP (2003) Jun N-terminal kinase pathway enhances signaling of monocytic differentiation of human leukemia cells induced by 1,25-dihydroxyvitamin D3. J Cell Biochem 89:1087–1101
Jiang C, Ting AT, Seed B (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391:82–86
Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK (1998) The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 391:79–82
Ohara M, Sawa T (1999) Current topics in the regulation of prostanoids-4. The feedback regulation by PPAR-gamma. Masui 48:146–151
Barbier O, Torra IP, Duguay Y, Blanquart C, Fruchart JC, Glineur C et al (2002) Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol 22:717–726
Blanquart C, Barbier O, Fruchart JC, Staels B, Glineur C (2003) Peroxisome proliferator-activated receptors: regulation of transcriptional activities and roles in inflammation. J Steroid Biochem Mol Biol 85:267–273
Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF (2005) Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol 191:331–336
Sundararajan S, Landreth GE (2004) Antiinflammatory properties of PPARgamma agonists following ischemia. Drug News Perspect 17:229–236
Pereira MP, Hurtado O, Cardenas A, Alonso-Escolano D, Bosca L, Vivancos J et al (2005) The nonthiazolidinedione PPARgamma agonist l-796,449 is neuroprotective in experimental stroke. J Neuropathol Exp Neurol 64:797–805
Shimazu T, Inoue I, Araki N, Asano Y, Sawada M, Furuya D et al (2005) A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke 36:353–359
Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE (2005) Peroxisome proliferator-activated receptor-gamma ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience 130:685–696
Zhao Y, Patzer A, Gohlke P, Herdegen T, Culman J (2005) The intracerebral application of the PPARgamma-ligand pioglitazone confers neuroprotection against focal ischaemia in the rat brain. Eur J Neurosci 22:278–282
Bordet R, Ouk T, Petrault O, Gele P, Gautier S, Laprais M et al (2006) PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochem Soc Trans 34:1341–1346
Ou Z, Zhao X, Labiche LA, Strong R, Grotta JC, Herrmann O et al (2006) Neuronal expression of peroxisome proliferator-activated receptor-gamma (PPARgamma) and 15d-prostaglandin j2-mediated protection of brain after experimental cerebral ischemia in rat. Brain Res 1096:196–203
Chu K, Lee ST, Koo JS, Jung KH, Kim EH, Sinn DI et al (2006) Peroxisome proliferator-activated receptor-gamma-agonist, rosiglitazone, promotes angiogenesis after focal cerebral ischemia. Brain Res 1093:208–218
Luo Y, Yin W, Signore AP, Zhang F, Hong Z, Wang S et al (2006) Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-gamma agonist rosiglitazone. J Neurochem 97:435–448
Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C et al (2006) Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol 530:70–80
Lin TN, Cheung WM, Wu JS, Chen JJ, Lin H, Liou JY et al (2006) 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol 26:481–487
Victor NA, Wanderi EW, Gamboa J, Zhao X, Aronowski J, Deininger K et al (2006) Altered PPARgamma expression and activation after transient focal ischemia in rats. Eur J Neurosci 24:1653–1663
Culman J, Nguyen-Ngoc M, Glatz T, Gohlke P, Herdegen T, Zhao Y (2012) Treatment of rats with pioglitazone in the reperfusion phase of focal cerebral ischemia: a preclinical stroke trial. Exp Neurol 238:243–253
Gonzales NR, Shah J, Sangha N, Sosa L, Martinez R, Shen L et al (2012) Design of a prospective, dose-escalation study evaluating the safety of pioglitazone for hematoma resolution in intracerebral hemorrhage (SHRINC). Int J Stroke 8(5):388–396
Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J (2006) 15d-prostaglandin J2 activates peroxisome proliferator-activated receptor-gamma, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J Cereb Blood Flow Metab 26:811–820
Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N et al (2007) Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol 61:352–362
