
Abstract
Spermatogonial stem cells (SSCs) are the resident stem cells in the testes of adult males and are responsible for maintaining near lifelong spermatogenesis in mammals. Yet, SSCs represent only a small fraction of adult germ cells. Due to the apparent paucity of SSCs, the authentic stem cell population within the adult testis has yet to be definitively identified. This chapter contrasts classical models of self-renewal and differentiation of mouse and human SSCs. Furthermore, we summarize recent findings, based on information now available through transplantation assays, that pertain to the molecular characterization and function of normal adult SSCs in rodent and human systems. We address both the intrinsic molecular mechanisms as well as the extrinsic stimuli that regulate SSC fate decisions. We also examine the composition and role of the spermatogonial stem cell niche. Finally, we discuss the unique ability of adult male germ cells to reprogram into embryonic-like stem cells in culture, resulting in the loss of lineage commitment and the acquisition of pluripotency.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sertoli Cell
- Seminiferous Tubule
- Spermatogonial Stem Cell
- Promyelocytic Leukemia Zinc Finger
- Undifferentiated Spermatogonia
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Spermatogonial stem cells (SSCs) are the resident stem cells in the testes of adult males and are responsible for maintaining lifelong spermatogenesis in mammals, yet represent only a tiny fraction of adult germ cells (e.g., about 0.03 % in mice) [1]. In humans, SSCs seem to be similarly scarce but only indirect estimates have been made, and these are based in part on ethically problematic experiments performed on prisoners who were dosed with radioisotopes in the 1960s [2]. Given the apparent paucity of SSCs, it should come as no surprise that, as yet, we are unable to definitively identify the authentic stem cell population within the testis. Nonetheless, remarkable technology developed by Ralph Brinster and others has enabled the discovery of critical molecular and functional features of SSCs, not only in mice but also in other species, making SSC biology a preeminent model for long-term self-renewing adult stem cells. In addition to maintaining genomic and epigenomic integrity for future generations, SSCs have the unusual property among other adult stem cell types of undergoing spontaneous programming in vitro to produce a pluripotent phenotype, a process that is poorly understood despite a number of recent controversial studies, particularly in humans [3–6]. The goal of this chapter is to present recent discoveries that pertain to the characterization and function of normal adult SSCs in mice and humans and also to address the current understanding of reprogramming of adult male germ cells.
Spermatogonial Stem Cells in Rodents
According to the classical view, known as the As model, mammalian SSCs are characterized by morphological criteria obtained from whole mount preparations of testicular seminiferous tubules. This model, initially proposed by Clermont and Bustos-Obregon [7], defines rodent SSCs as isolated Asingle (As) spermatogonia. These As spermatogonia are located on the basement membrane of the seminiferous tubules and are part of a larger subcategory of undifferentiated spermatogonia, Aundiff, which are recognized by their apparent lack of condensed heterochromatin in the nucleus. As spermatogonia either self-renew, dividing into two new SSCs, or begin to differentiate, forming Apaired (Apr) spermatogonia which remain connected by intercellular cytoplasmic bridges [8]. Apr spermatogonia continue on the path of differentiation to form longer chains of 4–32 cells, which are referred to as Aaligned (Aal) spermatogonia. These Aal spermatogonia continue to differentiate, ultimately giving rise to diploid spermatocytes.
More recently, functional and molecular features have essentially supplanted the classic morphological descriptions of putative SSCs. The minimum requirement for stem cell functionality is the ability to maintain the stem cell population while producing differentiating progeny. This functionality can only be definitively assessed by means of transplantation, which was first published as an assay in 1994 by Ralph Brinster and others [9, 10]. The transplantation assay demonstrates that donor SSCs, when injected into the seminiferous tubules of infertile mice, have the capacity to migrate to the proper microenvironment along the basement membrane and carry out long-term self-renewal and spermatogenesis. It was also shown that donor spermatozoa could generate normal offspring and were, thus, fully functional.
It had been widely assumed, in accordance with Clermont’s earlier model, that As spermatogonia, exclusively, are the true SSCs. However, due to the fact that there is no universally accepted As-specific marker and that SSCs can only be definitively identified in retrospect using the aforementioned functional transplantation assay, the smallest population that has been proven to have stem cell properties includes all undifferentiated spermatogonia (As, Apr, and Aal). Furthermore, it is equally unclear whether the stem cell population is limited to an even smaller subset of undifferentiated spermatogonia than the As spermatogonia.
Recent studies have presented convincing data suggesting that Clermont’s original model is likely flawed. Using an in vivo lineage tracing strategy, Nakagawa et al. described two functional populations of SSCs in the mouse testis; these were referred to as “actual stem cells,” which are self-renewing, and “potential stem cells,” which have the ability to self-renew but only do so under stress [11]. A recent study by this same group showed that the putative stem cell pool, as defined by the As model, is heterogeneous and that the actual stem cell population is contained within a subpopulation of As spermatogonia [12]. Other studies have cast doubt on the schema of self-renewal and differentiation suggested by the Clermont model, according to which differentiation is linear and nonreversible, and have shown that the commitment of spermatogonia to the differentiation pathway is indeed reversible [12, 13]. The extent to which this phenomenon is generally applicable to SSCs in other mammals, including humans, is not currently clear.
