
Abstract
The mammalian host has evolved to develop a diverse array of innate immune receptors and strategies to defend itself against infection by microbial pathogens. These germ-line-encoded and conserved microbial receptors, called pattern recognition receptors (PRRs), are associated with the membranes or within the cytosol of host cells. PRRs enable the host to rapidly respond to pathogen-associated molecular patterns (PAMPs), as a first line of defence against microbial intrusion. Signalling via PAMPs enables the host to mount a rapid and non-specific immune response that results in inflammation and ultimately the activation of the adaptive immune system.
The host has a variety of PRRs, including the membrane-bound toll-like receptors (TLRs) and the cytoplasmic nucleotide-binding oligomerisation domain (Nod)-like receptor (NLR) protein family. In this chapter, we will focus predominantly on Nod1 and Nod2, which are members of the NLR family of proteins, and the role they have in the initiation and development of an immune response to bacteria. We will discuss the various methods whereby bacteria are detected and can induce signalling via Nod receptors and the role of Nod proteins in human disease, especially Nod2’s role in Crohn’s disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Nod-Like Receptors
Mammalian cells express one or more types of cytosolic PRRs that play important roles in host defence against microbial pathogens, in addition to their recognition of “danger signals” from within eukaryotic cells. One family of mammalian PRRs is the nucleotide oligomerisation domain (Nod) family, which have a central role in host defence against microbial pathogens [1]. Since the initial discovery of the first Nod-like receptors (NLRs), being Nod1 and Nod2, this family of PRRs expanded to currently consist of 23 genes in humans and 34 genes in mice [2, 3]. Hence, due to the rapid expansion of this group of cytoplasmic PRRs, it has been renamed the nucleotide-binding domain (NBD) and leucine-rich repeat (LRR) containing, or NLR family [4].
The NLR family is comprised of cytoplasmic proteins that are suggested to be sentinel receptors at front-line mucosal surfaces as well as in immune cells [5]. NLR proteins share some common features, being a C-terminal LRR-containing domain and central NACHT NBD(s) [1, 6]. The NLR family is now divided into four subfamilies based on the composition of the n-terminal effector domain of these receptors [4]. The Nod proteins, Nod1 and Nod2, contain a caspase-activated recruitment domain (CARD) at their n-terminus and are classified within the NLRC subfamily. This chapter will predominantly focus on the expression, detection and immunoregulation initiated by Nod1 and Nod2 in response to bacterial pathogens and their contribution to the regulation of gastrointestinal homeostasis, inflammation and immunity.
Nod1 and Nod2
The mammalian Nod proteins, Nod1 and Nod2, are critical in the regulation of inflammation and host defence against bacterial infections [2]. Nod1 and Nod2 proteins are located within the cytosolic compartment of host cells, and it has been suggested that their role in controlling the intestinal microbiota may have been a major selective pressure throughout evolution [3]. Nods are comprised of three domains: the first being a central nucleotide-binding oligomerisation domain (Nod) that is required for self-oligomerisation of the receptor. Secondly, Nods have a C-terminal LRR domain that contains multiple LRRs whose function is to sense the bacterial pathogen-associated molecular pattern (PAMP), being peptidoglycan. Finally, all Nods have a CARD. The CARD is essential for the homodimerisation of the receptor, and the recruitment of downstream adaptor proteins through homophilic and heterophilic protein interactions that are required to facilitate the pro-inflammatory signalling cascade in response to bacterial PAMP recognition [7]. Nod1 contains only one CARD domain, whereas Nod2 contains two CARD domains.
Initially, Nod1 and Nod2 were proposed to be intracellular sensors of bacterial lipopolysaccharide (LPS) [8, 9] and were suggested to have a role in regulating apoptosis and the NF-kappaB pathway [10]. However, the initial finding that Nods detected LPS was incorrect due to contaminants contained within the LPS preparations used in these studies. Further refinement and purification of bacterial cell preparations resulted in the identification that Nods sense bacterial peptidoglycan fragments contained within the cell wall of bacteria; however, the motifs recognised by Nod1 and Nod2 differ [11, 12].
In the host, Nod1 is expressed ubiquitously by most cell types [13–15]. Nod1 detects a specific and conserved structure of peptidoglycan that is commonly found in almost all Gram-negative bacterial peptidoglycan, as well as some Gram-positive bacteria such as Bacillus subtilis and Listeria monocytogenes [16, 17]. The muropeptide structure detected by Nod1 is composed of a disaccharide moiety, N-acetylglucosamine–N-acetylmuramic acid (GlcNAc–MurNAc), linked to a tripeptide of which the terminal amino acid is meso-diaminopimelate (mDAP), also known as GM-TriDAP [16, 17]. Most Gram-negative organisms contain mDAP within their cell wall [18]. Furthermore, most Gram-positive bacteria contain a lysine residue at the terminal position of their peptidoglycan, rendering their peptidoglycan incapable of signalling via Nod1. Interestingly, recognition of peptidoglycan by Nod1 is host specific, as human Nod1 specifically detects the GM-TriDAP structure of peptidoglycan [17]. However, murine Nod1 is most responsive to a tetrapeptide muropeptide containing l-alanine–d-glutamate–mesoDAP–d-alanine (GM-TetraDAP) [19].
Nod2 was first identified approximately 10 years ago [20], and similar to Nod1, it also is composed of a NBD and multiple C-terminal LRRs; however, it has two N-terminal CARDs. Nod2 expression is mainly restricted to leukocytes consisting of T cells [21], neutrophils [22], macrophages [20] and dendritic cells. In addition, Nod2 is expressed at low levels by intestinal epithelial cell lines and primary intestinal epithelial cells [10, 23, 24]. The expression of Nod2 is basal within these cells; however, its expression can be induced by a variety of inflammatory signals such as LPS, tumour necrosis factor (TNF) and interferon gamma (IFN-gamma) [23–25]. In contrast to Nod1, Nod2 is considered to be a sensor of both Gram-negative and Gram-positive bacteria due to its ability to detect muramyl dipeptide (MDP), a component common to the peptidoglycan of both classes of bacteria [11, 12]. Therefore, Nod2 regulates the production of inflammatory mediators in response to all types of bacterial pathogens in order to maintain gut homeostasis [6].
More recently, the importance and impact of Nods on immune responses and in pathogenesis now extends beyond detecting bacteria. Nods have been implicated in the progression and development of gut homeostasis, chronic asthma, arthritis, dermatitis, IBD, multiple sclerosis [26], obesity [27], Chagas disease [28] and malaria [29, 30]. Conversely, over-activation of Nods can result in the development of autoinflammatory diseases such as Blau syndrome and sarcoidosis [31]. Collectively, these studies highlight that Nods may contribute to the progression and development of disease of various aetiologies; however, the mechanisms whereby they contribute to these diseases are not well understood and remain to be elucidated.
Nods Initiate a Pro-inflammatory Signalling Cascade
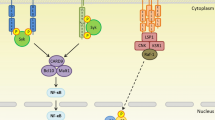
The detection of bacterial peptidoglycan by Nods initiates a signalling cascade that ultimately results in the production of pro-inflammatory cytokines and the development of an inflammatory innate immune response (see Fig. 10.1). The classical pathway of Nod signalling is as follows. Upon Nod recognition of peptidoglycan via its LRRs, it is speculated that Nod receptors self-oligomerise via their Nod domains. This activation and homodimerisation of Nods enables them to mediate the recruitment and oligomerisation of the RIP-like interacting CLARP kinase (RICK) [9, 32, 33], also known as receptor-interacting protein-2 (RIP-2), a member of the receptor-interacting protein kinase family [34]. RIP-2 subsequently interacts via an electrostatic interaction with the CARD domain(s) contained within Nod receptors [33, 35]. The interaction between the CARD of RIP-2 and Nods is specific and essential to the signalling process, as Nod1 signalling can be abolished when a truncated form of RIP-2 lacking the CARD is transfected into cells [20]. The homophilic CARD–CARD interaction between Nods and RIP-2 results in RIP-2 being subsequently K63-polyubiquitinated within its kinase domain by the E3 ubiquitin ligases cIAP1, cIAP2 and xIAP [36, 37]. This in turn initiates the K63-linked polyubiquitination of NEMO, a scaffolding protein and regulator of the IkappaB kinase (IKK) complex [38, 39]. Therefore, the polyubiquitination of RIP-2 is essential for the activation of IKK, which in the case of Nod2 signalling, subsequently mediates via polyubiquitinated NEMO the recruitment of the transforming growth factor-beta (TGF-beta)-associated kinase (TAK1) [32], in addition to the TAB1 and TAB2 complex. The interaction between Nods, IKK and TAK1 results in the phosphorylation and degradation of the IKK complex, and the degradation of IkappaB proteins by the proteasome [10, 20, 32, 33]. This in turn ultimately facilitates the dissociation of the NF-kappaB p50 and p65 complex and the phosphorylation and translocation of the p65 subunit into the nucleus, enabling it to bind to consensus binding sites within pro-inflammatory genes. This allows the transcription of pro-inflammatory molecules including CXCL5, CXCL8 and its murine homologue CXCL2 (or macrophage inflammatory protein-2, Mip-2) [17, 24, 40].