Hyong A, Jadhav V, Lee S, Tong W, Rowe J, Zhang JH et al (2008) Rosiglitazone, a PPAR gamma agonist, attenuates inflammation after surgical brain injury in rodents. Brain Res 1215:218–224
Yi JH, Park SW, Brooks N, Lang BT, Vemuganti R (2008) PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res 1244:164–172
Zhang Q, Hu W, Meng B, Tang T (2010) PPARgamma agonist rosiglitazone is neuroprotective after traumatic spinal cord injury via anti-inflammatory in adult rats. Neurol Res 32:852–859
Sauerbeck A, Gao J, Readnower R, Liu M, Pauly JR, Bing G et al (2011) Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp Neurol 227:128–135
Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE (2000) Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci 20:558–567
Landreth GE, Heneka MT (2001) Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging 22:937–944
Heneka MT, Landreth GE, Feinstein DL (2001) Role for peroxisome proliferator-activated receptor-gamma in Alzheimer’s disease. Ann Neurol 49:276
Breidert T, Callebert J, Heneka MT, Landreth G, Launay JM, Hirsch EC (2002) Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson’s disease. J Neurochem 82:615–624
Kielian T, Drew PD (2003) Effects of peroxisome proliferator-activated receptor-gamma agonists on central nervous system inflammation. J Neurosci Res 71:315–325
Duvanel CB, Honegger P, Pershadsingh H, Feinstein D, Matthieu JM (2003) Inhibition of glial cell proinflammatory activities by peroxisome proliferator-activated receptor gamma agonist confers partial protection during antimyelin oligodendrocyte glycoprotein demyelination in vitro. J Neurosci Res 71:246–255
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB (2004) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and INOS activation. J Neurochem 88:494–501
Gasparini L, Ongini E, Wenk G (2004) Non-steroidal anti-inflammatory drugs (NSAIDs) in Alzheimer’s disease: old and new mechanisms of action. J Neurochem 91:521–536
d’Abramo C, Massone S, Zingg JM, Pizzuti A, Marambaud P, Dalla Piccola B et al (2005) Role of peroxisome proliferator-activated receptor gamma in amyloid precursor protein processing and amyloid beta-mediated cell death. Biochem J 391:693–698
Heneka MT, Landreth GE, Hull M (2007) Drug insight: effects mediated by peroxisome proliferator-activated receptor-gamma in CNS disorders. Nat Clin Pract Neurol 3:496–504
Heneka MT, Reyes-Irisarri E, Hull M, Kummer MP (2011) Impact and therapeutic potential of PPARs in Alzheimer’s disease. Curr Neuropharmacol 9:643–650
Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L et al (2002) Peroxisome proliferator-activated receptor-gamma agonists prevent experimental autoimmune encephalomyelitis. Ann Neurol 51:694–702
Storer PD, Xu J, Chavis J, Drew PD (2005) Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J Neuroimmunol 161:113–122
Loria F, Petrosino S, Hernangomez M, Mestre L, Spagnolo A, Correa F et al (2010) An endocannabinoid tone limits excitotoxicity in vitro and in a model of multiple sclerosis. Neurobiol Dis 37:166–176
Luna-Medina R, Cortes-Canteli M, Alonso M, Santos A, Martinez A, Perez-Castillo A (2005) Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J Biol Chem 280:21453–21462
Dello Russo C, Gavrilyuk V, Weinberg G, Almeida A, Bolanos JP, Palmer J et al (2003) Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J Biol Chem 278:5828–5836
Cristiano L, Bernardo A, Ceru MP (2001) Peroxisome proliferator-activated receptors (PPARs) and peroxisomes in rat cortical and cerebellar astrocytes. J Neurocytol 30:671–683
Janabi N (2002) Selective inhibition of cyclooxygenase-2 expression by 15-deoxy-delta(12,14)(12,14)-prostaglandin J(2) in activated human astrocytes, but not in human brain macrophages. J Immunol 168:4747–4755
Roth AD, Leisewitz AV, Jung JE, Cassina P, Barbeito L, Inestrosa NC et al (2003) PPAR gamma activators induce growth arrest and process extension in B12 oligodendrocyte-like cells and terminal differentiation of cultured oligodendrocytes. J Neurosci Res 72:425–435