In addition to the above studies, the ability to characterize SSCs based on molecular markers that are present on the cell surface has greatly accelerated the field. In 1999, Shinohara et al. showed that α6-integrin and β1-integrin were expressed on the surface of SSCs [14]. Later, in 2003, Kubota et al. identified Thy1 (CD90) on mouse SSCs. Kubota showed that 95 % of the SSCs in the adult mouse testes are contained in the Thy1+ cell fraction [15]. Kanatsu-Shinohara previously found that mouse SSCs express CD9, though the CD9+ testis cell fraction was found to be enriched only 6.9-fold for SSCs [16]. In a more recent study, Kanatsu-Shinohara showed that SSCs are most concentrated in CD9+EPCAM−/low population [17]. GPR125 was also shown to be a marker for undifferentiated spermatogonia in the mouse [3]. Purification of SSCs has also been facilitated by the use of negative selection against molecules such as αV-integrin [18].
While cell surface markers are particularly useful for isolation of live SSCs, other signature genes have been identified, many of which are nuclear. These include, but are not limited to, PLZF, LIN28, NANOS2, and OCT4, which are all expressed by undifferentiated spermatogonia, but not specifically by As spermatogonia [19–24]. Conversely, KIT expression is absent in undifferentiated spermatogonia and marks the transition to differentiating type A spermatogonia [25]. In a recent paper, however, Oatley et al. showed that ID4 is expressed exclusively in As spermatogonia [26].
Spermatogonial Stem Cells in Humans
According to studies beginning with Clermont and Heller, primate spermatogonia were characterized morphologically as Adark (Ad) and Apale (Ap) spermatogonia, based on the distinct levels of chromatin condensation in the nuclei and the consequent intensity of the staining with hematoxylin [27, 28]. Both Ad and Ap spermatogonia were considered undifferentiated and it was suggested that the Ad spermatogonia are the reserve stem cells and the Ap spermatogonia are the actively renewing stem cells [27–30]. In this model of spermatogenesis, the Ap spermatogonia divide to form either new Ap spermatogonia or differentiated type B spermatogonia. The type B spermatogonia continue to divide, differentiating to form primary spermatocytes and spermatids. Other models of human SSC population dynamics suggest that the Ap spermatogonia, which undergo regular divisions, are actually transit-amplifying progenitors, whereas the Ad spermatogonia are the true SSCs [31, 32]. It has also been proposed that the Ad and Ap nuclear phenotypes may represent spermatogonia at distinct stages of the cell cycle as opposed to spermatogonia with differing stem cell fates [33]. Due to the difficulty of culturing human SSCs and the paucity of available assays, however, the true identity of the human SSC remains unknown, though it is most likely true that the human SSCs exist as a smaller subpopulation of the Ad or Ap spermatogonia [5].
In the last decade, however, progress has been made to define human SSCs using the same approaches as were used with rodent models. Izadyar et al. showed that putative SSCs in the adult human testis are phenotypically characterized as SSEA-4+, CD49f+, CD90+, GPR125+, and c-Kitneg/low [34]. The same study also found that about one-third of repopulating spermatogonia express OCT4 and NANOG, signifying the existence of populations of spermatogonia in the adult human testes with at least some characteristics of pluripotent cells.
In a 2010 study, the Dym group used human testicular material from deceased organ donors and confirmed that human spermatogonia express THY1, GFRα1, ITGα6 (although ITGα6 is also expressed in Sertoli cells), and PLZF, all of which are also markers of rodent SSCs [35]. Localized expression of GPR125 was observed in 1–2 spermatogonia per seminiferous tubule cross-section, and they proposed that GPR125 might be a marker of SSCs. In a more recent study, von Kopylow et al. shed substantial light on the original morphology-based model proposed by Clermont [36]. It was found that the gene expression profile of Ap and Ad spermatogonia differed in regard to expression of KIT, Ki-67, and DMRT1, while many putative SSC markers were common to Ap and Ad spermatogonia. Specifically, they found that KIT, Ki-67, and DMRT1 were restricted to subtypes which lacked nuclear rarefaction zones, i.e., types Ap and B spermatogonia only. Ad spermatogonia, however, were marked by high levels of exosome component 10 (EXOSC10) in the nuclear vacuole, which may reflect differential nuclear RNA metabolism in the Ad spermatogonial population; this feature was linked to the cell’s immature state. Thus, as additional molecular correlates of stemness in the human testis are validated, it is likely that the utility of morphological assessments will continue to decline.
While several groups reported that OCT4 expression is not conserved in human spermatogonia [6, 35], Bhartiya et al. suggested that the reason for the discrepancy between findings in rodents and humans may be that the antibodies and primer sets used were derived from the overlapping domain between OCT4A and OCT4B rather than from an exon specific to OCT4A [37]. A novel population of 5–10 μm cells was found to express nuclear OCT4A and also other pluripotent markers such as NANOG and TERT, suggesting that these cells may represent a distinct population of cells with pluripotent features in the testis. Given the numerous pitfalls associated with accurate and valid measurement of pluripotency genes and of possible markers of human SSCs, it remains to be seen whether genes such as OCT4, NANOG, and SOX2 are meaningfully expressed, either at the level of mRNA or protein in human SSCs. Such questions become particularly relevant when addressing the reprogramming of adult germ cells, as discussed in the final section of this chapter.