Pro-inflammatory signalling mediated by Nod1 and Nod2. Nod1 and Nod2 detect their peptidoglycan ligands, N-acetylglucosamine–N-acetylmuramic acid-l-Ala–d-Glu–meso-diaminopimelic acid (GM-triDAP) and muramyl dipeptide (MDP), respectively, to trigger inflammation. Through their recruit of Rip2, Nod1 and Nod2 trigger NFkappaB and MAPK pathways to drive inflammatory cytokine production. Nod1 has also been shown to active IRF7 downstream of TBK1 and IKK epsilon leading to type I interferon production (IFB-beta) (Courtesy of artist: Priya Alwis)
Although significant advances have been made to broaden our understanding of the mechanisms and pathway(s) of Nod activation and Nod-dependent pro-inflammatory responses, certain key components of this pathway are yet to be elucidated. For example, the mechanism and location(s) where Nods directly interact with peptidoglycan and RIP-2 remain unknown. It has been proposed that when Nods are in an inactive state, their LRR remain folded over the Nod region and upon sensing their ligand undergo conformational changes that allow the homodimerisation of the receptors [1, 41]. Furthermore, our knowledge of Nod binding partners is limited. Two different protein interaction screens identified the LRR- and PDZ domain-containing family member Erbin as a binding partner of Nod2; however, the cellular impact of this interaction and its role in Nod signalling remains unclear [42, 43]. Additional screens have also found other Nod2 interactors, including Grim19 [30] and, more recently, carbamoyl phosphate synthetase/aspartate transcarbamylase/dihydroorotase (CAD [44]), but their overall contribution to bacterial detection and Crohn’s disease pathogenesis is still unclear. Moreover, researchers have postulated that additional cytoplasmic host structures may be required for Nod signalling, similar to the inflammasome, and hence, the existence of a “Nodosome” or “Nod signalosome” has been proposed [45, 46]. Further studies are required to address these key steps in the process of Nod activation and ligand recognition.
Outcome of Nod Signalling
The Production of Antimicrobial Peptides
Signalling via the Nod1 and Nod2 receptor pathways results in the production of antimicrobial peptides, known as defensins, by epithelial cells. The production of defensins in response to Nod2 signalling is essential for the regulation of commensal organisms and maintaining gut homeostasis, as Nod2-defficient mice have an impaired regulation of bacterial load in their terminal ileum [47]. This study was performed by Petnicki-Ocwieja and colleagues, who isolated the intestinal crypts of wild-type C57BL/6 and Nod2 knockout mice and subsequently cultured them with bacteria. The supernatant obtained from wild-type crypts cultured with bacteria displayed potent antimicrobial activity against Escherichia coli, Salmonella and Listeria monocytogenes in a dose-dependent manner. Whereas supernatants from crypts isolated from Nod2 or RIP-2 knockout mice and stimulated with bacteria were hindered in their antimicrobial function [47]. The antimicrobial activity was attributed to Nod2 signalling inducing the production of alpha-defensins, which are small, cationic antimicrobial peptides produced by Paneth cells of the intestine [47]. In addition, a second study identified that both Nod2 and RIP-2 expressed by epithelial cells located within the intestinal ileal crypts facilitated the protection of mice against intestinal Helicobacter hepaticus-induced inflammation, due to the production of alpha-defensins which function in controlling the pathogen [48]. Furthermore, Nod2-dependent production of alpha-defensins protects mice against the intracellular pathogen Listeria, as Nod2 knockout mice display an inability to produce intestinal antimicrobial alpha-defensin peptides, known as cryptdins [49]. These findings have been corroborated in vivo by Wehkamp and colleagues, who identified that the production of alpha-defensins by Paneth cells in the intestine of Crohn’s disease patients was reduced, and this decrease was most pronounced in patients with mutations in Nod2, suggesting that Nod2 and alpha-defensins may have a role in regulating the integrity and homeostasis of the gastrointestinal tract [50]. It should be noted that a Nod2-independent, MyD88-dependent mechanism for the production of antimicrobial peptides by Paneth cells has been reported [51], and this system may potentially function in unison with Nod2 to regulate the level of microbial flora within the gut, which will be discussed in detail below.
Indeed, a similar role for Nod1 in maintaining the intestinal microbiota homeostasis has been reported. Bouskra et al., identified that Nod1 knockout mice have a greater total number of bacteria in their gut, possibly due to the lack of beta-defensin antimicrobials produced at the intestinal epithelial surface in the absence of Nod1 signalling [52]. The antimicrobial peptides human beta-defensins (HBDs) are small cationic, low molecular weight peptides with immunomodulatory properties required for host defence from bacterial pathogens [53]. HBDs are endogenously produced by epithelial cells and their expression can be upregulated during infection in a Nod1-dependent manner. We, and others, identified that the gastric pathogen Helicobacter pylori induced the production of HBD2 by human epithelial cells in an NF-kappaB and Nod1-dependent manner [54, 55]. Furthermore, we demonstrated that culture supernatants obtained from H. pylori-stimulated epithelial cells contained HBD2 which exerted potent antimicrobial activity against H. pylori, and that Nod1 was essential for the production of this functional antimicrobial [55]. Similarly, the ability of Nod2 to induce the expression of HBDs has been demonstrated using MDP [56]. Collectively, these studies identify the ability of Nods to induce the production of alpha- and beta-defensins that function to regulate the overall number of the intestinal microbiota and reduce the ability of pathogenic bacteria to colonise the gastrointestinal tract.
The Production of Type I Interferons (IFNs)
Work by Watanabe and colleagues identified a novel pathway of Nod1 signalling, resulting in the induction of type I interferons (IFNs), an immune response typically associated with a viral infection [57]. The authors demonstrated that H. pylori stimulation of gastrointestinal epithelial cells initiated Nod1 signalling, the activation of RIP-2 and its interaction with the TNF receptor-associated factor 3 (TRAF3). This resulted in the sequential activation of TANK-binding kinase 1 (TBK1), IκB kinase epsilon (IKK epsilon) and the IFN regulatory factor 7 (IRF7). Subsequently, IRF7 can activate the transcription factor complex IFN-stimulated gene factor 3 (ISGF3), composed of Stat1, Stat2 and IRF9, enabling it to bind to an IFN-stimulated response element (ISRE), resulting in the production of the pro-inflammatory cytokines CXCL10, also known as IP-10 and IFN-beta [57] (see Fig. 10.1). The authors propose that this will in turn result in the generation of a pro-inflammatory T helper1 (Th1) response as a result of H. pylori infection.
The Production of Inflammatory Cytokines and the Recruitment of Innate Immune Cells
One of the key outcomes of Nod signalling is the production of cytokines, resulting in the recruitment and activation of pro-inflammatory innate immune cells. Studies using knockout animals have clarified the contribution of Nods to pathogen-initiated inflammation. Work by Masumoto and colleagues identified that administration of a Nod1 ligand intraperitoneally to wild-type mice induced neutrophil recruitment and the production of CCL2, also known as monocyte chemotactic protein-1 (MCP-1), and CXCL2 in the serum of these animals. However, Nod1 knockout mice displayed an inability to produce CCL2 in their serum in response to Nod1 ligand administration. Their findings clearly identified a role for Nod1 in the production of pro-inflammatory cytokines that functions to recruit monocytes and dendritic cells to the site of infection, further enhancing the development of a cellular innate and adaptive immune response [58]. In addition, Nod1-mediated neutrophil recruitment is an important immune response against the enteric Gram-positive pathogen Clostridium difficile [59]. C. difficile is normally located within the intestinal tract of healthy individuals, where its levels are maintained by the intestinal microbiota; however, in antibiotic-treated individuals, it is the causative agent of pseudomembranous colitis [60]. Work by Hasegawa and colleagues identified that Nod1 knockout mice infected with C. difficile in their intestinal tract were more prone to lethality due to an impaired clearance of the pathogen, compared to wild-type controls [59]. The impaired clearance of C. difficile in Nod1 knockout animals was dependent on a defect in the ability of these animals to produce CXCL1 and induce the recruitment of neutrophils to the infected site.
Similarly, Nod2 signalling by pathogens results in the secretion of pro-inflammatory cytokines and the recruitment of inflammatory cells, and some examples are listed below. Clearance of the enteric pathogen Citrobacter rodentium is regulated by Nod2-induced production of CCL2, which enables the recruitment of inflammatory monocytes into the colon and the induction of an adaptive immune response [61]. Streptococcus pneumoniae infection of phagocytes results in the production of CCL2, which propagates the inflammatory response by inducing the recruitment of macrophages to the site of infection to assist in the clearance of the pathogen [62]. Furthermore, it was shown in vivo using Nod2 knockout mice that Nod2 was also required for the generation of an antibody response specific for S. pneumoniae [62]. Also, infection with the Nod2-signalling bacterium Mycobacterium, influences the production of TNFα and IL1-β by macrophages in addition to regulating the ability of macrophages to control the intracellular growth of this pathogen [63]. This finding was validated in vitro using siRNA to knockdown of Nod2 and observing that the lack of Nod2 enhanced growth of Mycobacterium in macrophages [63]. A genetic association of Nod2 in regulating M. leprae infection was identified by performing a genome analysis of patients with leprosy, revealing that these infected individuals had a single-nucleotide polymorphism (SNP) in Nod2 that may be attributed to the disease outcome [64]. Moreover, using RIP-2 knockout animals, and peripheral blood mononuclear cells from individuals homozygous for a Nod2 polymorphism, it was determined that Nod2 plays a key role in the production of the pro-inflammatory cytokines IL-10, IL-6 and IL-1β in response to Borrelia, the causative agent of Lyme disease [65]. Finally, Legionella pneumophila has been reported to activate both Nod1 and Nod2, resulting in the induction of NF-kappaB and IFN-beta [66]. These researchers also showed that Nod1 is essential for the clearance of L. pneumophila in vivo [66].