Bernardo A, Bianchi D, Magnaghi V, Minghetti L (2009) Peroxisome proliferator-activated receptor-gamma agonists promote differentiation and antioxidant defenses of oligodendrocyte progenitor cells. J Neuropathol Exp Neurol 68:797–808
De Nuccio C, Bernardo A, De Simone R, Mancuso E, Magnaghi V, Visentin S et al (2011) Peroxisome proliferator-activated receptor gamma agonists accelerate oligodendrocyte maturation and influence mitochondrial functions and oscillatory Ca(2+) waves. J Neuropathol Exp Neurol 70:900–912
Petrova TV, Akama KT, Van Eldik LJ (1999) Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-delta12,14-prostaglandin J2. Proc Natl Acad Sci USA 96:4668–4673
Bernardo A, Minghetti L (2006) PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des 12:93–109
Hamblin M, Chang L, Fan Y, Zhang J, Chen YE (2009) PPARs and the cardiovascular system. Antioxid Redox Signal 11:1415–1452
Wu QQ, Wang Y, Senitko M, Meyer C, Wigley WC, Ferguson DA et al (2011) Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARgamma, and HO-1. Am J Physiol Renal Physiol 300:F1180–F1192
Zhao X, Strong R, Zhang J, Sun G, Tsien JZ, Cui Z et al (2009) Neuronal PPARgamma deficiency increases susceptibility to brain damage after cerebral ischemia. J Neurosci 29:6186–6195
Vemuganti R (2008) Therapeutic potential of PPARgamma activation in stroke. PPAR Res 2008:461981
Culman J, Zhao Y, Gohlke P, Herdegen T (2007) PPAR-gamma: therapeutic target for ischemic stroke. Trends Pharmacol Sci 28:244–249
Fatehi-Hassanabad Z, Tasker RA (2011) Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) activation confers functional neuroprotection in global ischemia. Neurotox Res 19:462–471
Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M (1997) P300 functions as a coactivator for the peroxisome proliferator-activated receptor alpha. J Biol Chem 272:33435–33443
Gelman L, Zhou G, Fajas L, Raspe E, Fruchart JC, Auwerx J (1999) P300 interacts with the N- and C-terminal part of PPARgamma2 in a ligand-independent and -dependent manner, respectively. J Biol Chem 274:7681–7688
Li M, Pascual G, Glass CK (2000) Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol 20:4699–4707
Straus DS, Pascual G, Li M, Welch JS, Ricote M, Hsiang CH et al (2000) 15-deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in the NF- kappa B signaling pathway. Proc Natl Acad Sci USA 97:4844–4849
Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M et al (2000) Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of ikappaB kinase. Nature 403:103–108
Delerive P, Gervois P, Fruchart JC, Staels B (2000) Induction of ikappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-alpha activators. J Biol Chem 275:36703–36707
Cernuda-Morollon E, Rodriguez-Pascual F, Klatt P, Lamas S, Perez-Sala D (2002) PPAR agonists amplify inos expression while inhibiting NF-kappaB: implications for mesangial cell activation by cytokines. J Am Soc Nephrol 13:2223–2231
Heneka MT, Gavrilyuk V, Landreth GE, O’Banion MK, Weinberg G, Feinstein DL (2003) Noradrenergic depletion increases inflammatory responses in brain: effects on ikappaB and HSP70 expression. J Neurochem 85:387–398
Uryu S, Harada J, Hisamoto M, Oda T (2002) Troglitazone inhibits both post-glutamate neurotoxicity and low-potassium-induced apoptosis in cerebellar granule neurons. Brain Res 924:229–236
Zhao X, Ou Z, Grotta JC, Waxham N, Aronowski J (2006) Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain Res 1073–1074:460–469
Moore KJ, Rosen ED, Fitzgerald ML, Randow F, Andersson LP, Altshuler D et al (2001) The role of PPAR-gamma in macrophage differentiation and cholesterol uptake. Nat Med 7:41–47
Asada K, Sasaki S, Suda T, Chida K, Nakamura H (2004) Antiinflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. Am J Respir Crit Care Med 169:195–200
Patel SN, Serghides L, Smith TG, Febbraio M, Silverstein RL, Kurtz TW et al (2004) Cd36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J Infect Dis 189:204–213
Majai G, Sarang Z, Csomos K, Zahuczky G, Fesus L (2007) PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol 37:1343–1354