Microanatomy of the Spermatogonial Stem Cell Niche
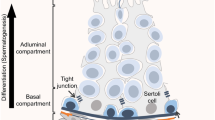
A stem cell niche is the specialized microenvironment that supports self-renewal and survival of the stem cell population. Stem cell niches are formed by contributions from surrounding support cells, which provide extrinsic stimuli to regulate self-renewal and differentiation both through secreted growth factors and extracellular matrix support. Spermatogenesis occurs within the seminiferous tubules of the testis, which are surrounded by the basement membrane (Fig. 1). The developing germ cells and Sertoli cells together form the seminiferous epithelium [38]. Tight junctions formed between the Sertoli cells create both a basal compartment, which houses all undifferentiated spermatogonia, and an adluminal compartment. Peritubular myoid cells line the outside of the basement membrane and provide structural support for the tubules. The interstitial region between the tubules consists predominantly of Leydig cells, which secrete testosterone, along with the vascular network, and also tissue macrophages. Each of the cell types mentioned, in addition to vascular contributions, have been implicated as contributors to the SSC niche [39–44].

Structure of the mouse seminiferous tubule and SSC niche. (a) Hematoxylin- and eosin-stained section of adult mouse testis. Red, dashed line shows area that is illustrated in (b). (b) Cartoon showing undifferentiated spermatogonia, including stem cells, are nurtured from within the seminiferous tubule by signals produced by Sertoli cells (turquoise) and also from the outside of the tubule by other somatic cell types, such as peritubular myoid cells (green) and others. Additional cell types of note that are present in the interstitial region include endothelial cells, macrophages, and Leydig cells
The Sertoli cell is the only somatic cell type within the seminiferous tubule; in addition to critical roles in fostering the latter stages of spermatogenesis, it is generally accepted that the Sertoli cell is the predominant participant in the SSC niche. Oatley et al. recently provided more direct evidence that the Sertoli cells regulate the SSC niche, showing that increasing the number of Sertoli cells in the testes of mice concomitantly increases the number of niches accessible for colonization by SSCs posttransplantation [45].
While the Sertoli cell is critical, somatic cell populations in the interstitial tissue likely contribute to the niche as well [39, 40]. Chiarini et al. showed that undifferentiated spermatogonia accumulate in areas of the seminiferous tubule where the basement membrane is more closely associated with the interstitial tissue. Additionally, Yoshida et al. (2007) implicated the vascular network of the testes in regulation of spermatogonia by showing that during the process of differentiation, undifferentiated spermatogonia migrate away from areas of the tubule that are associated with the interstitial vasculature [46]. The functional roles of vascular-derived instructions in SSC self-renewal have yet to be elucidated.
Extrinsic Factors Regulating Fate Decisions
Substantial progress has been made in identifying extrinsic stimuli that control the decision of SSCs to self-renew rather than differentiate and in using this knowledge to establish culture conditions that support the long-term propagation of SSCs in vitro [47–49]. The first, and arguably the most important, extrinsic regulator of SSC self-renewal and propagation to be found was glial cell line-derived neurotrophic factor (GDNF) [50]. Produced by Sertoli cells, GDNF is a member of the transforming growth factor beta (TGFβ) superfamily. Meng et al. (2000) were among the first to recognize the importance of GDNF signaling in the maintenance of undifferentiated spermatogonia. It was shown that spermatogenesis is disrupted in GDNF-deficient mice, while overexpression of GDNF in transgenic mice results in the accumulation of undifferentiated spermatogonia [50]. These findings ultimately enabled the successful creation of an in vitro culture system that could sustain SSCs long term. In 2004, Kubota et al. found that the addition of recombinant GDNF to serum-free medium did indeed promote the long-term expansion of mouse SSCs [49]. A recent study has shown that GDNF is required not only for the initial establishment of the stem spermatogonial pool but also for the maintenance of the SSC population in the normal adult testis [51]. GDNF was found to promote self-renewal over differentiation of replicating stem spermatogonia in the normal mature testis. GDNF is also known to signal via the GFRα1/RET co-receptor through activation of Src family kinases, Ras, and PI3K-Akt pathways and subsequently induces expression of target genes in SSCs [52–55].
In addition to GDNF, other growth factors that enhance SSC self-renewal have been identified. Kubota et al. (2004) found that while fibroblast growth factor 2 (FGF2) alone does not support SSC expansion, it does increase the rate of proliferation when added in conjunction with GDNF [49]. Kanatsu-Shinohara et al. (2005) also found that inclusion of either FGF2 or EGF in serum-free medium along with GDNF supports long-term expansion of SSCs [56]. Leukemia inhibitory factor (LIF) supports SSC growth in vitro and, thus, may also play a role in the regulation of SSC fate decisions in vivo, although it is not strictly required in vitro [57]. Of note, the Shinohara group recently demonstrated that activation of MAP2K1 downstream of FGF2 drives expression of ETV5 and BCL6B in SSCs [58].