The Production of Reactive Oxygen Species
In addition to driving innate immune cells to produce pro-inflammatory cytokines and facilitate the further recruitment of innate immune cells, Nod signalling can enhance the production of reactive oxygen species by innate immune cells. Moreover, Nod expression can be upregulated in the presence of pro-inflammatory cytokines, further enhancing the innate immune response initiated via Nod signalling. A study by Totemeyer and colleagues identified that IFN-gamma increased the expression of Nod2 within macrophages, heightening the production of the antimicrobial nitric oxide (NO) [67]. Furthermore, Nod1 stimulation with Gram-negative peptidoglycan or bacteria resulted in the expression of inducible nitric oxide synthase and NO production in combination with IFN-gamma in a diverse range of host cell types including bone marrow-derived dendritic cells [68] macrophages [19, 69], hepatocytes [70], mesothelial cells and smooth muscle cells [71]. An in vivo example of the requirement of Nods in the clearance of pathogens via NO production has been reported using Chlamydophila pneumoniae [72]. Clearance of C. pneumoniae in Nod1, Nod2 and RIP-2 knockout animals was impaired, due to an inability to induce iNOS expression and NO production, which subsequently resulted in delayed neutrophil recruitment to the lungs [72].
Enhanced Phagocytosis by Innate Immune Cells
Nod1 stimulation can promote and enhance the ability of innate immune cells such as macrophages and neutrophils to phagocytose pathogenic organisms. Indeed, peptidoglycan fragments from the Gram-negative organism Haemophilus influenzae were capable of inducing neutrophils to phagocytose opsonised Gram-positive S pneumoniae [73]. This finding was further corroborated using a murine infection model, whereby neutrophils from mice treated with purified peptidoglycan ligands or Nod1-signalling H. influenzae displayed increased killing of S. pneumonia in a Nod1-dependent manner. Moreover, the requirement for Nod1 to facilitate phagocytosis of bacterial pathogens by neutrophils was further validated in vivo, as Nod1 knockout mice administered with H. influenzae prior to infection with S. pneumoniae had an impaired ability to clear the pathogen [73]. In addition, a second study reported that peptidoglycan originating from the intestinal microbiota may facilitate in priming bone marrow-derived neutrophils to display enhanced killing of S. pneumoniae and Staphylococcus aureus [22]. The authors demonstrated by colonising germ-free mice with Escherichia coli containing radiolabelled peptidoglycan that during colonisation, peptidoglycan from intestinal organisms can translocate across the intestinal mucosa, entering the circulation where it can facilitate in the development of neutrophil function [22].
Autophagy
Autophagy is a cellular cytoplasmic process that targets intracellular components for degradation and occurs downstream of the early endosome pathway [74, 75]. The process of autophagy is essential for the clearance of cytosolic cargo, being either damaged host organelles or proteins, or as a defence mechanism for the degradation of internalised bacterial or viral pathogens [74, 75].
Three studies have recently reported the ability of Nods to regulate the intracellular degradation process of autophagy in response to bacterial pathogens. We identified that both Nod1 and Nod2 are required for autophagy in response to bacteria, using the invasive pathogen Shigella flexneri [76]. Furthermore, we identified that Nods interacted with ATG16L1, a component of the autophagosome, enabling its recruitment to the cellular site of bacterial entry into host cells to establish autophagy. Indeed, mutations in ATG16L1 are linked to susceptibility of Crohn’s disease, providing a possible physiological relevance for the requirement of Nods in bacterial-induced autophagy, discussed in further detail below. Other research groups have also established a requirement for Nods in the development of bacteria-induced autophagy and the regulation of an inflammatory response in Nod-stimulated human dendritic and colonic epithelial cells [77, 78]. However, there are some key differences between the findings reported by all three groups. Cooney et al. identified that Nod2 was required for the induction of autophagy in dendritic cells and promoting the generation of an adaptive immune response as a result of autophagy-induced increased antigen presentation within MHC II complexes [77]. This study also identified that Nod2-dependent autophagy required the autophagy-related proteins ATG5, ATG7 and ATG16L and was dependent on RIP-2 [77]. Similarly, Homer and colleagues also identified that Nod-2 dependent autophagy required RIP-2 [78]. Whereas, contrary to these findings, work by Travassos and colleagues showed that Nod1-induced autophagy of intracellular bacteria was RIP-2 independent, as ATG16 could co-localise with Nod1-signalling Shigella in RIP-2-deficient mouse embryonic fibroblasts (MEFs). This clear discrepancy in findings for the requirement of RIP-2 between groups may potentially be a difference in the cell type examined, or the pathogen model used, as Travassos focused on epithelial cells and murine macrophages using a Shigella pathogen model [76]. Whereas work by Cooney and Homer focused specifically on human dendritic cells stimulated with Nod-ligands [77] and Salmonella-infected colonic cells, respectively [78]. Further work is required to elucidate the exact role of RIP-2 in Nod-induced autophagy.
Development of the Gut Microbiota
The importance of Nods in the development of the intestinal microbiota is in part due to their location at the mucosal surfaces, in addition to their rapid ability to sense the presence of bacteria and produce antimicrobial peptides that function to control the bacterial burden at these sites. Using Nod2 knockout animals, it has been identified that this pattern recognition molecule (PRM) plays a key role in the composition of the intestinal microbiota during development [79]. Rehman and colleagues examined the faecal and ileal microbiota compositions in wild-type C57BL/6 and Nod2 knockout animals by generating a 16s ribosomal RNA clone library. This study identified that there was a shift in the composition of the ileal and faecal microbiota composition in Nod2 knockout animals when compared to C57BL/6 control mice [79]. Indeed, they identified that in the absence of Nod2, elevated total bacterial numbers were present within the faeces and terminal ileum of mice compared to their wild-type controls, and that Nod2 knockout mice displayed increased numbers of Bacteroidetes and Firmicutes compared to control mice [79]. In addition, a second study reported of a similar increase in the numbers of Bacteroides, Firmicutes and Bacillus spp. present in the terminal ilea of Nod2-deficient or RIP-2-deficient animals [47]. Interestingly, the regulation of Nod2 in the development of the gastrointestinal microbiota occurred early in the developmental stage of these animals, as an altered microbial composition was evident upon weaning of these mice [79].
The importance of the increased number of Firmicutes and Bacteroides spp. in these knockout animals is apparent when comparisons are made to the microbiota of Crohn’s disease patients. Individuals who were homozygous for the Nod2 SNP13, commonly associated with Crohn’s disease, displayed elevated numbers of Firmicutes and Bacteroides in their ileum compared to their healthy counterparts, suggesting that Nod2 regulation of the intestinal microbiota is associated with a genetic predisposition to Crohn’s disease [79]. This genetic alteration may account for the dysregulation in the intestinal microbiota of Crohn’s disease patients, as Nod2 may be required to suppress the levels of opportunistic pathogens in these individuals [79].
Role in Intestinal Development
In addition to controlling the bacterial composition of the intestinal tract, Nod signalling induced by the microbial flora also contributes to the development of lymphoid follicles within the intestinal tract. Bouskra et al. identified that Nod1 signalling by the microbiota present within intestinal crypts resulted in the production of defensins and CCR6 signalling, ultimately facilitating the development and formation of lymphoid follicles [52]. Using bone marrow chimaeras, it was determined that Gram-negative bacterial commensals signalling via Nod1 present within intestinal epithelial cells, and not haematopoietic cells, were responsible for the development of intestinal lymphoid follicles within animals [52]. The result of Nod1 signalling in epithelial cells by the intestinal microbiota subsequently enabled the host to generate polymeric IgA antibodies, that are immunoreactive against the intestinal flora, and progress the development of Peyer’s patches and mesenteric lymph nodes that drain the intestinal tissue [52]. Ultimately, these studies identified that the impaired development of intestinal lymphoid follicles in Nod1 knockout animals resulted in an altered microbial flora, in addition to identifying a direct function of microbial Nod1 signalling in the development of the lymphoid compartment and the generation of secondary lymphoid tissues [52]. These findings have been implicated as having a role in shaping the development of the mucosal lymphoid compartment of Crohn’s disease individuals.
