
Abstract
The glass transition of food solids has received considerable attention and its relationships with the behaviour of food solids in various processes and food storage are well established. The glass transition properties for food components have been obtained primarily from calorimetric measurements, and their limit has been in identifying a transition temperature range with no particular information on the kinetics of changes associated with the transition. On the other hand, theories on the fragility of glass-forming materials have advanced with some reference to food and pharmaceutical applications. Information on enthalpy relaxations and their use to derive the fragility of glass formers in food is also available. Understanding glass-transition-related relaxations and their coupling with the engineering properties of food materials is a challenging and developing area of food materials science. The glass formation of complex solid food systems and their stability is of the utmost importance in the development of advanced nutrient delivery systems. Our studies have shown that knowledge of the macroscopic glass-transition behaviour of food systems may often be misleading in the prediction of characteristics of food components and their storage stability. For example, the glass transition and relaxation times determined for mixtures of carbohydrates and proteins vary and need to be interpreted carefully when coupled with measurements of the flow properties of powders or reaction kinetics. We have found that the contact time of particles for liquid bridging in stickiness measurements may be governed by the mobility of selected molecular species forming food solids. This has shown varying relaxation times of reactive components which may affect physicochemical properties and kinetics in food processing and storage. The new information can advance innovations in food formulation by mapping the engineering properties of food components and their mixes and the engineering of novel nutrient delivery systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
The engineering properties of solids in food materials are highly dependent on their physical state, i.e. amorphous non-crystalline, crystalline or liquid. Variations of food material properties and states may occur as a result of changes in external thermodynamic conditions, such as pressure and temperature, and within materials because of changes in plasticiser or solvent (water) contents. The food polymer science approach introduced by Levine and Slade (1986) has been successful in explaining the time-dependent characteristics of amorphous food components and cryostabilisation in the manufacture of frozen foods. However, complex food systems are composed of numerous miscible, partially miscible, immiscible, partially crystalline and partially amorphous components. This makes the understanding and control of the properties of individual food systems in various processing and storage conditions very challenging and different from the behaviour of synthetic polymers and somewhat similar pharmaceutical preparations with more definite characteristics.
Glass-transition data have been published for numerous food components, primarily carbohydrates (Roos 1993) and proteins (Aguilera et al. 1993), as well as food solids, such as milk (Jouppila et al. 1997) and apples (Bai et al. 2001), which typically include carbohydrates, proteins, lipids and minor components in complex and often cellular, local and specific structures. The glass-transition data of complex food systems with highly heterogeneous microstructures can be problematic as most glass transitions measured result from those of single or miscible components. More importantly, water as a plasticiser can be differently distributed within various components. Typical glass transition measurements use differential scanning calorimetry (DSC), which gives global glass transitions for food solids but cannot measure changes in individual components responsible for thermal properties. The mechanical properties of food systems have been measured by dynamic mechanical analysis (DMA) and related to dielectric properties measured by dielectric analysis (DEA) (Moates et al. 2001). These measurements give information on dielectric or mechanical relaxations alpha-relaxation shows relaxation times above the glass transition. Glass-transition measurements using DSC primarily determine the glass-transition temperature, T g , its temperature range and the magnitude of the change in heat capacity over the glass transition (ΔCp). DSC may also be used to determine enthalpy relaxations associated with a glass transition and to show the dynamic effects of molecular packaging and aging on the material response to plasticisation at a glass transition. Enthalpy relaxations reflect changes in molecular mobility around the glass transition and their effects on the translational diffusion of the non-crystalline phase components above the glass transition. Hence, rates of diffusion-controlled reactions involving glass components may show increasing rates above the T g . A typical diffusion-controlled property above the glass transition is the crystallisation of the non-crystalline components, such as lactose in milk-based powders. It has also been recognised that the flow properties and stickiness of powders are affected by the glass transition (Bhandari and Howes 1999). Glass-transition measurements emphasise information about the temperature range over which dramatic changes in material properties may occur, but no information on the extent of changes in material characteristics in specific conditions, e.g., during food processing and storage, can be obtained.