Two recent studies, using gene expression profiling, found that Csf1r, the receptor for Colony Stimulating Factor 1 (CSF1), is highly expressed in undifferentiated spermatogonia isolated from mouse testes [42, 59]. The ligand, CSF1, was thus implicated as a potential extrinsic factor in the regulation of SSC proliferation. When added to cultures of undifferentiated spermatogonia, which were also supplemented with GDNF and FGF2, CSF1 did not enhance proliferative activity but did increase SSC content. These data indicate that CSF1 exposure alters the balance of SSC self-renewal versus differentiation and demonstrate that CSF1 influences SSC self-renewal without affecting proliferation of non-stem spermatogonia. Because CSF1 alone (i.e., without GDNF) did not support cluster formations, it was speculated that CSF1 likely acts in collaboration with or through GDNF. CSF1 expression was observed in both Leydig cells and select myoid cells, suggesting that these cells, too, contribute to the SSC niche [42, 59].
The Wnt family of proteins, which comprises secreted glycoproteins, is another group of cell-extrinsic signals that have been implicated in SSC maintenance in vitro [60, 61]. Yeh et al. (2011) showed that Wnt5a, in particular, supports SSC maintenance and enhances survival of stem spermatogonia in vitro, while Wnt3a may target progenitors [60]. Because the effects of Wnt5a were eliminated by the inhibition of a β-catenin-independent signaling pathway and also because germ cells with active β-catenin signaling lacked SSC activity, these data suggest that Wnt5a supports SSC self-renewal independently of β-catenin. Interestingly, it was also shown that Wnt5a is expressed by Sertoli cells and that SSCs express the cognate receptors. In contrast, Golestaneh et al. found that Wnt3a induces cell proliferation of spermatogonia [61]. It was suggested that Wnt3a acts through the β-catenin-dependent pathways. Unfortunately, direct comparison of these studies is difficult due to substantial methodological differences.
Intrinsic Molecular Mechanisms Regulating Spermatogonial Stem Cell Maintenance
In the SSC system, germ cell-intrinsic factors have essential roles in the maintenance of stem cells and, thus, contribute to the niche in a cell-autonomous manner. Because GDNF is generally regarded as the most important extrinsic factor in the regulation of SSC self-renewal, the study of cell-intrinsic mechanisms involved in SSC maintenance have focused on those pathways that are regulated by GDNF. To date, numerous genes have been found to intrinsically regulate SSC maintenance. These include POU3F1, ETV5, BCL6B, LHX1, and NANOS2 [53, 62–66]. Wu et al. recently demonstrated that POU3F1 is an intrinsic regulator of GDNF-induced survival and self-renewal of mouse SSCs [63, 64]. The Brinster group showed that siRNA silencing of POU3F1 induces apoptosis in cultured THY1+ spermatogonia and, in transplantation assays, greatly reduces that number of colonies formed in the testes of recipient mice [63, 64]. These studies strongly suggest that POU3F1 is an integral intrinsic regulator of SSC survival and likely acts as a suppressor of apoptosis-related genes.
ETV5 is another gene that has been strongly implicated as an upstream regulator of SSC fate in the GDNF-signaling cascade [62, 63]. Wu et al. (2011) demonstrated that ETV5 knockdown and GDNF withdrawal both dramatically reduced the expression of BCL6B, LHX1, Brachyury, and CXCR4. These data provide evidence to the fact that ETV5 is an upstream of effector of all four genes and is itself regulated via GDNF activation [63]. Loss of BCL6B, a transcriptional repressor, has been shown to upregulate genes associated with apoptosis [63]. LHX1 knockdown by siRNA impairs SSC maintenance in vitro [53]. NANOS2, a zinc finger RNA-binding protein, has an expression pattern consistent with undifferentiated spermatogonia, including As, Apr, and some Aal [65]. While NANOS2 was initially thought to be unaffected via GDNF, a recent paper demonstrated that the GDNF signaling pathway induces NANOS2 expression [62, 66]. Disruption of NANOS2 results in rapid depletion of undifferentiated spermatogonia, while overexpression results in accumulation of undifferentiated spermatogonia and reduction in the number of differentiating spermatogonia [65].
In parallel to GDNF-activated signaling pathways, additional cell-intrinsic factors have been identified in the self-renewal and survival of the SSC population. One of these factors is promyelocytic leukemia zinc finger protein (PLZF), a transcriptional repressor [19, 20]. It was previously shown that male mice lacking PLZF expression undergo progressive germ cell loss and testis atrophy, strongly suggesting that PLZF is a cell-intrinsic factor that is necessary for the maintenance of germ cell lineage [19, 20]. Hobbs et al. (2010) then showed that PLZF−/− spermatogonial progenitor cells can be maintained in long-term culture [18]. Similarly, Wu et al. (2011) found that PLZF silencing did not affect the ability of SSCs to self-renew in vitro [63]. However, PLZF promotes in vivo SSC self-renewal indirectly by repressing mTORC1 activity, which inhibits normal spermatogonial progenitor cell response to GDNF [18].