The Development of Adaptive Immune Responses
Innate immune responses initiated by PRRs such as Nods are broad, have been conserved throughout evolution and ultimately result in the recruitment of pro-inflammatory cells such as dendritic cells, macrophages or neutrophils to the site of infection. These activated innate immune cells produce cytokines, as described in the aforementioned sections, and facilitate further the recruitment of adaptive inflammatory immune cells. Activation of the adaptive immune system results in the generation of a pathogen-specific response and involves T cells that are responsible for the progression of inflammation, or B cells that are required for the generation of a humoral, antibody-mediated response.
To date, most of the studies examining Nod-dependent adaptive immune responses have used Nod ligands as an adjuvant in conjunction with model antigens, such as the chicken egg ovalbumin protein, OVA. A study by Fritz and colleagues was one of the first to report the requirement for Nod1 in priming antigen-specific T cell development and humoral antibody responses in vivo [80]. Indeed, using Nod1-deficient mice, and Nod1-deficient animals reconstituted with bone marrow from Nod1-competent animals, this study reported that Nod1 stimulation in non-haematopoietic cells was responsible for priming antigen-specific Th2 immunity in response to injection with the Nod1 ligand FK156 and ovalbumin as an antigen [80]. The Th2 immune response observed was characterised by the presence of IL-4- and IL-5-producing CD4+ T cells and antibodies of the IgG1 subtype [80]. This study has subsequently been validated and expanded upon by Magalhães and colleagues, who showed that both Nod1 and Nod2 activation results in the development of a Th2-dependent immune response in vivo and that RIP-2 is essential for the establishment of Nod-dependent adaptive immune responses [81]. Again, this study relied on the injection of Nod agonists into wild-type and Nod knockout animals, and adaptive immune responses to OVA were determined. The authors identified that wild-type mice administered with both FK156 and OVA displayed elevated IL-4- and IL-5-producing cells in their spleens in addition to IgG1 antibody responses, which were abrogated in RIP-2-deficient animals [81]. Indeed, using bone marrow chimeric mice, a second study by the same authors identified that these responses are dependent on Nod1 and Nod2 expression by cells within the stromal compartment, and not by dendritic cells, considered as the most potent antigen-presenting cell in the host [82]. Therefore, the initial Nod1-signalling response triggered in epithelial cells can direct the hosts dendritic cells to initiate the development of a T helper 1 (Th1) inflammatory response and T helper 2 (Th2) humoral response, resulting in antibody production. Furthermore, Nod1 can function in combination with toll-like receptor (TLR) stimulation to enhance the adaptive immune response and initiate the development of a Th1, Th2 and Th17 responses [80].
In addition to Nod1, Nod2 signalling is also capable of facilitating the development of the adaptive arm of the immune response. Nod2-deficient T cells have an impaired ability to induce the production of IL-2 and IFN-gamma and function similarly to T cells that lack CD28, a costimulatory molecule required to enable the clonal expansion of T cells and the generation of Th1 immunity [83]. However, it should be noted that this finding was observed using a model of the intracellular parasite, Toxoplasma gondii [83]. Furthermore, Nod2 expressed by human dendritic cells seems to be required for bacterial processing and handling, via autophagy, and the generation of CD4+ T cell responses, possibly due to presentation of antigen via MHC class II to T cells [77]. This finding was validated using human dendritic cells with impaired Nod2 function displaying an inferior ability to induce antigen-specific T cell responses [77].
Th17 CD4+ T cells are one of the most recently discovered adaptive immune cells and are characterised by their secretion of IL-17 which functions in recruiting neutrophils to the site of inflammation [84]. In addition, Th17 cells also produce IL-22, which facilitates the production of antimicrobial peptides and tissue repair factors by epithelial cells [85]. Recently, the role of Nods in the generation of innate Th17 immune cells during microbial infection was determined [86]. Using the animal models of Citrobacter rodentium- and S. typhimurium-induced colitis, it was identified that both Nod1 and Nod2 were essential for the induction of mucosal Th17 immune responses early during infection, being 4 days and 24 h postinfection with the respective pathogens. The Th17 immune response initiated by innate Th17 cells (iTh17) was dependent on the expression of IL-6, and these cells were essential for the development of mucosal immunity against both bacterial pathogens [86]. Using Nod1 and Nod2 knockout animals, it was determined that Nods were essential for the generation of Th17 responses to mucosal pathogens, as these knockout animals displayed an increase in the burden of infection and reduced pathology, when compared to wild-type control mice [86]. This is the first identification of the requirement for Nods in the development of the third arm of the adaptive immune response, being the rapid development of iTh17 immunity, which may function to fill the immune gap until the mature adaptive immune response develops [86].
Finally, Nods have been identified as having a role in regulating the development of an adaptive immune response, via the generation and regulation of Foxp3 expressing suppressive T regulatory cells (Tregs) [21]. Tregs function to maintain self-tolerance in the host and suppress autoreactive T cells in the periphery via their production of the immunosuppressive cytokine IL-10 [87]. Peripheral blood mononuclear cells from Crohn’s disease patients expressing a mutation in their Nod2 gene had a low production of the immunosuppressive cytokine IL-10, indicating a potential role for Nod2 in facilitating the suppression of immunoreactive T cells located within the periphery [88]. Furthermore, Rahman and colleagues reported the ability of the Nod2 agonist MDP to activate NF-kappaB in Tregs, hence protecting them from Fas-mediated apoptosis or programmed cell death [21]. Moreover, this study reported of a deficiency in Tregs located within the lamina propria of patients who were homozygous for nod2 variants, providing a possible explanation for the chronic inflammatory response observed in these Crohn’s disease patients [21].
How Bacteria Signal via Nod1 and Nod2
Bacteria utilise numerous mechanisms to release their peptidoglycan and facilitate its entry into host cells. Similarly, the host uses a variety of mechanisms to enable the uptake of peptidoglycan into the cytoplasm to initiate Nod signalling. These mechanisms are discussed in detail below and are summarised in Fig. 10.2.

Mechanisms that facilitate entry of Nod ligands into the cytoplasm of target cells. There are a number of ways that peptidoglycan has been shown to enter into the cytoplasm to interact with either Nod1 or Nod2. These include (1) phagocytosis, (2) endocytosis, (3) entry through pores made by pore-forming toxins, (4) delivery through outer membrane vesicles (OMVs), (5) peptide transporters and (6) bacterial secretion systems (Courtesy of artist: Priya Alwis)
Invasion
A number of bacteria have been reported to be capable of inducing Nod signalling, with a dependence on the bacteria being viable or actively invading host cells (see Fig. 10.2). One of the earliest examples identifying that invasive pathogens could initiate NF-kappaB-dependent IL-8 production potentially via intracellular Nod1 was demonstrated using invasive Shigella flexneri [9]. This study also reported that NF-kappaB-dependent IL-8 production was not induced by noninvasive nor heat-killed Shigella [9]. This finding was subsequently confirmed, as viable and invasive Chlamydophila pneumoniae could induce Nod1 signalling and the secretion of IL-8 by human endothelial cells, whereas this was not evident when epithelial cells were stimulated with heat-inactivated organisms [89]. Furthermore, a third study demonstrated that transfection of heat-killed bacteria into epithelial cells enabled Nod1 signalling to occur [89]. This same group also subsequently identified that invasive Listeria monocytogenes was capable of inducing IL-8 production by human endothelial cells, in a Nod1 and p38 MARK-dependent manner [90]. Since then, numerous studies have reported that Nod1 signalling, resulting in NF-kappaB-dependent IL-8 production, can be induced in host epithelial and haematopoietic cells by the invasive enteric pathogens Escherichia coli [91], Listeria [92], Salmonella enterica serovar Typhimurium [68] and Mycobacterium avium ssp. paratuberculosis, which is associated with Crohn’s disease [93]. In addition, Moraxella catarrhalis, a lung pathogen, is capable of invading bronchial epithelial and primary small airway epithelial cells, resulting in the generation and secretion of IL-8 in a Nod1-dependent manner [94].
Similarly, invasive pathogens can signal via Nod2, as is the case for Mycobacterium tuberculosis [95, 96] and Mycobacterium bovis infected macrophages [96]. The requirement for Nod2 in the recognition of M. tuberculosis and cytokine production was demonstrated using mononuclear cells isolated from individuals homozygous for the 3020insC Nod2 mutation, revealing a defective cytokine production in response to infection [95]. Intracellular Salmonella can also activate Nod2, resulting in killing of the pathogen, and mutations in Nod2 enable intracellular Salmonella to survive within host cells [23]. In addition, S. pneumoniae can transiently invade epithelial and endothelial cells, resulting in further upregulation of Nod2 expression in vivo and in vitro [97]. The authors reported that RIP2 and the signal-transducing molecules IRAK, IRAK2, TRAF6, NIK, TAB2, and TAK1 are involved in this process [97].