The DMA and DEA measurements of material properties show relaxation times above the glass transition. The relaxation time at the onset temperature of the change in heat capacity measured by DSC is 100 s and corresponds to the viscosity of 1012 Pa s typical of the solid, glassy state of materials (Roos 1995; Angell 1991). Furthermore, it is well known that the glass transition and material properties related to the glass transition are time-dependent (Roos 1995), i.e., rates of various changes above the T g may correlate with the structural relaxation times. The changes in relaxation times above the glass transition according to the Williams-Landel-Ferry relationship was emphasised by Levine and Slade (1986), and the ‘fragility’ concept developed by Angell (1991) has aimed at showing the structural relaxation properties of ‘strong’ and ‘fragile’ glass formers above the glass transition. Our approach has been to develop knowledge on the dielectric and mechanical properties, and the corresponding structural relaxation times, of food systems at temperatures around and above the calorimetric glass transition. The present review will address the glass transition, relaxations and engineering properties of food components and real food systems taking into account the complexity of food systems and the effects of various components and their miscibility on food processing and storage stability.
Relaxation Times and Fluidness
The glass transition is an important property of food solids in such processes as dehydration, extrusion and freezing and helps to understand the properties of confectionary, edible films and frozen foods, for example. More importantly, several delivery systems for sensitive food components and pharmaceuticals use non-crystalline solids as protective matrices, or the active components need to remain non-crystalline for rapid release and uptake. It is also important to note that reactions that control the stability of biological materials require the presence of a non-crystalline state of reactants and may therefore be controlled by the physical state and plasticisation of the substances. This often means understanding the properties of food solids at low or intermediate water contents where water is a strong plasticiser (Levine and Slade 1986). Non-crystalline solids form stable glassy states, and vitrification is often a prerequisite for the success of stabilisation of dehydrated and frozen materials. Thermal and water plasticisation that results in a glass transition and induce translational molecular mobility causes structural ʻfluidnessʼ as structural relaxation times decrease at and above the glass transition. Fluid, fluidness and fluidity are used here as terms referring to the characteristics of glass-forming materials above their glass transitions and associated material-specific changes in structural relaxation times in their super-cooled liquid states. This is somewhat different from the ‘fragility’ concept developed by Angell (1991), which is defined as the steepness of the T g -scaled Arrhenius plot near the glass transition or sharpness of the glass transition (Angell 2002). Such fragility and material fluidness show similarities, but fluidness refers clearly to a super-cooled liquid state while non-crystalline, solid glasses are generally mechanically fragile without translational mobility or fluidness. Fluidness can also relate to various important properties of glass-forming food materials, including reactivity, collapse, caking, stickiness and various other phenomena which occur above the glass transition. It also takes into account the non-Arrhenius behaviour and temperature dependence of the activation energy of super-cooled liquids at and above the glass transition.
Glass transition is a universal property like other changes in the state of materials, i.e. it refers to the non-equilibrium liquid and solid forms of the super-cooled state typical of inorganic glass formers, synthetic polymers and sugars (Angell 2002), and it is a property of materials existing below their equilibrium melting temperature with no well-defined structure. The glass transition in all materials shows complexity due to simultaneous rapid changes in kinetic and thermodynamic processes. Therefore, individual materials may show an indefinite number of molecular arrangements and glass structures with varying levels of molecular packing and order. The extent of molecular packing and order is a result of the rate of molecular ‘freezing’ towards the glassy state during cooling or annealing or aging processes at conditions allowing slow molecular arrangements in the vicinity of the glass transition. In food systems, the molecular arrangements may also change as a result of fluctuations in temperature, water content and water activity, and it may vary within the food microstructure. The non-equilibrium properties of glass-forming materials underpin, for example, the enthalpy and volume relaxations occurring around the glass transition as well as aging processes below the glass transition.