FOXO1, another transcription factor, was recently found to be essential to both SSC homeostasis and spermatogenesis [67]. As a specific marker of a subcategory of spermatogonia with stem cell potential in addition to mouse gonocytes, it was revealed that FOXO1 is closely associated with the “stemness” of the spermatogonia. This group also showed that FOXO1 is an important effector of PI3K-Akt signaling in SSCs, thus revealing novel FOXO-dependent mechanisms that affect SSC fate decisions [67]. Thus, a plethora of signals are emerging as regulators of SSCs under normal physiologic conditions.
Loss of Lineage Commitment: Culture-Induced Acquisition of Pluripotency
As opposed to SSC self-renewal which can be demonstrated in vivo or in vitro, reprogramming of adult germ cells into a pluripotent state is generally considered a culture-induced phenomenon, wherein a unipotent germ cell converts into an ES-like state (Fig. 2). In contrast, reprogramming in vivo either in adult mice or in men is an extremely rare event (<1 in ~11,000 in wild-type laboratory mice and <1 in ~16,000 in humans) [68, 69]. The basis for studying reprogramming of SSCs in vitro rests upon (1) the availability of technology to derive and maintain SSC lines in vitro which we regard as germ lineage-committed, non-pluripotent cells and (2) the identification and functional validation of cells that have actually undergone reprogramming to a pluripotent state, concomitant with the loss of most germ cell features. Multiple studies in mice have shown that the resultant pluripotent cells are highly similar but not identical to ES cells with respect to gene expression, function, and epigenetic features [3, 70–73].

Reprogramming of adults mouse SSCs in culture. (a) Cultures of SSCs exhibit variably sized grape-like clusters (arrows) of cells that are tightly associated with each other but loosely attached to underlying feeder cells. (b) Spontaneous reprogramming of SSCs yields embryonic stem-cell line colonies with sharp, refractile borders that can be maintained as such if transferred to culture conditions designed from mouse embryonic stem cells. Reprogrammed cells rapidly differentiate when maintained in suboptimal conditions
The reprogramming of spermatogonia in vitro is akin to induced pluripotency in which a different type of stable precursor (e.g., fibroblasts) is reprogrammed into a pluripotent state, with an unambiguous distinction between the precursors (e.g., fibroblasts or spermatogonia) and the resultant pluripotent cell type [74]. However, such an unambiguous distinction requires that the precursors be clearly defined, most critically, by functional assays for long-term self-renewal both in vitro and in vivo. Unfortunately, these stringent criteria are not met in many cases.
Following the seminal observations by Shinohara et al. (2004) that SSCs derived from neonatal mice could reprogram in vitro after long-term culture, the same group demonstrated that even after single cell cloning of SSCs, such potency was retained [70]. In 2007, we showed, using GPR125 to track germ cells, that even adult SSC lines in long-term culture retain the ability to reprogram spontaneously [3]. As per standard criteria for pluripotency, the reprogrammed cells derived from adult SSC lines were shown not only to form teratomas in immunocompromised mice but also to contribute to chimeric tissues upon blastocyst injection, even though gene expression was not identical to that of ES cells. Guan et al. (2006) demonstrated pluripotent cells could be derived from the adult testis but the precursor population was less defined in that study due to the absence of a long-term SSC culture phase [75]. Subsequently, the Scholer group showed, using OCT4-GFP reporter cells, that the culture-induced reprogramming of adult SSCs was highly dependent upon plating density [72].
While the origination of pluripotent stem cells from long-term cultures of cells with testis-repopulating activity strongly argues that SSCs are the substrate for conversion, it has not been clearly demonstrated whether all spermatogonia are similarly potent or alternatively whether only a subset give rise to pluripotent colonies. Izadyar (2008) presented data that the OCT4+/KIT+ fraction of spermatogonia were enriched for cells that could be reprogrammed which is interesting, because KIT expression has been considered marker for commitment to differentiation of adult spermatogonia [72]. Intriguingly, Morimoto et al. (2012) recently found that whereas freshly isolated CD9+ testis cells (enriched for SSCs) could produce ES-like colonies upon transfection of the Yamanaka factors (Oct4, Klf4, Sox2, and Myc), cultured SSCs could not, suggesting that in vitro propagation of cells has a negative influence on reprogramming [76].
The first evidence of culture-based reprogramming of human spermatogonia came from the Skutella group who found that testicular cells expressing germ cell markers rapidly upregulated OCT4 during the first week in culture [6]. Subsequently, colonies of putative pluripotent cells were formed continuously during the following weeks in culture. Upon differentiation, the pluripotent cells were able to form functional tissues in vitro and limited teratomas in immunocompromised mice. Despite substantial increases in expression of pluripotency genes, the levels were nonetheless significantly lower than those observed in human ES cells. Subsequently, the Scholer group questioned these findings and concluded that the testis-derived cells thought to be pluripotent were actually more closely related to fibroblasts [77, 78]. An additional caveat is that the Conrad et al. study lacked a long-term self-renewal phase of SSCs in culture prior to reprogramming, without which it is difficult to be sure of the identity of the precursors to the cells that underwent reprogramming.