Uptake of Bacteria by Phagocytosis and the Degradation of Their Peptidoglycan by Lysosomes
The cellular process of phagocytosis enables the uptake of large particles by host phagocytic cells, predominantly by macrophages. Phagocytosis of Gram-negative and Gram-positive pathogens by host phagocytes enables intracellular Nods capable of detecting bacterial peptidoglycan and initiating a pro-inflammatory response, via the degradation of the internalised pathogen by phagolysosomal fusion. The host produces enzymes that can degrade peptidoglycan to subunits that are capable of being recognised by Nod1 and Nod2. The most prevalent host enzyme responsible for peptidoglycan degradation is lysozyme, which is found in host mucosal secretions and in granules contained within phagocytes. An analysis of lysosomal extracts obtained from human innate immune cells revealed that lysosome-degraded peptidoglycan components were capable of activating the Nod-signalling pathway in host cells [98]. Therefore, the authors of this study proposed that intracellular peptidoglycan traffics to the lysosome, where it is degraded into smaller soluble subunits, enabling more efficient recognition by the intracellular Nod receptors [98].
Release of Peptidoglycan by Hydrolases or Remodelling That Can Be Internalised by Host Cells
Bacteria are required to constantly remodel their peptidoglycan layer during the process of bacterial growth and division. During the process of cellular remodelling and biosynthesis, bacteria shed their peptidoglycan into their extracellular environment, and this shedding is particularly high during the stage of exponential growth [99]. Furthermore, bacterial MDP can be released after degradation of ingested bacteria by host lysozyme [100].
Some bacteria are very efficient at recycling their peptidoglycan and minimising the amount that is released into the surrounding environment during this remodelling process. For example, E. coli is very efficient at recycling its peptidoglycan as it only releases approximately 6 % of its total peptidoglycan content [101], whereas Bacilli can release between 30 and 50 % of their peptidoglycan during remodelling, which may have a function in its pathogenesis [102]. Indeed, it has been suggested that some pathogens such as Neisseria gonorrhoeae and Bordetella pertussis release peptidoglycan to facilitate invasive disease due to destruction of the epithelial cell barrier [2]. Released peptidoglycan can ultimately be taken up by host cells via endocytosis or peptide transporters, as discussed below.
Bacterial Secretion Systems
Some of the more virulent H. pylori strains harbour a cag pathogenicity island (cagPAI), which encodes for a type 4 secretion system (T4SS). It was well established that H. pylori cagPAI-positive bacteria harbouring a T4SS are able to induce IκB degradation and the nuclear translocation of NF-kappaB [103, 104]. Work by Viala and colleagues identified that H. pylori cagPAI-positive bacteria are able to translocate their peptidoglycan into host epithelial cells via the T4SS [40] (see Fig. 10.2). We have furthered these findings and identified that H. pylori cagPAI-positive bacteria can also activate p38 and extracellular signal-related kinase (ERK) MAPKs and AP-1 in a Nod1-dependent manner, identifying a novel pathway of Nod signalling in response to Gram-negative bacterial infection [105]. Moreover, we reported that the T4SS of H. pylori interacts with integrins located within lipid rafts on the host cell membrane to facilitate Nod1 signalling [106]. A second pathogen, C. rodentium, which expresses a functional type III secretion system, has similarly been reported to initiate the production of cytokines via both Nod1 and Nod2 pathways [107].
Endocytosis and Peptide Transporters
Non-phagocytic epithelial cells are capable of endocytosing peptidoglycan in a clathrin-mediated and dynamin-dependent process. Lee and colleagues used HEK293T cells to show that the internalisation of Nod1 ligands was pH dependent and was optimal at pH 5.5–6, suggesting that the intracellular location of peptidoglycan was within early endosomes. Similarly, Nod2-stimulating peptidoglycan was capable of entering human epithelial cell lines in an identical manner to Nod1-signalling peptidoglycan [108]. In addition, a putative transporter for Nod1 ligands was suggested to exist within early endosomes, being SLC15A4, which was confirmed by knocking down expression of this transporter using siRNA. Indeed, the expression of this putative transporter, whose optimal function is at pH 5.5–6, was highly expressed in tissues obtained from IBD patients, suggesting a potential role for Nod detection of peptidoglycan in IBD-affected individuals [108]. Furthermore, a second study also reported the mechanism of Nod2-signalling peptidoglycan entry into host cells, being macrophages [109]. The authors expanded on the current knowledge that MDP is internalised into acidified vesicles in macrophages and identified that MDP enters macrophages in a clathrin- and dynamin-mediated manner [109]. It was previously identified that MDP could cross the plasma membrane of host cells and enter the cytoplasm via a plasma membrane transporter hPepT1 [110, 111], initially identified as a transporter of oligopeptides [112]. Two studies reported that the hPepT1 transporter was responsible for transporting Nod2-stimulating MDP, but not Nod1-inducing peptidoglycan, into the cytoplasm of intestinal epithelial cells [110, 111]. However, a second group reported that MDP uptake and subsequent Nod2-dependent signalling in macrophages did not require the peptide transporter PepT1 [109]. The contribution of the hPepT1 transporter in facilitating MDP translocation into the cytoplasm may be a cell-specific phenomenon, and further research is required to clarify the requirement of this transporter within different cells. Collectively, these studies indicate that the endocytic pathway enables the uptake of Nod-stimulating peptidoglycan ligands into host cells in a pH-dependent manner.
Outer Membrane Vesicles
Almost all Gram-negative bacteria secrete outer membrane vesicles (OMVs) or “blebs” as part of their normal growth. OMVs are spherical, bilayered membrane nano-structures ranging from 20 to 300 nm in size, which are released naturally both in vitro and in vivo [113]. We have recently identified that OMVs from H. pylori, Pseudomonas aeruginosa and N. gonorrhoea contain peptidoglycan [114]. We have shown that Gram-negative bacterial OMVs enter non-phagocytic host epithelial cells via lipid rafts, rendering their peptidoglycan-containing cargo accessible to Nod1. Indeed, depletion of lipid rafts on the surface of epithelial cells impaired the ability of OMVs to enter and signal via Nod1, and this was restored once lipid rafts were replenished on the host cellular membrane [114]. Similarly, a second study also has shown the ability of OMVs from E. coli to enter host cells in a lipid raft-dependent manner and release toxin within the host cell [115]. In addition, work by Bielig and colleagues confirmed the ability of Gram-negative bacterial OMVs to induce Nod-dependent responses using the pathogen Vibrio cholerae [116].
Furthermore, oral administration of H. pylori OMVs to wild-type and Nod1 knockout animals revealed that OMVs could initiate rapid innate immune responses within the gastric tissue of immunocompetent animals, whereas no inflammatory responses were observed in Nod1 knockout mice [114]. Moreover, oral OMV administration to wild-type animal resulted in the development of an OMV-specific antibody response that was absent in Nod1 knockout animals. Collectively, these findings identified a mechanism whereby Gram-negative mucosal pathogens can initiate Nod1-dependent innate and adaptive immune responses in the absence of cellular invasion or bacterial secretion systems.
Direct Binding of Ligands to Nod Receptors
Since the discovery of Nod1 and Nod2 and the identification of peptidoglycan as the bacterial product triggering the activation of these receptors, scientists have debated whether or not these molecules directly bound their respective ligands. Early work had suggested that this was likely the case. Indeed, mouse Nod1 prefers the tetrapeptide, l-Ala–d-Glu–mesoDAP–d-Ala, whereas human Nod1 is better triggered by l-Ala–d-Glu–mesoDAP; therefore, by swapping their LRR domains, the sensing specificities of these two molecules could be switched [19].
Recently, three papers demonstrated direct in vitro binding of their ligands. The first paper showed using surface plasmon resonance and atomic force microscopy that Nod1 can directly bind to l-Ala-γ–d-Glu–meso-diaminopimelic acid but not MDP, the Nod2 ligand. Following this, two papers showed the direct interaction of Nod2 with MDP, one using chips coated with MDP self-assembled monolayers and surface plasmon resonance to measure Nod2–MDP interactions [117] and the other, binding of recombinant Nod2 to biotinylated MDP [118]. The significance of these studies is that now these kinds of assays can be used to develop screens for identifying novel inhibitors or activators of Nod proteins, which might reveal interesting small molecules for the treatment of Crohn’s disease.
Role of Nods in Crohn’s Disease
Nod2 was the first susceptibility gene linked to Crohn’s disease. In 2001, two groups reported that mutations in Nod2, and in particular, a frame-shift mutation caused by a cytosine insertion at nucleotide position 3020, were linked to Crohn’s disease susceptibility [8, 119]. For Nod1, its link to inflammatory bowel disease has been less clear. Although some studies have shown associations of Nod1 polymorphisms with Crohn’s disease [120], this has not been supported in other populations [121].
Nod2 Genetics and Crohn’s Disease
The original paper by Hugot et al., which first described Nod2 as susceptibility loci for Crohn’s disease, identified two variants of Nod2, termed SNP8 and SNP12, in addition to the frame-shift mutation, termed SNP13. While SNP13 lies within the LRR region of Nod2 and results in impaired sensing of MDP [11, 12], SNP12 is proximal to SNP13 within the LRR and SNP8 is within the NOD domain. Interestingly, these two mutations have a variable affect on MDP sensing and at high MDP doses, approach wild-type levels of cell activation [122]. On the other hand, individuals homozygous for the frame-shift mutation are completely unable to detect MDP at any dose [122]. With this in mind, these findings call into question whether a lack of ability to detect MDP really underlies Crohn’s disease pathogenesis. Alternatively, these findings may suggest that treating patients with MDP, especially Nod2 heterozygous and compound heterozygous individuals, might ameliorate disease.