Zheng Z, Lee JE, Yenari MA (2003) Stroke: molecular mechanisms and potential targets for treatment. Curr Mol Med 3:361–372
Danton GH, Dietrich WD (2003) Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol 62:127–136
Wen YD, Zhang HL, Qin ZH (2006) Inflammatory mechanism in ischemic neuronal injury. Neurosci Bull 22:171–182
Mehta SL, Manhas N, Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54:34–66
Nakka VP, Gusain A, Mehta SL, Raghubir R (2008) Molecular mechanisms of apoptosis in cerebral ischemia: multiple neuroprotective opportunities. Mol Neurobiol 37:7–38
Brea D, Sobrino T, Ramos-Cabrer P, Castillo J (2009) Inflammatory and neuroimmunomodulatory changes in acute cerebral ischemia. Cerebrovasc Dis 27(Suppl 1):48–64
Candelario-Jalil E (2009) Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs 10:644–654
Guo MF, Yu JZ, Ma CG (2011) Mechanisms related to neuron injury and death in cerebral hypoxic ischaemia. Folia Neuropathol 49:78–87
Zhang YQ, Zhang YN, Wu J, Zhu XY, Xu CQ (2005) Effect of peroxisome proliferation activated receptor-gamma on neuronal cell death induced by hypoxia and ischemia in rats in vitro and in vivo. Zhonghua Yi Xue Za Zhi 85:684–688
Clemens JA (2000) Cerebral ischemia: gene activation, neuronal injury, and the protective role of antioxidants. Free Radic Biol Med 28:1526–1531
Starkov AA, Chinopoulos C, Fiskum G (2004) Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 36:257–264
Tuttolomondo A, Di Sciacca R, Di Raimondo D, Arnao V, Renda C, Pinto A et al (2009) Neuron protection as a therapeutic target in acute ischemic stroke. Curr Top Med Chem 9:1317–1334
Chen SD, Yang DI, Lin TK, Shaw FZ, Liou CW, Chuang YC (2011) Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci 12:7199–7215
Girnun GD, Domann FE, Moore SA, Robbins ME (2002) Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol 16:2793–2801
Jung TW, Lee JY, Shim WS, Kang ES, Kim SK, Ahn CW et al (2007) Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J Neurol Sci 253:53–60
Chance B, Sies H, Boveris A (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59:527–605
Moreno S, Mugnaini E, Ceru MP (1995) Immunocytochemical localization of catalase in the central nervous system of the rat. J Histochem Cytochem 43:1253–1267
Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW (2001) Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med 7:135–145
Gu W, Zhao H, Yenari MA, Sapolsky RM, Steinberg GK (2004) Catalase over-expression protects striatal neurons from transient focal cerebral ischemia. Neuroreport 15:413–416
Mahadik SP, Makar TK, Murthy JN, Ortiz A, Wakade CG, Karpiak SE (1993) Temporal changes in superoxide dismutase, glutathione peroxidase, and catalase levels in primary and peri-ischemic tissue. Monosialoganglioside (GM1) treatment effects. Mol Chem Neuropathol 18:1–14
Ye R, Li N, Han J, Kong X, Cao R, Rao Z et al (2009) Neuroprotective effects of ginsenoside RD against oxygen-glucose deprivation in cultured hippocampal neurons. Neurosci Res 64:306–310
Ricart KC, Fiszman ML (2001) Hydrogen peroxide-induced neurotoxicity in cultured cortical cells grown in serum-free and serum-containing media. Neurochem Res 26:801–808
Amantea D, Marrone MC, Nistico R, Federici M, Bagetta G, Bernardi G et al (2009) Oxidative stress in stroke pathophysiology validation of hydrogen peroxide metabolism as a pharmacological target to afford neuroprotection. Int Rev Neurobiol 85:363–374
Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF et al (1997) Reduction of cuzn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci 17:4180–4189
Chan PH (1994) Oxygen radicals in focal cerebral ischemia. Brain Pathol 4:59–65
Sambrano GR, Steinberg D (1995) Recognition of oxidatively damaged and apoptotic cells by an oxidized low density lipoprotein receptor on mouse peritoneal macrophages: role of membrane phosphatidylserine. Proc Natl Acad Sci USA 92:1396–1400
Fadok VA, Warner ML, Bratton DL, Henson PM (1998) Cd36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3). J Immunol 161:6250–6257