Following the study by Conrad et al., several studies have found evidence for the ability of normal human testicular cells to undergo apparent reprogramming, although the cell of origin and mechanism are not entirely clear [4, 5, 79, 80]. However, no study to date has demonstrated reprogramming of validated human SSCs from long-term, self-renewing cultures that have been maintained for longer than several months. Kossack et al. (2009) observed appearance of ES-like colonies within several weeks of culture of testicular cells and found not only expression of OCT4 and SOX2 but also the ability of stem cells to differentiate robustly in vitro, but no teratomas were formed in vivo [4]. Subsequently, the van Pelt group also showed in vitro differentiation into all three germ layers but not teratoma formation by ES-like cells derived from testicular cell cultures that had been maintained up to 8 weeks but not thereafter [79, 80]. Since teratoma formation is one of the few assays for pluripotency available for human cells in vivo, the observed reprogramming may have been incomplete or inadvertently produced an intermediate cellular state. Subsequently, the same group concluded that similarly derived ES-like cells were not, in fact, pluripotent due to the absence of spontaneous tri-lineage differentiation. In contrast, Golestaneh et al. (2009) discovered that ES-like cells appear after only 4 days of culture of testicular cells from organ donors; within 4 weeks, lines of pluripotent stem cells were obtained that could form teratomas in vivo [5]. Unfortunately, none of the aforementioned studies was able to unequivocally identify the precursor for the reprogrammed cells, which would require a combination of single cell cloning and subsequent functional characterization of both the putative SSCs (using germ cell transplantation assays) and their ES-like progeny (through formation of teratomas).
Conclusions
The rapid progress of the SSC field beyond morphological criteria and into a phase of functional and molecular studies has ushered in a new era. With the ability to rigorously define this cell type, various groups are moving forward with strategies to address urgent clinical problems, such as treatment-related infertility, using SSCs. Of course, such approaches will require that the level of data produced from the aforementioned rodent studies are at least matched, where possible, using human tissue. At the same time, it is urgent to understand the mechanisms behind reprogramming not only for safety-related reasons in SSC-based cell therapy but also if reprogrammed germ cells are ever to be used for disease modeling or other translational purposes.
Abbreviations
- SSCs:
-
Spermatogonial stem cells
References
Tegelenbosch RA, de Rooij DG. A quantitative study of spermatogonial multiplication and stem cell renewal in the C3H/101 F1 hybrid mouse. Mutat Res. 1993;290(2):193–200.
Heller CG, Clermont Y. Spermatogenesis in man: an estimate of its duration. Science. 1963;140(3563):184–6.
Seandel M, et al. Generation of functional multipotent adult stem cells from GPR125+ germline progenitors. Nature. 2007; 449(7160):346–50.
Kossack N, et al. Isolation and characterization of pluripotent human spermatogonial stem cell-derived cells. Stem Cells. 2009;27(1):138–49.
Golestaneh N, et al. Pluripotent stem cells derived from adult human testes. Stem Cells Dev. 2009;18(8):1115–26.
Conrad S, et al. Generation of pluripotent stem cells from adult human testis. Nature. 2008;456(7220):344–9.
Clermont Y, Bustos-Obregon E. Re-examination of spermatogonial renewal in the rat by means of seminiferous tubules mounted “in toto”. Am J Anat. 1968;122(2):237–47.
de Rooij DG, Russell LD. All you wanted to know about spermatogonia but were afraid to ask. J Androl. 2000;21(6):776–98.
Brinster RL, Zimmermann JW. Spermatogenesis following male germ-cell transplantation. Proc Natl Acad Sci U S A. 1994;91(24):11298–302.
Brinster RL, Avarbock MR. Germline transmission of donor haplotype following spermatogonial transplantation. Proc Natl Acad Sci U S A. 1994;91(24):11303–7.
Nakagawa T, Nabeshima Y, Yoshida S. Functional identification of the actual and potential stem cell compartments in mouse spermatogenesis. Dev Cell. 2007;12(2):195–206.
Nakagawa T, et al. Functional hierarchy and reversibility within the murine spermatogenic stem cell compartment. Science. 2010; 328(5974):62–7.
Barroca V, et al. Mouse differentiating spermatogonia can generate germinal stem cells in vivo. Nat Cell Biol. 2009;11(2):190–6.
Shinohara T, Avarbock MR, Brinster RL. Beta1- and alpha6-integrin are surface markers on mouse spermatogonial stem cells. Proc Natl Acad Sci U S A. 1999;96(10):5504–9.
Kubota H, Avarbock MR, Brinster RL. Spermatogonial stem cells share some, but not all, phenotypic and functional characteristics with other stem cells. Proc Natl Acad Sci U S A. 2003;100(11): 6487–92.
Kanatsu-Shinohara M, Toyokuni S, Shinohara T. CD9 is a surface marker on mouse and rat male germline stem cells. Biol Reprod. 2004;70(1):70–5.
Kanatsu-Shinohara M, et al. Dynamic changes in EPCAM expression during spermatogonial stem cell differentiation in the mouse testis. PLoS One. 2011;6(8):e23663.
