
Abstract
In this work we report the results of the study on luminescent lanthanide-containing silica-based hybrid materials. The preparation of organic–inorganic hybrids was realized in several stages. At first, the prior modification of the organic ligand (ethylenediaminetetraacetic (EDTA), diethylenetriaminepentaacetic (DTPA) acids, or p-tert-butyl-calix[4]arene (TBC)) with triethoxysylil group was carried out. Then the obtained precursors were reacted with an equimolar amount of lanthanide salt. At last, a sol-gel process was held and organic–inorganic hybrid materials were synthesized. All samples were characterized by elemental analysis, IR-, NMR-spectroscopy, and SEM. Under UV-excitation typical 4f-luminescence was observed in Ln-EDTA-APTMS/SiO2, Ln-DTPA-APTMS/SiO2 and Ln-TBC-TESPIC/SiO2 (Ln = Nd(III), Eu(III), Tb(III), and Yb(III)). The factors determining luminescence efficiency in studied systems are discussed.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Design and study of new luminescent compounds as active components of nanomaterials is one of the challenges for scientists working on light-emitting devices, luminescent thin films, OLEDs, optical amplifiers, solar energy conversion, alternative light sources, and other. Among a large number of luminescent compounds lanthanide-containing ones should be noted as unique luminescent probes. The emission spectra of lanthanide ions is described as narrow bands covering the wide range from the UV (Gd(III)) to visible (Pr(III), Sm(III), Eu(III), Tb(III), Dy(III), Tm(III)) and near-infrared (Pr(III), Nd(III), Ho(III), Er(III), Yb(III)). The lifetimes of lanthanide emission are rather large, e.g., 0.2–0.5 ms for Nd(III), 1–11 ms for Eu(III) [1–3]. These unique properties of the lanthanide emission (4f-emission) are the result of the forbidden transitions between f-orbitals of the inner 4f-shell, shielded by the filled 5s—and 5p-shells. Because of this shielding the influence of the local environment of the lanthanide ion is limited.
Since all lanthanide ions have low molar absorption coefficients (<10 L mol−1 cm−1), the direct excitation in absorption bands of f–f-transitions leads to weak 4f-emission. One of the efficient method to improve 4f-luminescent characteristics is sensibilization of lanthanide luminescence in complexes with strongly absorbing organic ligands [3]. Under excitation at the absorption bands of organic ligands much more energy can be absorbed by the organic chromophores than the lanthanide ion itself. Then the excitation energy is transferred from the organic ligands to the lanthanide ion by intramolecular energy transfer mechanism. The scheme of the commonly accepted mechanism of energy transfer from the organic ligands to the lanthanide [4, 5] is presented in Fig. 21.1.

Schematic energy diagram showing main photophysical processes in lanthanide complexes
The organic ligands absorbing energy are excited to one of the vibrational levels of the first excited singlet state (S0 → S1). The molecule undergoes fast internal conversion to the lowest vibrational level of the S1-state. The excited singlet state can be deactived radiatively to the ground state exhibiting molecular fluorescence (S1 → S0) or go through nonradiative intersystem crossing from the singlet state S1 to the triplet state T1. The triplet state T1 can be deactivated radiatively to the ground state S0 by the spinforbidden transition T1 → S0. This results in molecular phosphorescence. A nonradiative transition from the triplet state to an excited state of the lanthanide ion can occur. After this indirect excitation by energy transfer, the lanthanide ion may undergo a radiative transition to a lower 4f state by characteristic narrow band luminescence or may be deactivated by nonradiative or back transfer processes. Such indirect excitation leads to a large Stoke’s shift and efficient 4f-luminescence and gives many advantages to lanthanide complexes as luminescent probes. On the other hand, studying the 4f-luminescence some information on the local structure around lanthanide ion can be gained: number of metal-ion sites, composition and symmetry of the first coordination sphere, solution state of the Ln(III) ion, or donor–acceptor distances [6–10].
Design of highly luminescent lanthanide complexes implies the improvement of two main factors that determine the efficiency of 4f-luminescence: energy transfer and minimization of non-radiative processes. Concerning the first one there is an energy match criteria suggested on simplified model that the only existing pathway of energy transfer is from ligand triplet state. The energy difference between the singlet S1 and triplet T1 ΔE(S1–T1) should be at about 5000 cm−1 and between triplet state and emissive level ΔE(T1−Ln3+)—in the range of 2500–3500 cm−1 [11, 12]. A close match between the energy of the triplet state and the energy of the receiving 4f-level of the lanthanide ion is not desirable because energy back transfer of the lanthanide ion to the triplet state can occur [13, 14].