Yamada Y, Doi T, Hamakubo T, Kodama T (1998) Scavenger receptor family proteins: roles for atherosclerosis, host defence and disorders of the central nervous system. Cell Mol Life Sci 54:628–640
Aderem A, Underhill DM (1999) Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 17:593–623
Stolzing A, Grune T (2004) Neuronal apoptotic bodies: phagocytosis and degradation by primary microglial cells. FASEB J 18:743–745
Nicholson AC (2004) Expression of cd36 in macrophages and atherosclerosis: the role of lipid regulation of PPARgamma signaling. Trends Cardiovasc Med 14:8–12
Vinals M, Bermudez I, Llaverias G, Alegret M, Sanchez RM, Vazquez-Carrera M et al (2005) Aspirin increases CD36, SR-BI, and ABCA1 expression in human THP-1 macrophages. Cardiovasc Res 66:141–149
Hebbel RP, Miller WJ (1984) Phagocytosis of sickle erythrocytes: immunologic and oxidative determinants of hemolytic anemia. Blood 64:733–741
Schwartz RS, Tanaka Y, Fidler IJ, Chiu DT, Lubin B, Schroit AJ (1985) Increased adherence of sickled and phosphatidylserine-enriched human erythrocytes to cultured human peripheral blood monocytes. J Clin Invest 75:1965–1972
Connor J, Pak CC, Schroit AJ (1994) Exposure of phosphatidylserine in the outer leaflet of human red blood cells. Relationship to cell density, cell age, and clearance by mononuclear cells. J Biol Chem 269:2399–2404
Haslett C, Savill JS, Whyte MK, Stern M, Dransfield I, Meagher LC (1994) Granulocyte apoptosis and the control of inflammation. Philos Trans R Soc Lond B Biol Sci 345:327–333
Ren Y, Silverstein RL, Allen J, Savill J (1995) Cd36 gene transfer confers capacity for phagocytosis of cells undergoing apoptosis. J Exp Med 181:1857–1862
Navazo MD, Daviet L, Savill J, Ren Y, Leung LL, McGregor JL (1996) Identification of a domain (155–183) on CD36 implicated in the phagocytosis of apoptotic neutrophils. J Biol Chem 271:15381–15385
Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM (1998) PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93:241–252
Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA et al (2005) Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 25:1647–1653
Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M et al (2004) Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res 94:609–616
Sussan TE, Jun J, Thimmulappa R, Bedja D, Antero M, Gabrielson KL et al (2008) Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One 3:e3791
Maruyama A, Tsukamoto S, Nishikawa K, Yoshida A, Harada N, Motojima K et al (2008) Nrf2 regulates the alternative first exons of CD36 in macrophages through specific antioxidant response elements. Arch Biochem Biophys 477:139–145
Olagnier D, Lavergne RA, Meunier E, Lefevre L, Dardenne C, Aubouy A et al (2011) Nrf2, a PPARgamma alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PLoS Pathog 7:e1002254
Savill J (1997) Recognition and phagocytosis of cells undergoing apoptosis. Br Med Bull 53:491–508
Boullier A, Bird DA, Chang MK, Dennis EA, Friedman P, Gillotre-Taylor K et al (2001) Scavenger receptors, oxidized LDL, and atherosclerosis. Ann N Y Acad Sci 947:214–222, discussion 222–213
Husemann J, Loike JD, Anankov R, Febbraio M, Silverstein SC (2002) Scavenger receptors in neurobiology and neuropathology: their role on microglia and other cells of the nervous system. Glia 40:195–205
Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM (1998) Oxidized ldl regulates macrophage gene expression through ligand activation of PPARgamma. Cell 93:229–240
Eto M, Yoshikawa H, Fujimura H, Naba I, Sumi-Akamaru H, Takayasu S et al (2003) The role of cd36 in peripheral nerve remyelination after crush injury. Eur J Neurosci 17:2659–2666
Janabi M, Yamashita S, Hirano K, Sakai N, Hiraoka H, Matsumoto K et al (2000) Oxidized LDL-induced NF-kappa b activation and subsequent expression of proinflammatory genes are defective in monocyte-derived macrophages from cd36-deficient patients. Arterioscler Thromb Vasc Biol 20:1953–1960
Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G et al (2005) The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci 25:2504–2512
Woo MS, Wang X, Faustino JV, Derugin N, Wendland MF, Zhou P et al (2012) Genetic deletion of CD36 enhances injury after acute neonatal stroke. Ann Neurol 72:961–970
Majno G (1975) The healing hand: man and wound in the ancient world. Harvard University Press, Cambridge, MA
Giulian D, Chen J, Ingeman JE, George JK, Noponen M (1989) The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci 9:4416–4429
Marcus SL, Miyata KS, Zhang B, Subramani S, Rachubinski RA, Capone JP (1993) Diverse peroxisome proliferator-activated receptors bind to the peroxisome proliferator-responsive elements of the rat hydratase/dehydrogenase and fatty acyl-coa oxidase genes but differentially induce expression. Proc Natl Acad Sci USA 90:5723–5727
Aperlo C, Pognonec P, Saladin R, Auwerx J, Boulukos KE (1995) Cdna cloning and characterization of the transcriptional activities of the hamster peroxisome proliferator-activated receptor happar gamma. Gene 162:297–302
Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cesario RM, Jow L et al (1997) Sensitization of diabetic and obese mice to insulin by retinoid x receptor agonists. Nature 386:407–410
Mukherjee R, Jow L, Croston GE, Paterniti JR Jr (1997) Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid x receptor agonists and antagonists. J Biol Chem 272:8071–8076
Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD et al (1997) Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid x receptor. Proc Natl Acad Sci U S A 94:237–241
Diab A, Hussain RZ, Lovett-Racke AE, Chavis JA, Drew PD, Racke MK (2004) Ligands for the peroxisome proliferator-activated receptor-gamma and the retinoid x receptor exert additive anti-inflammatory effects on experimental autoimmune encephalomyelitis. J Neuroimmunol 148:116–126
Okuma Y, Uehara T, Miyazaki H, Miyasaka T, Nomura Y (1998) The involvement of cytokines, chemokines and inducible nitric oxide synthase (INOS) induced by a transient ischemia in neuronal survival/death in rat brain. Nihon Yakurigaku Zasshi 111:37–44
Forster C, Clark HB, Ross ME, Iadecola C (1999) Inducible nitric oxide synthase expression in human cerebral infarcts. Acta Neuropathol 97:215–220
Wang Q, Tang XN, Yenari MA (2007) The inflammatory response in stroke. J Neuroimmunol 184:53–68
Iadecola C, Alexander M (2001) Cerebral ischemia and inflammation. Curr Opin Neurol 14:89–94
Stowe AM, Adair-Kirk TL, Gonzales ER, Perez RS, Shah AR, Park TS et al (2009) Neutrophil elastase and neurovascular injury following focal stroke and reperfusion. Neurobiol Dis 35:82–90
Ishrat T, Sayeed I, Atif F, Hua F, Stein DG (2010) Progesterone and allopregnanolone attenuate blood–brain barrier dysfunction following permanent focal ischemia by regulating the expression of matrix metalloproteinases. Exp Neurol 226:183–190
Jin R, Yang G, Li G (2010) Molecular insights and therapeutic targets for blood–brain barrier disruption in ischemic stroke: critical role of matrix metalloproteinases and tissue-type plasminogen activator. Neurobiol Dis 38:376–385
Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G et al (2000) Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. J Biol Chem 275:32681–32687
Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R et al (1998) Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 395:137–143
Moi P, Chan K, Asunis I, Cao A, Kan YW (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci USA 91:9926–9930
Ishii T, Itoh K, Yamamoto M (2002) Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol 348:182–190
Itoh K, Wakabayashi N, Katoh Y, Ishii T, O’Connor T, Yamamoto M (2003) Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 8:379–391
Giudice A, Montella M (2006) Activation of the Nrf2-are signaling pathway: a promising strategy in cancer prevention. Bioessays 28:169–181
Lee JM, Shih AY, Murphy TH, Johnson JA (2003) NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex i inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem 278:37948–37956