Vitrification of materials at the glass transition differs significantly from melting and crystallisation, which are well-defined thermodynamic changes in systems with high chemical purity, such as metals. Glass transition has no latent heat and is a property of pure materials and of mixtures of two or more miscible food components. A glass transition in food systems at a well-defined water content can be a specific, complex property of numerous food components. However, most glass-transition data available are limited to binary mixtures of single food components and water or food solids with poor compositional characterisation. The glassy state and properties of super-cooled liquids above the glass transition have been of great interest across materials science because of the fundamental effects of the glass transition on the physicochemical properties of materials, and particularly the effects of glass transition on the material fluidness above the glass transition. The non-equilibrium characteristics of the super-cooled liquid and glassy states of materials underscore the importance of understanding the time-dependent nature of glass-forming food solids. The relative rates of change in the glassy and super-cooled liquid states are related to structural relaxation times that indicate molecular mobility and its variations at different temperatures, pressures and levels of water plasticisation. Such relaxation times may be derived from measurements of dielectric and rheological properties, such as complex moduli and dielectric loss and permittivity. Relative relaxation times may also use measurements of viscosity and other molecular-mobility-dependent parameters.
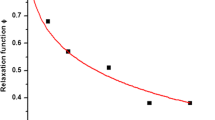
Williams et al. (1955) pointed out a general problem in the use of a common reference temperature, such as 25 °C, to describe changes in glass-forming material properties. They found that mechanical and dielectric relaxation times above the glass transition followed a simple, universal relationship when relaxation times of various glass-forming materials were compared. A common, arbitrarily chosen reference temperature of approximately 50 K above the glass-transition temperature, T g , was used as a reference temperature, T s . Although the reference temperature T g = T s could be used, T s was preferred because of the difficulties of measurements of relaxation times at the glass transition, the problems of varying T g values caused by residual solvents and plasticisers, and thermal history effects on the T g measurements. The Williams–Landel–Ferry (WLF) relationship (4.1) (Williams et al. 1955) was derived from the general behaviour of most inorganic and organic glass formers that showed a similar temperature dependence of relaxation times and viscosity over a temperature range of T g to T g + 100 K (Fig. 4.1). This was different from the parent Vogel–Tammann–Fulcher (VTF) relationship (4.2), which used a reference temperature, T o , well below the T g (Williams et al. 1955).

Viscosity (a) and relaxation time, τ (e.g. dielectric relaxation) (b) above the glass transition temperature, T g , predicted by the Williams–Landel–Ferry (WLF) relationship with the universal constants C 1 = −17.44 and C 2 = 51.6 K
It is important to note that the VTF relationship has a form that is similar to that of the Arrhenius equation, but the VTF model uses a T 0 as a reference temperature instead of 0 K and allows determination of apparent activation energy. Also, the WLF equation can be written in the form of the VTF relationship, as shown by Eqs. 4.1–4.3, but T 0 is replaced by T s or T g and the constant B is replaced by a T−T s or T−T g -dependent parameter that includes the constants C 1 and C 2. An assumption of B = 2.303 C 1 C 2 has been suggested (Angell 1997), but this assumption is valid only at large T−T g values of =100 °C, where deviation from the Arrhenius temperature dependence of structural relaxation times is minimal. The activation energy in the WLF equation is highly temperature and material dependent (Williams et al. 1955) and follows the deviation of the temperature dependence of structural relaxation times from the Arrhenius behaviour. As discussed by Angell (1997), the deviation from the Arrhenius behaviour in the WLF equation is measured by the C 2 constant. Although the problems of using universal reference temperatures are well known, as described by Williams et al. (1955), the fragility concept introduced by Angell (1991) uses individual T g values on the absolute temperature scale and plots of relaxation times against T g /T. Such plotting uses 0 K as a reference temperature for all materials and identifies the T g at T g /T = 1 for each material independent of its specific numeric value on the absolute temperature scale. This shows the logarithmic values of the structural relaxation times against T g -multiplied activation energies (log τ vs. T g × 1/T), indicating that the fragility is T g value dependent and that materials with a similar fluidness but different T g also have different fragilities. Therefore, materials with the same activation energy in the vicinity of T g also have different fragilities if their T g values differ (Fig. 4.2). The fragility concept has successfully pointed out differences in the structural relaxation times of various materials above the T g , but it does not seem to measure the fluidness of biological materials close to the glass transition, which is important for understanding glass-former properties in the food and pharmaceutical industries.