The main reason of deactivation processes of excited 4f-states is vibronic coupling with the ligand and solvent molecules. High energy vibrations, such as O–H, N–H, C–H, quench the lanthanide luminescence, especially for Nd(III), Er(III), and Yb(III) emitting in near IR range. There is a number of synthetic methods for elimination of radiationless losses of energy, such as perhalogenation (usually, perfluorination [15–17]) or deuteration [18–20] of ligand, confinement of coordination of solvent molecules to lanthanide ion: synthesis in deuterated media [21, 22], in presence of surfactants [23–25] or heteroligand chelation [26], insertion of the lanthanide ion in pre-organized environment, e.g., macrocyclic ligands (pophyrins [27–29], calix[n]arenes [30–33], for instance), metall-organic frameworks [34, 35]. Generally, the use of these methods sometimes requires difficult experimental technique and increases the cost of the lanthanide-based material for its practical use. Recent studies on organic–inorganic hybrids containing lanthanide ions have demonstrated their technological potential. In lanthanide-containing hybrid materials usually the complex is embedded in an organic (e.g., poly(methyl methacrylate), poly(vinyl alcohol), poly (N-vinylcarbazole) [36–38]), or inorganic (e.g., SiO2-, Al2O3-, TiO2-based sol-gel materials [39–41]) polymeric host matrix. The benefit of such organic–inorganic materials is its superior mechanical and thermal stability and luminescence output than the molecular lanthanide complex. In this context, the sol–gel method is a powerful tool for embedding lanthanide complex into an inorganic host and processing of hybrid materials as well as nanocomposites, nanoparticles with interesting properties.
In this work, in order to obtain hybrid materials containing lanthanide complexes covalently bonded to a silica polymeric network the following strategy was applied. At first, the organic ligands were modified by reaction with amino- or isocyano-derivatives of trialkoxysilane. Then the stage of complex formation with Ln(III) ion was held. And finally a hybrid material was obtained by sol–gel process. As organic ligands aminopolycarboxylic (ethylenediaminetetraacetic (EDTA) and diethylenetriaminepentaacetic (DTPA)) acids and p-tert-butylcalix[4]arene (TBC) were used. The advantage of EDTA and DTPA is high stability of Ln(III) complexes [42], that is important for acid- or base-catalyzed sol–gel synthesis. The aminopolycarboxylic acids possess enough donor atoms to fill high coordination number of Ln(III) ions. The influence of the silica host on the 4f-luminescence can be also studied due to the absence of strong light-absorbing functional groups in EDTA and DTPA. In contrast with aminopolycarboxylates p-tert-butylcalix[4]arene (TBC) have strong absorption band in the UV-region and it is one of the suitable ligands for sensitization of Ln(III) luminescence due to aromatic fragments playing a role of “photoantenna” [43, 44]. Furthermore, p-tert-butyl radicals at the upper rim of calix[4]arene stabilize the “cone” conformation of the macrocycle and provide its conformational stability in chemical reactions. The hydroxyl groups on the lower rim of calix[4]arene quite easily undergo chemical modifications with substitution of mobile hydrogen atoms that allows to provide covalent bonding of ligand/complex species to an inorganic carrier.
All synthesized compounds and materials have been characterized by elemental analysis, ESI-mass spectrometry, IR- and 1H NMR-spectroscopy. Elemental analysis (C, H, N) was carried out on Perkin–Elmer CHN-240 analyzer. The content of the lanthanide ions was determined by complexometric titration. ESI-Mass spectra of compounds were recorded on Waters ESI TOF Premier spectrometer. 1H NMR spectra were obtained on Bruker Avance AV 400 (400 MHz) spectrometer in D2O solutions at pH ≈ 10 (with the addition of NaOD) for aminopolycarboxylic acids and in CDCl3 ion the case of calixarene and its complexes. IR spectra were recorded on Perkin–Elmer Frontier FT-IR spectrometer in KBr pellets.