Kraft AD, Johnson DA, Johnson JA (2004) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci 24:1101–1112
Shih AY, Li P, Murphy TH (2005) A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci 25:10321–10335
Leonard MO, Kieran NE, Howell K, Burne MJ, Varadarajan R, Dhakshinamoorthy S et al (2006) Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J 20:2624–2626
Kraft AD, Lee JM, Johnson DA, Kan YW, Johnson JA (2006) Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J Neurochem 98:1852–1865
Satoh T, Okamoto SI, Cui J, Watanabe Y, Furuta K, Suzuki M et al (2006) Activation of the keap1/Nrf2 pathway for neuroprotection by electrophilic [correction of electrophillic] phase II inducers. Proc Natl Acad Sci USA 103:768–773
Zhao J, Kobori N, Aronowski J, Dash PK (2006) Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci Lett 393:108–112
van Muiswinkel FL, Kuiperij HB (2005) The nrf2-are signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord 4:267–281
Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P et al (2005) Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem 280:22925–22936
Park EY, Cho IJ, Kim SG (2004) Transactivation of the PPAR-responsive enhancer module in chemopreventive glutathione s-transferase gene by the peroxisome proliferator-activated receptor-gamma and retinoid x receptor heterodimer. Cancer Res 64:3701–3713
Cho HY, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP et al (2010) Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med 182:170–182
Bowie A, O’Neill LA (2000) Oxidative stress and nuclear factor-kappab activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol 59:13–23
Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW et al (2006) Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 116:984–995
Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB (2010) Oxidative stress, inflammation, and cancer: how are they linked? Free Radic Biol Med 49:1603–1616
Higgins LG, Hayes JD (2011) Mechanisms of induction of cytosolic and microsomal glutathione transferase (GST) genes by xenobiotics and pro-inflammatory agents. Drug Metab Rev 43:92–137
Polvani S, Tarocchi M, Galli A (2012) PPARgamma and oxidative stress: con(beta) catenating Nrf2 and foxo. PPAR Res 2012:641087
Smith SA, Monteith GR, Holman NA, Robinson JA, May FJ, Roberts-Thomson SJ (2003) Effects of peroxisome proliferator-activated receptor gamma ligands ciglitazone and 15-deoxy-delta 12,14-prostaglandin J2 on rat cultured cerebellar granule neuronal viability. J Neurosci Res 72:747–755
Rohn TT, Wong SM, Cotman CW, Cribbs DH (2001) 15-deoxy-delta12,14-prostaglandin J2, a specific ligand for peroxisome proliferator-activated receptor-gamma, induces neuronal apoptosis. Neuroreport 12:839–843
Kondo M, Shibata T, Kumagai T, Osawa T, Shibata N, Kobayashi M et al (2002) 15-deoxy-delta(12,14)-prostaglandin J(2): the endogenous electrophile that induces neuronal apoptosis. Proc Natl Acad Sci USA 99:7367–7372
Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L (2000) Inhibition of ikappaB kinase and ikappaB phosphorylation by 15-deoxy- delta(12,14)-prostaglandin J(2) in activated murine macrophages. Mol Cell Biol 20:1692–1698
Cernuda-Morollon E, Pineda-Molina E, Canada FJ, Perez-Sala D (2001) 15-deoxy-delta 12,14-prostaglandin J2 inhibition of nf-kappaB-DNA binding through covalent modification of the p50 subunit. J Biol Chem 276:35530–35536
Campo PA, Das S, Hsiang CH, Bui T, Samuel CE, Straus DS (2002) Translational regulation of cyclin d1 by 15-deoxy-delta(12,14)-prostaglandin J(2). Cell Growth Differ 13:409–420
Oliva JL, Perez-Sala D, Castrillo A, Martinez N, Canada FJ, Bosca L et al (2003) The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J2 binds to and activates H-Ras. Proc Natl Acad Sci USA 100:4772–4777
Park EJ, Park SY, Joe EH, Jou I (2003) 15d-PGJ2 and rosiglitazone suppress Janus kinase-stat inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J Biol Chem 278:14747–14752