More recently, deep sequencing of GWAS-identified loci found five new variants of Nod2 associated with Crohn’s disease [123]. Functional analyses of two of these Nod2 variants showed diminished responsiveness to MDP compared to wild-type Nod2 transfected cells, yet not as profound as cells transfected with the Nod2 frame-shift mutant [123]. Further studies into how these Nod2 variants are impaired in their activity will certainly shed light onto our understanding of Crohn’s disease pathogenesis.
Lessons Learned from Colitis Models
No animal model can mimic all aspects of human disease. Nevertheless, much insight into how Nod2 potentially regulates intestinal homeostasis has been gained from models of colitis in Nod2-deficient mice. The most commonly used chemical models are the dextran sulphate sodium (DSS) and trinitrobenzene sulfonic acid (TNBS)-induced models, where the chemical insult damages the epithelial layer leading to intestinal inflammation that is driven by exposure of damaged mucosal tissue to the commensal microbiota. Bacteria-induced models of colitis are milder and perhaps more physiologically relevant models of induced colitis, which have been extensively used in the past few years. Again, while no model is perfect, different aspects governing the regulation of intestinal inflammation can be revealed by these different models.
In the DSS and TNBS models, Nod2 triggering has been shown to be important to protect mice from severe disease. Treatment of wild-type mice with the Nod2 ligand, MDP, ameliorates colitis induced by DSS, and this effect was gone in DSS-treated Nod2-deficient animals [36, 124]. Moreover, in a TNBS colitis model, a Lactobacillus strain producing a highly active Nod2 ligand within its peptidoglycan was also able to ameliorate colitis in a Nod2-dependent fashion [125]. In the TNBS model, Nod2-deficient mice have been reported to develop more severe colitis [126, 127]. Interestingly, one group showed that the protective Nod2-dependent signals emanate from the bone marrow since chimeric mice with a Nod2-deficient hematopoietic compartment were as susceptible as Nod2-deficient mice [127]. However, it is not yet known how these bone marrow-derived Nod2 signals are protective in colitis and, indeed, what these factors might be.
The role of Nod2 in host defence against microbial infection and subsequent colitis has been examined in different bacterial infection models. Nod2-deficient mice are more susceptible to Helicobacter hepaticus infection, demonstrating increased intestinal inflammation and increased frequency of IFN-gamma secreting Th1 cells in Peyer’s patches. Interestingly, over-expression of alpha-defensins in Nod2-deficient Paneth cells was able to dampen intestinal inflammation [48]. Nod2-deficient mice were also shown to display a delayed intestinal clearance of Citrobacter rodentium due to reduced CCL2 expression in the colon and resulting in impaired recruitment of inflammatory monocytes [61]. Our group, on the other hand, found only in the background of Nod1 deficiency that Nod2-deficient mice were more susceptible to C. rodentium infection [86] and, indeed, Salmonella enterica serovar Typhimurium infection [128]. The reason for this discrepancy is unclear but likely points to the ever-growing understanding that differences in commensal microbiota between animal facilities impact disease susceptibility. Finally, a knock-in mouse strain carrying the frame-shift mutation in Nod2, which is associated with human Crohn’s disease, was generated recently and, similar to Nod2-deficient mice, this mutant Nod2 strain exhibits severely impaired sensing of MDP and increased susceptibility to the enteric organism Enterococcus faecalis [129]. It will be interesting in the future to explore the mechanisms of intestinal homeostasis dysregulation in this mouse model.
Conclusion and Perspectives
Nod1 and Nod2 were the first characterised NLR family of cytosolic PRRs. As described in this chapter, much research has focused on how these receptors detect their ligand, peptidoglycan, and how this triggers an inflammatory response. More recent findings have highlighted that Nod signalling at the intestinal mucosa, especially Nod2, is critical for the maintenance of the integrity of the gut barrier and the regulation of inflammatory pathways that control both homeostasis and protection against intrusion by microbial pathogens. The challenge for the future will be to understand how dysregulation of Nod2 signalling leads to inflammation in Crohn’s disease patients. What is still unclear is what cell type is critical for Nod2-dependent homeostatic regulation, be it hematopoietic cells or epithelial cells of the intestine, including Paneth cells. Moreover, a key question for the future will be to understand whether Nod2’s ability to detect MDP is linked to intestinal homeostasis. As pointed out above, while cells derived from patients with the homozygous frame-shift mutation in Nod2 cannot detect MDP, cells with other mutations still retain some ability to respond to this bacterial product. In the future, understanding the pathogenic implications of these Crohn’s disease-associated mutations in Nod2, beyond the frame-shift mutation, will help to unravel the mysteries behind this disease as well as point to new avenues for treatment.
References
Strober W, Murray PJ, Kitani A, Watanabe T (2006) Signalling pathways and molecular interactions of NOD1 and NOD2. Nat Rev Immunol 6(1):9–20, Epub 2006/02/24
Clarke TB, Weiser JN (2011) Intracellular sensors of extracellular bacteria. Immunol Rev 243(1):9–25, Epub 2011/09/03
Ausubel FM (2005) Are innate immune signaling pathways in plants and animals conserved? Nat Immunol 6(10):973–979
Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK et al (2008) The NLR gene family: a standard nomenclature. Immunity 28(3):285–287, Epub 2008/03/18
Philpott DJ, Girardin SE (2010) Nod-like receptors: sentinels at host membranes. Curr Opin Immunol 22(4):428–434
Werts C, Rubino S, Ling A, Girardin SE, Philpott DJ (2011) Nod-like receptors in intestinal homeostasis, inflammation, and cancer. J Leukoc Biol 90(3):471–482, Epub 2011/06/10
Inohara N, Nunez G (2003) NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol 3(5):371–382
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R et al (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411(6837):603–606
Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR et al (2001) CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep 2(8):736–742
Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S et al (1999) Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem 274(21):14560–14567
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G et al (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278(11):8869–8872, Epub 2003/01/16
Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J et al (2003) Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 278(8):5509–5512
Fritz JH, Girardin SE, Fitting C, Werts C, Mengin-Lecreulx D, Caroff M et al (2005) Synergistic stimulation of human monocytes and dendritic cells by toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur J Immunol 35(8):2459–2470
Enoksson M, Ejendal KF, McAlpine S, Nilsson G, Lunderius-Andersson C (2011) Human cord blood-derived mast cells are activated by the Nod1 agonist M-TriDAP to release pro-inflammatory cytokines and chemokines. J Innate Immun 3(2):142–149, Epub 2010/11/26
Qiu F, Maniar A, Diaz MQ, Chapoval AI, Medvedev AE (2011) Activation of cytokine-producing and antitumor activities of natural killer cells and macrophages by engagement of Toll-like and NOD-like receptors. Innate Immun 17(4):375–387, Epub 2010/08/05
Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L et al (2003) An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 4(7):702–707, Epub 2003/06/11
Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J et al (2003) Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300(5625):1584–1587, Epub 2003/06/07
Chamaillard M, Girardin SE, Viala J, Philpott DJ (2003) Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol 5(9):581–592
Magalhaes JG, Philpott DJ, Nahori MA, Jehanno M, Fritz J, Bourhis LL et al (2005) Murine Nod1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep 6(12):1201–1207
Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G (2001) Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem 276(7):4812–4818
Rahman MK, Midtling EH, Svingen PA, Xiong Y, Bell MP, Tung J et al (2010) The pathogen recognition receptor NOD2 regulates human FOXP3+ T cell survival. J Immunol 184(12):7247–7256
Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN (2010) Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med 16(2):228–231
Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK (2003) CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 124(4):993–1000
Gutierrez O, Pipaon C, Inohara N, Fontalba A, Ogura Y, Prosper F et al (2002) Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem 277(44):41701–41705
Rosenstiel P, Fantini M, Brautigam K, Kuhbacher T, Waetzig GH, Seegert D et al (2003) TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 124(4):1001–1009, Epub 2003/04/03
Shaw PJ, Barr MJ, Lukens JR, McGargill MA, Chi H, Mak TW et al (2011) Signaling via the RIP2 adaptor protein in central nervous system-infiltrating dendritic cells promotes inflammation and autoimmunity. Immunity 34(1):75–84