Viscosity, η (a) and relaxation time, τ, as predicted by the Williams–Landel–Ferry (WLF) relationship ( 4.1 ) (b) above the glass transition temperature, T g , shown in ‘T g -scaled’ Arrhenius plots using the abscissa of T g /T according to Angell (1991) with experimental viscosity data for glucose (Parks and Gilkey 1929), glycerol (Segur and Oberstar 1951) and water (Hallett 1963). In accordance with Angell (1991), fragility is shown as the F½ = (1− T g /T) fragility or ‘steepness index’, m = [mmin(1 + F½)/(1−F½)], where Fmin is 16 for τmin and 17 for ηmin. FWLF refers to the WLF-predicted τ at T − T g = 100 K. The apparent differences in F½ and FWLF shown result only from differences in the individual T g values of the substances. Plotting of the data against viscosity against T − T g gives a single curve (Fig. 4.1)
The use of viscosity and relaxation time models assumes that the viscosity of the super-cooled liquid state approaches 1012 Pa s at the glass transition and the dielectric relaxation time becomes approximately 100 s at the onset of the calorimetric glass transition (Angell 1991). The WLF constants, C 1 and C 2, may be assigned ‘universal’ values of −17.44 and 51.6, respectively, when T s = T g is used as the reference temperature (Williams et al. 1955). Although the WLF relationship may fit to viscosity and relaxation time data above the glass transition, it is obvious that changes around the glass transition occur gradually showing an upward concavity when plotted against T−T g , as shown by Peleg (1996). This problem, however, is not apparent, and downward concavity is obtained when the data are plotted against 1/T−T g (4.4). In such plots, ηg approaches the high viscosity of 1012 Pa s for the glassy state when T−T g approaches 0 K. When viscosity or relaxation time data are shown isothermally against water activity or water content, the Fermi model proposed by Peleg (1996) may be preferred because of its simplicity and parameters may be used to measure the broadness and extent of the changes in stiffness at the glass transition:
where a T is the ratio of the relaxation times, τ and τ s, or viscosities, η and η s , at temperature T and a reference temperature, T s , respectively, and C 1, C 2, and B are constants. T g refers to the onset temperature of glass transition in a differential scanning calorimetry heating scan.
The use of the glass-transition-anchored reference temperature showing the effects of the glass transition of various substances to their structural relaxation times is important for comparisons of various glass-forming materials as mobility-related properties change above the glass transition. The changes appear very different if T g /T plotting instead of a specific, individual glass-transition-related reference temperature is used for the various glass-forming materials. The WLF equation is a simple approach because it makes it possible to plot relaxation times (or viscosity) against (T − T g ). As shown by Eq. 4.4 and pointed out by Angell (1997), the WLF constant C 1 is a ‘scaling parameter’ defining the number of logarithmic decades for the change in relaxation time or viscosity above the glass transition. For a relaxation time of 100 s at the onset of glass transition, this means that the relaxation time approaches 10−C 1 −2 s as T − T g approaches infinity, and the use of C 1 = 16 was suggested by Angell (1997). Alternatively, the viscosity approaches 10−C 1 −12 when T − T g approaches infinity, with C 1 = 17. The C 2 of the WLF relationship can be taken as the ‘fluidness’ parameter, which becomes different depending on the changes in structural relaxation times above the material-specific T g .