Hobbs RM, et al. Plzf regulates germline progenitor self-renewal by opposing mTORC1. Cell. 2010;142(3):468–79.
Costoya JA, et al. Essential role of plzf in maintenance of spermatogonial stem cells. Nat Genet. 2004;36(6):653–9.
Buaas FW, et al. Plzf is required in adult male germ cells for stem cell self-renewal. Nat Genet. 2004;36(6):647–52.
He Z, et al. Gfra1 silencing in mouse spermatogonial stem cells results in their differentiation via the inactivation of RET tyrosine kinase. Biol Reprod. 2007;77(4):723–33.
Zheng K, et al. The pluripotency factor LIN28 marks undifferentiated spermatogonia in mouse. BMC Dev Biol. 2009;9:38.
Dann CT, et al. Spermatogonial stem cell self-renewal requires OCT4, a factor downregulated during retinoic acid-induced differentiation. Stem Cells. 2008;26(11):2928–37.
Shen R, Xie T. NANOS: a germline stem cell’s guardian angel. J Mol Cell Biol. 2010;2(2):76–7.
Schrans-Stassen BH, et al. Differential expression of c-kit in mouse undifferentiated and differentiating type A spermatogonia. Endocrinology. 1999;140(12):5894–900.
Oatley MJ, et al. Inhibitor of DNA binding 4 is expressed selectively by single spermatogonia in the male germline and regulates the self-renewal of spermatogonial stem cells in mice. Biol Reprod. 2011;85(2):347–56.
Clermont Y. Spermatogenesis in man. A study of the spermatogonial population. Fertil Steril. 1966;17(6):705–21.
Clermont Y. Renewal of spermatogonia in man. Am J Anat. 1966;118(2):509–24.
Clermont Y. Kinetics of spermatogenesis in mammals: seminiferous epithelium cycle and spermatogonial renewal. Physiol Rev. 1972;52(1):198–236.
Clermont Y. The cycle of the seminiferous epithelium in man. Am J Anat. 1963;112:35–51.
Ehmcke J, Wistuba J, Schlatt S. Spermatogonial stem cells: questions, models and perspectives. Hum Reprod Update. 2006;12(3):275–82.
Ehmcke J, Schlatt S. A revised model for spermatogonial expansion in man: lessons from non-human primates. Reproduction. 2006;132(5):673–80.
Hermann BP, et al. Spermatogonial stem cells in higher primates: are there differences from those in rodents? Reproduction. 2010;139(3):479–93.
Izadyar F, et al. Identification and characterization of repopulating spermatogonial stem cells from the adult human testis. Hum Reprod. 2011;26(6):1296–306.
He Z, et al. Isolation, characterization, and culture of human spermatogonia. Biol Reprod. 2010;82(2):363–72.
von Kopylow K, et al. Differential marker protein expression specifies rarefaction zone-containing human adark spermatogonia. Reproduction. 2012;143(1):45–57.
Bhartiya D, et al. Newer insights into premeiotic development of germ cells in adult human testis using Oct-4 as a stem cell marker. J Histochem Cytochem. 2010;58(12):1093–106.
Oatley JM, Brinster RL. The germline stem cell niche unit in mammalian testes. Physiol Rev. 2012;92(2):577–95.
Chiarini-Garcia H, Raymer AM, Russell LD. Non-random distribution of spermatogonia in rats: evidence of niches in the seminiferous tubules. Reproduction. 2003;126(5):669–80.
Chiarini-Garcia H, et al. Distribution of type A spermatogonia in the mouse is not random. Biol Reprod. 2001;65(4):1179–85.
Mullaney BP, Skinner MK. Basic fibroblast growth factor (bFGF) gene expression and protein production during pubertal development of the seminiferous tubule: follicle-stimulating hormone-induced sertoli cell bFGF expression. Endocrinology. 1992;131(6):2928–34.
Oatley JM, et al. Colony stimulating factor 1 is an extrinsic stimulator of mouse spermatogonial stem cell self-renewal. Development. 2009;136(7):1191–9.
Spinnler K, et al. Glial cell line-derived neurotrophic factor is constitutively produced by human testicular peritubular cells and may contribute to the spermatogonial stem cell niche in man. Hum Reprod. 2010;25(9):2181–7.
Tadokoro Y, et al. Homeostatic regulation of germinal stem cell proliferation by the GDNF/FSH pathway. Mech Dev. 2002;113(1):29–39.
Oatley MJ, Racicot KE, Oatley JM. Sertoli cells dictate spermatogonial stem cell niches in the mouse testis. Biol Reprod. 2011;84(4):639–45.
Yoshida S, Sukeno M, Nabeshima Y. A vasculature-associated niche for undifferentiated spermatogonia in the mouse testis. Science. 2007;317(5845):1722–6.
Kanatsu-Shinohara M, et al. Long-term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod. 2003;69(2):612–6.
Kubota H, Avarbock MR, Brinster RL. Culture conditions and single growth factors affect fate determination of mouse spermatogonial stem cells. Biol Reprod. 2004;71(3):722–31.
Kubota H, Avarbock MR, Brinster RL. Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc Natl Acad Sci U S A. 2004;101(47):16489–94.