Zhao L, Hu P, Zhou Y, Purohit J, Hwang D (2011) NOD1 activation induces proinflammatory gene expression and insulin resistance in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 301(4):E587–E598, Epub 2011/06/23
Silva GK, Gutierrez FR, Guedes PM, Horta CV, Cunha LD, Mineo TW et al (2010) Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J Immunol 184(3):1148–1152, Epub 2010/01/01
Finney CA, Lu Z, LeBourhis L, Philpott DJ, Kain KC (2009) Disruption of Nod-like receptors alters inflammatory response to infection but does not confer protection in experimental cerebral malaria. Am J Trop Med Hyg 80(5):718–722
Sorbara MT, Philpott DJ (2011) Peptidoglycan: a critical activator of the mammalian immune system during infection and homeostasis. Immunol Rev 243(1):40–60, Epub 2011/09/03
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE (2006) Nod-like proteins in immunity, inflammation and disease. Nat Immunol 7(12):1250–1257
Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G et al (2008) A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J 27(2):373–383, Epub 2007/12/15
Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF et al (2000) An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem 275(36):27823–27831
Meylan E, Tschopp J (2005) The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci 30(3):151–159
Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA et al (2002) RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416(6877):194–199
Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M (2009) Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30(6):789–801, Epub 2009/05/26
Krieg A, Correa RG, Garrison JB, Le Negrate G, Welsh K, Huang Z et al (2009) XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A 106(34): 14524–14529, Epub 2009/08/12
Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA (2007) NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem 282(50):36223–36229, Epub 2007/10/20
Abbott DW, Wilkins A, Asara JM, Cantley LC (2004) The Crohn’s disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol 14(24):2217–2227, Epub 2004/12/29
Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP et al (2004) Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5(11):1166–1174, Epub 2004/10/19
Tanabe T, Chamaillard M, Ogura Y, Zhu L, Qiu S, Masumoto J et al (2004) Regulatory regions and critical residues of NOD2 involved in muramyl dipeptide recognition. EMBO J 23(7):1587–1597, Epub 2004/03/27
Kufer TA, Kremmer E, Banks DJ, Philpott DJ (2006) Role for Erbin in bacterial activation of Nod2. Infect Immun 74(6):3115–3124
McDonald C, Chen FF, Ollendorff V, Ogura Y, Marchetto S, Lecine P et al (2005) A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol Chem 280(48):40301–40309, Epub 2005/10/06
Richmond AL, Kabi A, Homer CR, Marina-Garcia N, Nickerson KP, Nesvizhskii AI et al (2012) The nucleotide synthesis enzyme CAD inhibits NOD2 antibacterial function in human intestinal epithelial cells. Gastroenterology 142(7):1483–1492.e6. Epub 2012/03/06
Magalhaes JG, Sorbara MT, Girardin SE, Philpott DJ (2010) What is new with Nods? Curr Opin Immunol 23(1):29–34
Tattoli I, Travassos LH, Carneiro LA, Magalhaes JG, Girardin SE (2007) The Nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol 29(3):289–301
Petnicki-Ocwieja T, Hrncir T, Liu YJ, Biswas A, Hudcovic T, Tlaskalova-Hogenova H et al (2009) Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci U S A 106(37):15813–15818, Epub 2009/10/07
Biswas A, Liu YJ, Hao L, Mizoguchi A, Salzman NH, Bevins CL et al (2010) Induction and rescue of Nod2-dependent Th1-driven granulomatous inflammation of the ileum. Proc Natl Acad Sci U S A 107(33):14739–14744, Epub 2010/08/04
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G et al (2005) Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307(5710):731–734, Epub 2005/02/05
Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M et al (2004) NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 53(11):1658–1664
Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV (2008) Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A 105(52):20858–20863, Epub 2008/12/17
Bouskra D, Brezillon C, Berard M, Werts C, Varona R, Boneca IG et al (2008) Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456(7221):507–510, Epub 2008/11/07
Ganz T (2003) Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3(9):710–720, Epub 2003/09/02
Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG et al (2006) Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J Biol Chem 281(17):11637–11648, Epub 2006/03/04
Grubman A, Kaparakis M, Viala J, Allison C, Badea L, Karrar A et al (2010) The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides. Cell Microbiol 12(5):626–639, Epub 2009/12/31
Voss E, Wehkamp J, Wehkamp K, Stange EF, Schroder JM, Harder J (2006) NOD2/CARD15 mediates induction of the antimicrobial peptide human beta-defensin-2. J Biol Chem 281(4):2005–2011
Watanabe T, Asano N, Fichtner-Feigl S, Gorelick PL, Tsuji Y, Matsumoto Y et al (2010) NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J Clin Invest 120(5):1645–1662, Epub 2010/04/15
Masumoto J, Yang K, Varambally S, Hasegawa M, Tomlins SA, Qiu S et al (2006) Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med 203(1):203–213, Epub 2006/01/19
Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G et al (2011) Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 186(8):4872–4880
Rupnik M, Wilcox MH, Gerding DN (2009) Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7(7):526–536, Epub 2009/06/17
Kim YG, Kamada N, Shaw MH, Warner N, Chen GY, Franchi L et al (2011) The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34(5):769–780, Epub 2011/05/14
Davis KM, Nakamura S, Weiser JN (2011) Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J Clin Invest 121(9):3666–3676
Brooks MN, Rajaram MVS, Azad AK, Amer AO, Valdivia-Arenas MA, Park J-H et al (2010) NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell Microbiol 13(3):402–418
Imielinski M, Baldassano RN, Griffiths A, Russell RK, Annese V, Dubinsky M et al (2009) Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet 41(12):1335–1340, Epub 2009/11/17
Oosting M, Berende A, Sturm P, ter Hofstede HJM, de Jong DJ, Kanneganti T-D et al (2010) Recognition of Borrelia burgdorferi by NOD2 is central for the induction of an inflammatory reaction. J Infect Dis 201(12):1849–1858
Berrington WR, Iyer R, Wells RD, Smith KD, Skerrett SJ, Hawn TR (2010) NOD1 and NOD2 regulation of pulmonary innate immunity to Legionella pneumophila. Eur J Immunol 40(12):3519–3527
Totemeyer S, Sheppard M, Lloyd A, Roper D, Dowson C, Underhill D et al (2006) IFN-gamma enhances production of nitric oxide from macrophages via a mechanism that depends on nucleotide oligomerization domain-2. J Immunol 176(8):4804–4810, Epub 2006/04/06
Le Bourhis L, Magalhaes JG, Selvanantham T, Travassos LH, Geddes K, Fritz JH et al (2009) Role of Nod1 in mucosal dendritic cells during Salmonella pathogenicity island 1-independent Salmonella enterica serovar Typhimurium infection. Infect Immun 77(10):4480–4486
Mosa A, Trumstedt C, Eriksson E, Soehnlein O, Heuts F, Janik K et al (2009) Nonhematopoietic cells control the outcome of infection with Listeria monocytogenes in a nucleotide oligomerization domain 1-dependent manner. Infect Immun 77(7):2908–2918
Scott MJ, Chen C, Sun Q, Billiar TR (2010) Hepatocytes express functional NOD1 and NOD2 receptors: A role for NOD1 in hepatocyte CC and CXC chemokine production. J Hepatol 53(4):693–701
Moreno L, McMaster SK, Gatheral T, Bailey LK, Harrington LS, Cartwright N et al (2010) NOD1 is a dominant pathway for NOS2 induction in vascular smooth muscle cells: comparison with TLR4 responses in macrophages. Br J Pharmacol 160:1997–2007
Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, Slepenkin AV et al (2009) The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog 5(4):e1000379, Epub 2009/04/11
Lysenko ES, Clarke TB, Shchepetov M, Ratner AJ, Roper DI, Dowson CG et al (2007) Nod1 signaling overcomes resistance of S. pneumoniae to opsonophagocytic killing. PLoS Pathog 3(8):e118
Simonsen A, Tooze SA (2009) Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol 186(6):773–782, Epub 2009/10/03
Deretic V, Levine B (2009) Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5(6):527–549
Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG et al (2009) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11(1):55–62
Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P et al (2010) NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16(1):90–97, Epub 2009/12/08
Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C (2010) ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn’s disease pathogenesis. Gastroenterology 139(5):1630–1641.e2
Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF et al (2011) Nod2 is essential for temporal development of intestinal microbial communities. Gut 60(10):1354–1362
Fritz JH, Le Bourhis L, Sellge G, Magalhaes JG, Fsihi H, Kufer TA et al (2007) Nod1-mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 26(4):445–459
Magalhães JG, Lee J, Geddes K, Rubino S, Philpott DJ, Girardin SE (2011) Essential role of Rip2 in the modulation of innate and adaptive immunity triggered by Nod1 and Nod2 ligands. Eur J Immunol 41(5):1445–1455