The classification of glass-forming materials as ‘strong’ and ‘fragile’, as developed by Angell (1991), aims at using a fragility parameter (slope), m, as a measure of the deviation from the Arrhenius temperature dependence above at their respective glass-transition temperature. However, as shown in Fig. 4.2, the fragility parameter fails to compare glass-forming materials if they show differences in T g . Also, the same substance can have different fragilities depending on the level of plasticisation simply due to the artefact of differences in T g values. Strong liquids are those following the Arrhenius relationship, e.g. SiO2, while fragility increases with increasing deviation from the linearity of relaxation times against the reciprocal temperature (Fig. 4.2). Fragility may also be derived from the parameter D of the modified VTF relationship (4.5) as FVTF = 1/D, which varies from 0 to 1. Fragility has also been defined as F½ fragility (Angell 2002), as shown in Fig. 4.2 for viscosity:
The fragility approach with experimental data shows that SiO2 is a strong glass former with Arrhenius behaviour above the T g . Organic glass formers and water appear to be highly fragile glass formers. Although the fragility indexes in Fig. 4.2 for glycerol, glucose and trehalose are different, they show similar changes in viscosity and relaxation times above the T g and should be noted as being equally fluid (Fig. 4.3). The difference in the apparent fragility of these materials, for example, is a serious problem and a limitation of the fragility approach, which results from the differences in the individual T g values and the T g /T scaling. Water shows the highest fluidness, and water-plasticised food components may be assumed to increase in fluidness with increasing water plasticisation. Despite these limitations of the fragility concept, it appears quite obvious that the fluidness of food materials increases with increasing water content. Alternatively, as suggested by the ‘Arrhenius or VTF’-type WLF relationship of Eq. 4.4, a plot of log τ against 1/T−T g shows differences in the structural relaxation times of glass-forming materials around and above their glass transitions. The relationships also indicate that a decreasing C 2 in Eq. 4.4 increases the activation energy and the temperature dependence of the viscosity or structural relaxation times around the glass transition. This is particularly important over the temperature range of 10–20 K typical of the glass transition.

Fluidness plot for common glass-forming materials showing experimental viscosity data of Fig. 4.2 for SiO2, glucose, glycerol and water in their liquid and super-cooled liquid states. The constant C 1 = 17.44 of the WLF equation was used as the ‘scaling’ parameter and C 2 was used as the fluidness parameter
DEA and DMA studies of food materials have shown a significant decrease in relaxation times around the glass transition when derived from the respective frequency-dependent dielectric loss or loss modulus α-relaxation temperatures. Measurements of the relaxation times and effects of food composition on the relaxation properties of amorphous components in complex foods is fundamental for understanding food properties in processes and storage at high solid contents or low temperatures. Our studies have shown that the T g value of a glass former and particularly the presence of other components and water in food systems are more significant factors affecting food fluidness and properties than the fragility of a single glass former, as defined by Angell (1991).
Relaxation Times in Food Systems
According to Angell (1991, 2002) relaxation times, such as dielectric relaxation times and calorimetric relaxations at the onset T g , are typically 100 s and assumed to approach 10−14 s at high temperatures (Fig. 4.2). The structural relaxation times of food systems decrease to 10−3 s at 20–30 °C above the T g . This corresponds to a decrease in viscosity from 1012 to 105 Pa s, which agrees with critical viscosities for collapse in freeze-drying (Bellows and King 1973) and stickiness in spray-drying (Downton et al. 1982), as well-known examples.
The WLF relationship has been useful in relating the viscosities of super-cooled liquids to their stickiness (Downton et al. 1982) and times to crystallisation of amorphous sugars (Roos and Karel 1990). The assumption made is that the universal WLF constants apply and that the viscosity of the glass-forming liquids above the T g follows the WLF relationship, as shown in Fig. 4.1. The data in Fig. 4.1 show stickiness at a surface viscosity of 106 to 108 Pa s, corresponding to a dielectric relaxation time of 10−4 to 10−2 s and a surface contact time of 1–10 s at the ‘sticky point’ (Downton et al. 1982). These values were shown to apply to a non-crystalline 7:1 mixture of sucrose and fructose (Downton et al. 1982). The sticky points were found at approximately 20 °C above the onset temperature of the glass transition, T g , measured by DSC (Roos and Karel 1990). Several other studies have confirmed the relationships of the glass transition and stickiness of amorphous solids (Boonyai et al. 2004).
Dielectric and mechanical analyses of food systems allow for the determination of relaxation times at the α-relaxation, which can be related to the stickiness characteristics of food powders as well as other mechanical changes in food solids, such as collapse of structure or viscosity and diffusion. Dielectric analyses of fructose and glucose show that the α-relaxation time decreases from 102 s at the onset of the calorimetric T g to 10−2 to 10−3 s at the sticky point at T−T g of 20 °C.