Diamant M, Heine RJ (2003) Thiazolidinediones in type 2 diabetes mellitus: current clinical evidence. Drugs 63:1373–1405
Belcher G, Lambert C, Edwards G, Urquhart R, Matthews DR (2005) Safety and tolerability of pioglitazone, metformin, and gliclazide in the treatment of type 2 diabetes. Diabetes Res Clin Pract 70:53–62
Lincoff AM, Wolski K, Nicholls SJ, Nissen SE (2007) Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA 298:1180–1188
Dana SL, Hoener PA, Bilakovics JM, Crombie DL, Ogilvie KM, Kauffman RF et al (2001) Peroxisome proliferator-activated receptor subtype-specific regulation of hepatic and peripheral gene expression in the zucker diabetic fatty rat. Metabolism 50:963–971
Nedergaard M (1987) Transient focal ischemia in hyperglycemic rats is associated with increased cerebral infarction. Brain Res 408:79–85
Demchuk AM, Morgenstern LB, Krieger DW, Linda Chi T, Hu W, Wein TH et al (1999) Serum glucose level and diabetes predict tissue plasminogen activator-related intracerebral hemorrhage in acute ischemic stroke. Stroke 30:34–39
Thal SC, Engelhard K, Werner C (2005) New cerebral protection strategies. Curr Opin Anaesthesiol 18:490–495
Martini SR, Kent TA (2007) Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab 27:435–451
Lee J, Reding M (2007) Effects of thiazolidinediones on stroke recovery: a case-matched controlled study. Neurochem Res 32:635–638
Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK et al (2005) Secondary prevention of macrovascular events in patients with type 2 diabetes in the proactive study (prospective pioglitazone clinical trial in macrovascular events): a randomised controlled trial. Lancet 366:1279–1289
Betteridge DJ, DeFronzo RA, Chilton RJ (2008) Proactive: time for a critical appraisal. Eur Heart J 29:969–983
Scheen AJ (2012) Outcomes and lessons from the proactive study. Diabetes Res Clin Pract 98:175–186
Wilcox R, Bousser MG, Betteridge DJ, Schernthaner G, Pirags V, Kupfer S et al (2007) Effects of pioglitazone in patients with type 2 diabetes with or without previous stroke: results from proactive (prospective pioglitazone clinical trial in macrovascular events 04). Stroke 38:865–873
Wilcox R, Kupfer S, Erdmann E (2008) Effects of pioglitazone on major adverse cardiovascular events in high-risk patients with type 2 diabetes: results from prospective pioglitazone clinical trial in macro vascular events (proactive 10). Am Heart J 155:712–717
Nissen SE, Wolski K (2007) Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med 356:2457–2471
Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, Ali F, Sholley C, Worrall C et al (2010) Risk of acute myocardial infarction, stroke, heart failure, and death in elderly medicare patients treated with rosiglitazone or pioglitazone. JAMA 304:411–418
Charbonnel B, Dormandy J, Erdmann E, Massi-Benedetti M, Skene A (2004) The prospective pioglitazone clinical trial in macrovascular events (PROactive): can pioglitazone reduce cardiovascular events in diabetes? Study design and baseline characteristics of 5238 patients. Diabetes Care 27:1647–1653
Mannucci E, Monami M, Lamanna C, Gensini GF, Marchionni N (2008) Pioglitazone and cardiovascular risk. A comprehensive meta-analysis of randomized clinical trials. Diabetes Obes Metab 10:1221–1238
Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A et al (2008) Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the periscope randomized controlled trial. JAMA 299:1561–1573
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Zhao, X., Aronowski, J. (2014). The Role of PPARγ in Stroke. In: Chen, J., Hu, X., Stenzel-Poore, M., Zhang, J. (eds) Immunological Mechanisms and Therapies in Brain Injuries and Stroke. Springer Series in Translational Stroke Research, vol 6. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8915-3_17
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8915-3_17
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8914-6
Online ISBN: 978-1-4614-8915-3
eBook Packages: MedicineMedicine (R0)