Magalhaes JG, Rubino SJ, Travassos LH, Le Bourhis L, Duan W, Sellge G et al (2011) Nucleotide oligomerization domain-containing proteins instruct T cell helper type 2 immunity through stromal activation. Proc Natl Acad Sci USA 108(36):14896–14901, Epub 2011/08/23
Shaw MH, Reimer T, Sanchez-Valdepenas C, Warner N, Kim YG, Fresno M et al (2009) T cell-intrinsic role of Nod2 in promoting type 1 immunity to Toxoplasma gondii. Nat Immunol 10(12):1267–1274, Epub 2009/11/03
Ouyang W, Kolls JK, Zheng Y (2008) The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28(4):454–467, Epub 2008/04/11
Colonna M (2009) Interleukin-22-producing natural killer cells and lymphoid tissue inducer-like cells in mucosal immunity. Immunity 31(1):15–23, Epub 2009/07/17
Geddes K, Rubino SJ, Magalhães JG, Streutker C, Le Bourhis L, Cho JH et al (2011) Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat Med 17(7):837–844
Wing K, Sakaguchi S (2010) Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol 11(1):7–13, Epub 2009/12/18
Noguchi E, Homma Y, Kang X, Netea MG, Ma X (2009) A Crohn’s disease-associated NOD2 mutation suppresses transcription of human IL10 by inhibiting activity of the nuclear ribonucleoprotein hnRNP-A1. Nat Immunol 10(5):471–479, Epub 2009/04/08
Opitz B, Forster S, Hocke AC, Maass M, Schmeck B, Hippenstiel S et al (2005) Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ Res 96(3):319–326
Opitz B, Puschel A, Beermann W, Hocke AC, Forster S, Schmeck B et al (2006) Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J Immunol 176(1):484–490
Kim JG, Lee SJ, Kagnoff MF (2004) Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by toll-like receptors. Infect Immun 72(3):1487–1495
Boneca IG, Dussurget O, Cabanes D, Nahori MA, Sousa S, Lecuit M et al (2007) A critical role for peptidoglycan N-deacetylation in Listeria evasion from the host innate immune system. Proc Natl Acad Sci U S A 104(3):997–1002
Pott J, Basler T, Duerr CU, Rohde M, Goethe R, Hornef MW (2009) Internalization-dependent recognition of Mycobacterium avium ssp. paratuberculosis by intestinal epithelial cells. Cellular microbiology 11(12):1802–1815
Slevogt H, Seybold J, Tiwari KN, Hocke AC, Jonatat C, Dietel S et al (2007) Moraxella catarrhalis is internalized in respiratory epithelial cells by a trigger-like mechanism and initiates a TLR2- and partly NOD1-dependent inflammatory immune response. Cell Microbiol 9(3):694–707
Ferwerda G, Girardin SE, Kullberg BJ, Le Bourhis L, de Jong DJ, Langenberg DM et al (2005) NOD2 and toll-like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog 1(3):279–285
Brooks MN, Rajaram MV, Azad AK, Amer AO, Valdivia-Arenas MA, Park JH et al (2011) NOD2 controls the nature of the inflammatory response and subsequent fate of Mycobacterium tuberculosis and M. bovis BCG in human macrophages. Cell Microbiol 13(3):402–418
Opitz B, Puschel A, Schmeck B, Hocke AC, Rosseau S, Hammerschmidt S et al (2004) Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem 279(35):36426–36432
Iyer JK, Coggeshall KM (2011) Cutting edge: primary innate immune cells respond efficiently to polymeric peptidoglycan, but not to peptidoglycan monomers. J Immunol 186(7):3841–3845
Park JT, Uehara T (2008) How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol Mol Biol Rev 72(2):211–227. table of contents, Epub 2008/06/07
Meylan E, Tschopp J, Karin M (2006) Intracellular pattern recognition receptors in the host response. Nature 442(7098):39–44
Goodell EW, Schwarz U (1985) Release of cell wall peptides into culture medium by exponentially growing Escherichia coli. J Bacteriol 162(1):391–397, Epub 1985/04/01
Mauck J, Chan L, Glaser L (1971) Turnover of the cell wall of Gram-positive bacteria. J Biol Chem 246(6):1820–1827, Epub 1971/03/25
Hirata Y, Maeda S, Ohmae T, Shibata W, Yanai A, Ogura K et al (2006) Helicobacter pylori induces IkappaB kinase alpha nuclear translocation and chemokine production in gastric epithelial cells. Infect Immun 74(3):1452–1461
Maeda S, Yoshida H, Ogura K, Mitsuno Y, Hirata Y, Yamaji Y et al (2000) H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 119(1):97–108
Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL (2009) Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol 183(12):8099–8109, Epub 2009/12/17
Hutton ML, Kaparakis-Liaskos M, Turner L, Cardona A, Kwok T, Ferrero RL (2010) Helicobacter pylori exploits cholesterol-rich microdomains for induction of NF-kappaB-dependent responses and peptidoglycan delivery in epithelial cells. Infect Immun 78(11):4523–4531, Epub 2010/08/18
LeBlanc PM, Yeretssian G, Rutherford N, Doiron K, Nadiri A, Zhu L et al (2008) Caspase-12 modulates NOD signaling and regulates antimicrobial peptide production and mucosal immunity. Cell Host Microbe 3(3):146–157, Epub 2008/03/11
Lee J, Tattoli I, Wojtal KA, Vavricka SR, Philpott DJ, Girardin SE (2009) pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J Biol Chem 284(35):23818–23829
Marina-Garcia N, Franchi L, Kim YG, Hu Y, Smith DE, Boons GJ et al (2009) Clathrin- and dynamin-dependent endocytic pathway regulates muramyl dipeptide internalization and NOD2 activation. J Immunol 182(7):4321–4327, Epub 2009/03/21
Vavricka SR, Musch MW, Chang JE, Nakagawa Y, Phanvijhitsiri K, Waypa TS et al (2004) hPepT1 transports muramyl dipeptide, activating NF-kappaB and stimulating IL-8 secretion in human colonic Caco2/bbe cells. Gastroenterology 127(5):1401–1409, Epub 2004/11/03
Ismair MG, Vavricka SR, Kullak-Ublick GA, Fried M, Mengin-Lecreulx D, Girardin SE (2006) hPepT1 selectively transports muramyl dipeptide but not Nod1-activating muramyl peptides. Can J Physiol Pharmacol 84(12):1313–1319, Epub 2007/05/10
Fei YJ, Kanai Y, Nussberger S, Ganapathy V, Leibach FH, Romero MF et al (1994) Expression cloning of a mammalian proton-coupled oligopeptide transporter. Nature 368(6471):563–566, Epub 1994/04/07
Kuehn MJ, Kesty NC (2005) Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev 19(22):2645–2655
Kaparakis M, Turnbull L, Carneiro L, Firth S, Coleman HA, Parkington HC et al (2010) Bacterial membrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol 12(3):372–385, Epub 2009/11/06
Kesty NC, Mason KM, Reedy M, Miller SE, Kuehn MJ (2004) Enterotoxigenic Escherichia coli vesicles target toxin delivery into mammalian cells. EMBO J 23(23):4538–4549
Bielig H, Rompikuntal PK, Dongre M, Zurek B, Lindmark B, Ramstedt M et al (2011) NOD-like receptor activation by outer membrane vesicles from Vibrio cholerae non-O1 non-O139 strains is modulated by the quorum-sensing regulator HapR. Infect Immun 79(4):1418–1427, Epub 2011/01/26
Grimes CL, Ariyananda Lde Z, Melnyk JE, O’Shea EK (2012) The innate immune protein Nod2 binds directly to MDP, a bacterial cell wall fragment. J Am Chem Soc 134(33):13535–13537, Epub 2012/08/04
Mo J, Boyle JP, Howard CB, Monie TP, Davis BK, Duncan JA (2012) Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem 287(27):23057–23067, Epub 2012/05/03
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J et al (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411(6837):599–603, Epub 2001/06/01
McGovern DP, Hysi P, Ahmad T, van Heel DA, Moffatt MF, Carey A et al (2005) Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet 14(10):1245–1250, Epub 2005/03/26
Van Limbergen J, Nimmo ER, Russell RK, Drummond HE, Smith L, Anderson NH et al (2007) Investigation of NOD1/CARD4 variation in inflammatory bowel disease using a haplotype-tagging strategy. Hum Mol Genet 16(18):2175–2186, Epub 2007/07/07
van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T et al (2005) Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet 365(9473):1794–1796, Epub 2005/05/25
Rivas MA, Beaudoin M, Gardet A, Stevens C, Sharma Y, Zhang CK et al (2011) Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat Genet 43(11):1066–1073, Epub 2011/10/11
Watanabe T, Asano N, Murray PJ, Ozato K, Tailor P, Fuss IJ et al (2008) Muramyl dipeptide activation of nucleotide-binding oligomerization domain 2 protects mice from experimental colitis. J Clin Invest 118(2):545–559, Epub 2008/01/12
Macho Fernandez E, Valenti V, Rockel C, Hermann C, Pot B, Boneca IG et al (2011) Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut 60(8):1050–1059, Epub 2011/04/08
Barreau F, Meinzer U, Chareyre F, Berrebi D, Niwa-Kawakita M, Dussaillant M et al (2007) CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS One 2(6):e523, Epub 2007/06/15
Penack O, Smith OM, Cunningham-Bussel A, Liu X, Rao U, Yim N et al (2009) NOD2 regulates hematopoietic cell function during graft-versus-host disease. J Exp Med 206(10):2101–2110, Epub 2009/09/10
Geddes K, Rubino S, Streutker C, Cho JH, Magalhaes JG, Le Bourhis L et al (2010) Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun 78(12):5107–5115, Epub 2010/10/06
Kim YG, Shaw MH, Warner N, Park JH, Chen F, Ogura Y et al (2011) Cutting edge: Crohn’s disease-associated Nod2 mutation limits production of proinflammatory cytokines to protect the host from Enterococcus faecalis-induced lethality. J Immunol 187(6):2849–2852, Epub 2011/08/19
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Kaparakis-Liaskos, M., Philpott, D.J. (2013). Nod1 and Nod2 and the Immune Response to Bacteria. In: D'Amato, M., Rioux, J. (eds) Molecular Genetics of Inflammatory Bowel Disease. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8256-7_10
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8256-7_10
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8255-0
Online ISBN: 978-1-4614-8256-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)