The crystallisation of amorphous sugars occurs time-dependently above the glass transition. Dielectric α-relaxations suggest that an α-relaxation time of 1–2 s at 10 °C above the onset T g corresponds to a time to crystallisation of amorphous lactose (Roos and Karel 1992) of 10 days, and α-relaxation times of approximately 10−2 s correspond to crystallisation within 30 h.
The relaxation time data show that there is a significant increase in the mobility of carbohydrate components in food materials within the glass-transition temperature range measured by DSC. The most important range to be considered in using the T g data of sugar-containing food materials in relating glass transition to processing characteristics and storage stability is within the T−T g of 20 °C, but it might be different for complex food matrices.
Dielectric and Mechanical Relaxations of Food Solids
Amorphous food components have been studied for glass transitions, dielectric relaxations, mechanical relaxations, spectroscopic properties and various other characteristics showing changes in molecular mobility at and around the glass transition. There are, however, very few studies on the properties of food solids with varying carbohydrate and protein compositions (Fig. 4.4). Our recent approach in studies of the stickiness properties of food solids has been to use materials, particularly dairy-based systems, with various carbohydrate and protein compositions (Silalai and Roos 2010, 2011a, b). These materials have been studied for their sticky points and dielectric and mechanical relaxations at various water activities, aw. The results have revealed significant differences in the glass transition and relaxation behaviour of systems containing low molecular weight sugars with maltodextrins or proteins. We have shown that carbohydrates and proteins may form phase-separated regions in food systems, e.g. food powders. In a carbohydrate–protein system, the carbohydrate phase typically showed an almost protein-content-independent glass transition in a DSC study, and the glass transition approached that of the carbohydrate at increasing water contents (Silalai and Roos 2010). The sticky point was found for skim milk–milk protein solids systems to occur at increasing temperatures above the T g with rising protein content (Silalai and Roos 2010). The results also showed that although the T g was at an almost constant, aw-dependent temperature, stickiness developed at the higher temperature the higher was the protein content leading to a larger temperature difference between the sticky point and glass transition (Silalai and Roos 2011a). The α-relaxation time corresponding to the sticky point decreased with increasing aw, but there were increases in the temperature difference of the sticky point to T g with increasing aw. This could be related to aw-dependent interactions of the components in water-plasticised systems and availability of the carbohydrate phase for the formation of liquid bridges at particle surfaces (Silalai and Roos 2011a).

Food components with high solid contents, e.g. in food powders. Miscible components, such as sugars, maltodextrins and water, form amorphous structures showing glass formation according to the component properties (a); carbohydrate and protein systems are plasticised by water but show phase separation of protein, and the glass-forming properties are determined primarily by the carbohydrate phase (b); high molecular weight components, such as starch, may show partial crystallinity and phase separation from a continuous, low molecular weight, glass-forming carbohydrate matrix (c); a lipid phase is phase-separated from a continuous, glass-forming carbohydrate phase (d)
Skim milk–maltodextrin solid systems showed quite different properties from those of skim milk–protein systems (Silalai and Roos 2011b). Skim milk–maltodextrin solid systems formed a carbohydrate-rich phase with a maltodextrin-content-dependent T g (described in Fig. 4.3a, b). The sticky point occurred at approximately 20 °C above the maltodextrin-content- and molecular-size-dependent T g . These studies showed that stickiness in carbohydrate-protein powders is affected by the carbohydrate, and the sticky point is higher the higher the protein content. In skim milk–maltodextrin solid systems, a higher maltodextrin content gave a higher T g and the sticky point was at a higher temperature but at an approximately constant T−T g . Similar differences in glass-forming properties and behaviour-affecting characteristics of various other food systems, as shown in Fig. 4.4, may be expected.
Conclusion
Understanding glass-transition-related relaxations and their coupling with the engineering properties of food materials is essential for the design of complex food and nutrient delivery systems. The macroscopic glass-transition behaviour of food systems may often be misleading in the prediction of characteristics of food components and their storage stability because relaxation times determined for mixtures of carbohydrates and proteins vary and need to be interpreted carefully. The fragility concept, because of its limitations, cannot explain the glass-forming properties of food systems, but studies of relaxations around and above the glass transition give new information on fluidness that can advance innovations in food formulation by mapping the engineering properties of food components and their mixes and the engineering of novel nutrient delivery systems.