Meng X, et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science. 2000;287(5457):1489–93.
Savitt J, et al. The in vivo response of stem and other undifferentiated spermatogonia to the reversible inhibition of glial cell line-derived neurotrophic factor signaling in the adult. Stem Cells. 2012;30(4):732–40.
Lee J, et al. Akt mediates self-renewal division of mouse spermatogonial stem cells. Development. 2007;134(10):1853–9.
Oatley JM, Avarbock MR, Brinster RL. Glial cell line-derived neurotrophic factor regulation of genes essential for self-renewal of mouse spermatogonial stem cells is dependent on Src family kinase signaling. J Biol Chem. 2007;282(35):25842–51.
Naughton CK, et al. Glial cell-line derived neurotrophic factor-mediated RET signaling regulates spermatogonial stem cell fate. Biol Reprod. 2006;74(2):314–21.
Braydich-Stolle L, et al. Role of Src family kinases and N-Myc in spermatogonial stem cell proliferation. Dev Biol. 2007;304(1): 34–45.
Kanatsu-Shinohara M, et al. Long-term culture of mouse male germline stem cells under serum-or feeder-free conditions. Biol Reprod. 2005;72(4):985–91.
Kanatsu-Shinohara M, et al. Leukemia inhibitory factor enhances formation of germ cell colonies in neonatal mouse testis culture. Biol Reprod. 2007;76(1):55–62.
Ishii K, et al. FGF2 mediates mouse spermatogonial stem cell self-renewal via upregulation of Etv5 and Bcl6b through MAP2K1 activation. Development. 2012;139(10):1734–43.
Kokkinaki M, et al. The molecular signature of spermatogonial stem/progenitor cells in the 6-day-old mouse testis. Biol Reprod. 2009;80(4):707–17.
Yeh JR, Zhang X, Nagano MC. Wnt5a is a cell-extrinsic factor that supports self-renewal of mouse spermatogonial stem cells. J Cell Sci. 2011;124(Pt 14):2357–66.
Golestaneh N, et al. Wnt signaling promotes proliferation and stemness regulation of spermatogonial stem/progenitor cells. Reproduction. 2009;138(1):151–62.
Oatley JM, et al. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc Natl Acad Sci U S A. 2006;103(25):9524–9.
Wu X, et al. Spermatogonial stem cell self-renewal requires ETV5-mediated downstream activation of brachyury in mice. Biol Reprod. 2011;85(6):1114–23.
Wu X, et al. The POU domain transcription factor POU3F1 is an important intrinsic regulator of GDNF-induced survival and self-renewal of mouse spermatogonial stem cells. Biol Reprod. 2010;82(6):1103–11.
Sada A, et al. The RNA-binding protein NANOS2 is required to maintain murine spermatogonial stem cells. Science. 2009;325(5946):1394–8.
Sada A, et al. NANOS2 acts downstream of glial cell line-derived neurotrophic factor signaling to suppress differentiation of spermatogonial stem cells. Stem Cells. 2012;30(2):280–91.
Goertz MJ, et al. Foxo1 is required in mouse spermatogonial stem cells for their maintenance and the initiation of spermatogenesis. J Clin Invest. 2011;121(9):3456–66.
Krausz C, Looijenga LH. Genetic aspects of testicular germ cell tumors. Cell Cycle. 2008;7(22):3519–24.
Stevens L, Mackensen J. Genetic and environmental influences on teratocarcinogenesis in mice. J Natl Cancer Inst. 1961;27:443–53.
Kanatsu-Shinohara M, et al. Generation of pluripotent stem cells from neonatal mouse testis. Cell. 2004;119(7):1001–12.
Izadyar F, et al. Generation of multipotent cell lines from a distinct population of male germ line stem cells. Reproduction. 2008;135(6):771–84.
Ko K, et al. Induction of pluripotency in adult unipotent germline stem cells. Cell Stem Cell. 2009;5(1):87–96.
Seandel M, et al. Niche players: spermatogonial progenitors marked by GPR125. Cell Cycle. 2008;7(2):135–40.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Guan K, et al. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature. 2006;440(7088):1199–203.
Morimoto H, et al. In vitro transformation of mouse testis cells by oncogene transfection. Biol Reprod. 2012;86(5):148; 1–11.
Ko K, et al. Human adult germline stem cells in question. Nature. 2010;465(7301):E1; discussion E3.
Ko K, et al. Brief report: evaluating the potential of putative pluripotent cells derived from human testis. Stem Cells. 2011;29(8):1304–9.
Mizrak SC, et al. Embryonic stem cell-like cells derived from adult human testis. Hum Reprod. 2010;25(1):158–67.
Sadri-Ardekani H, et al. Propagation of human spermatogonial stem cells in vitro. JAMA. 2009;302(19):2127–34.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science + Business Media New York
About this chapter
Cite this chapter
Sachs, C., Seandel, M. (2013). Spermatogonial Stem Cells in Adult Mice and Men. In: Sell, S. (eds) Stem Cells Handbook. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-7696-2_14
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7696-2_14
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-7695-5
Online ISBN: 978-1-4614-7696-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)