References
Aguilera JM, Levi G, Karel M (1993) Effect of water content on the glass transition and caking of fish protein hydrolyzates. Biotechnol Prog 9:651–654
Angell CA (1991) Thermodynamic aspects of the glass transition in liquids and plastic crystals. Pure Appl Chem 63:1387–1392
Angell CA (1997) Why C1 = 16–17 in the WLF equation is physical – and the fragility of polymers. Polymer 38:6261–6266
Angell CA (2002) Liquid fragility and the glass transition in water and aqueous solutions. Chem Rev 102:2627–2650
Bai Y, Rahman MS, Perera CO, Smith B, Melton LD (2001) State diagram of apple slices: glass transition and freezing curves. Food Res Int 34:89–95
Bellows RJ, King CJ (1973) Product collapse during freeze drying of liquid foods. AIChE Symp Ser 69(132):33–41
Bhandari B, Howes T (1999) Implication of glass transition for the drying and stability of dried foods. J Food Eng 40:71–79
Boonyai P, Bhandari B, Howes T (2004) Stickiness measurement techniques for food powders: a review. Powder Technol 145:34–46
Downton DP, Flores-Luna JL, King CJ (1982) Mechanism of stickiness in hygroscopic, amorphous powders. Ind Eng Chem Fundam 21:447–451
Hallett J (1963) The temperature dependence of the viscosity of supercooled water. Proc Phys Soc 82:1046–1050
Jouppila K, Kansikas J, Roos YH (1997) Glass transition, water plasticization, and lactose crystallization in skim milk powder. J Dairy Sci 80:3152–3160
Levine H, Slade L (1986) A polymer physico-chemical approach to the study of commercial starch hydrolysis products (SHPs). Carbohydr Polym 6:213–244
Moates GK, Noel TR, Parker R, Ring SG (2001) Dynamic mechanical and dielectric characterisation of amylose-glycerol films. Carbohydr Polym 44:247–253
Parks GS, Gilkey WA (1929) Studies on glass IV. Some viscosity data on liquid glucose and glucose-glycerol solutions. J Phys Chem 33:1428–1437
Peleg M (1996) On modeling changes in food and biosolids at and around their glass transition temperature range. Crit Rev Food Sci Nutr 36:49–67
Roos Y (1993) Melting and glass transitions of low molecular weight carbohydrates. Carbohydr Res 238:39–48
Roos YH (1995) Phase transitions in foods. Academic, San Diego
Roos Y, Karel M (1990) Differential scanning calorimetry study of phase transitions affecting the quality of dehydrated materials. Biotechnol Prog 6:159–163
Roos Y, Karel M (1992) Crystallization of amorphous lactose. J Food Sci 57:775–777
Segur JB, Oberstar HE (1951) Viscosity of glycerol and its aqueous solutions. Ind Eng Chem 43:2117–2120
Silalai N, Roos YH (2010) Roles of water and solids composition in the control of glass transition and stickiness of milk powders. J Food Sci 75:E285–E296
Silalai N, Roos YH (2011a) Coupling of dielectric and mechanical relaxations with glass transition and stickiness of milk solids. J Food Eng 104(3):445–454
Silalai N, Roos YH (2011b) Mechanical α-relaxations and stickiness of maltodextrin-milk solids systems around glass transition. J Sci Food Agric 91(14):2529–2536
Williams ML, Landel RF, Ferry JD (1955) The temperature dependence of relaxation mechanisms in amorphous polymers and other glass-forming liquids. J Am Chem Soc 77:3701–3707
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Roos, Y.H. (2013). Relaxations, Glass Transition and Engineering Properties of Food Solids. In: Yanniotis, S., Taoukis, P., Stoforos, N., Karathanos, V. (eds) Advances in Food Process Engineering Research and Applications. Food Engineering Series. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-7906-2_4
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7906-2_4
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-7905-5
Online ISBN: 978-1-4614-7906-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)