Aminopolycarboxylic acids were modified by reaction of dianhydride of relevant acid with (3-aminopropyl)-trimethoxysilane (APTMS) according to modified method described in [45]. Aminopolycarboxylic acid dianhydride (5 mmol) was dissolved in 150 ml of anhydrous DMF with slight heating. Then an equimolar amount of APTMS was added dropwise. The reaction mixture was stirred at 80 °C for 48 h. Then 100 ml of THF were added, formed white precipitate was filtered and recrystallized from solvent mixture DMF-benzene (1:1). EDTA-APTMS: Yield 83 %. M.p. > 250 °C (dec.). Elemental analysis: found for C16H31N3O10Si %: C, 42.35; H, 6.91; N, 9.11; calculated, %: C, 42.37; H, 6.89; N, 9.27. ESI-MASS, m/z: 453 ([M]−), 393 ([M–2CH3-OCH3]−), 334 ([M–2CH3–OCH3–CH2COOH]−). 1H NMR, δ (D2O): 0.71 t (2H, –CH2–Si, J = 8.29 Hz), 1.64 m (2H, –CH 2 –CH2–Si, J = 7.53 Hz), 3.00 t (2H, –CH 2 –H2–CH2–Si, J = 7.15 Hz), 3.20–3.35 m (4H, N–CH2–CH2–N), 3.09 s (2H, –CH 2 –CONH–), 3.52 s (6H, –CH2–COO−). DTPA-APTMS: Yield 79 %. M.p. > 250 °C (dec.). Elemental analysis: found for C20H38N4O12Si, %: C, 43.47; H, 6.79; N 10.31; calculated, %: C, 43.31; H, 6.91; N 10.10. ESI-MASS, m/z: 554 ([M]−), 493 ([M–2CH3–OCH3]−), 391 ([M–(CH2)3Si(OMe)3]−). 1H NMR, δ (D2O): 0.59 t (2H, –CH2–Si, J = 7.79 Hz), 1.67 m (2H, –CH 2 –CH2–Si, J = 7.60 Hz), 2.90 t (2H, –CH 2 –CH2–CH2-Si, J = 6.02 Hz), 2.97 t (4H, N–CH2–CH2–N, J = 5.77 Hz), 3.09 t (4H, N–CH2–CH2–N, J = 5.77 Hz), 3.33 c (8H, –CH2–COO−), 3.49 c (2H, –CH 2 –CONH–).
The second step was synthesis of the lanthanide complexes with modified aminopolycarboxylic acids. Complex with paramagnetic ion Lu(III) was obtained for NMR spectroscopic studies. EDTA-APTMS (or DTPA-APTMS) (0.10 mmol) was dissolved in 10 ml of 0.01 M NaOH (pH = 8.0 ± 0.5) under heating. Then 0.10 mmol of lanthanide (III) chloride was added and stirred at 80 °C for 5 h. Then the solution was filtered, evaporated, and the solid residue was isolated by adding 20 mL of acetone, centrifuging and washing with ethanol and water several times. Elemental analysis: Nd-EDTA-APTMS, found for C13H25N3NaNdO12Si, %: C, 25.69; H, 4.27; N, 6.63; Nd, 23.78; calculated, %: C, 25.57; H, 4.13; N, 6.88; Nd, 23.62. Eu-EDTA-APTMS, found for C13H25N3NaEuO12Si, %: C, 25.39; H, 4.01; N, 6.97; Eu, 24.39; calculated, %: C, 25.25; H, 4.07; N, 6.80; Eu, 24.57. Tb-EDTA-APTMS, found for C13H25N3NaTbO12Si, %: C, 25.11; H, 3.93; N, 6.91; Tb, 25.57; calculated, %: C, 24.97; H, 4.03; N, 6.72; Tb, 25.41. Yb-EDTA-APTMS, found for C13H25N3NaYbO12Si, %: C, 24.29; H, 3.99; N, 6.77; Yb, 27.18; calculated, %: C, 24.42; H, 3.94; N, 6.57; Yb, 27.06. Lu-EDTA-APTMS, found for C13H25N3NaLuO12Si, %: C, 24.51; H, 3.81; N, 6.38; Lu, 27.35; calculated, %: C, 24.34; H, 3.93; N, 6.55; Lu, 27.28. ESI-MASS, m/z: 618 ([M–Na]−), 518 ([M–(CH2)3SiOONa]−). 1H NMR, δ (D2O): 0.79 t (2H, –CH2-Si, J = 7.78 Hz), 1.81 m (2H, –CH 2 –CH2–Si, J = 7.71 Hz), 3.04 t (2H, –CH 2 –CH2–CH2–Si, J = 7.61 Hz), 2.75 s (4H, N–CH2–CH2–N), 3.00-3.50 m (8H, –CH2–COO−, –CH 2 –CONH–). Nd-DTPA-APTMS, found for C17H27NdN4NaO12Si, %: C, 30.34; H, 4.19; N, 8.13; Nd, 21.22; calculated, %: C, 30.26; H, 4.03; N, 8.30; Nd, 21.38. Eu-DTPA-APTMS found for C17H27EuN4NaO12Si, %: C, 29.83; H, 3.81; N, 8.39; Eu, 22.41; calculated, %: C, 29.92; H, 3.99; N, 8.21; Eu, 22.27. Tb-DTPA-APTMS, found for C17H27TbN4NaO12Si, %: C, 29.86; H, 3.77; N, 8.29; Tb, 22.89; calculated, %: C, 29.62; H, 3.95; N, 8.13; Tb, 23.05. Yb-DTPA-APTMS, found for C17H27YbN4NaO12Si, %: C, 29.22; H, 3.75; N, 7.79; Yb, 24.81; calculated, %: C, 29.02; H, 3.87; N, 7.96; Yb, 24.60. Lu-DTPA-APTMS, found for C17H27LuN4NaO12Si, %: C, 28.79; H, 3.99; N, 7.81; Lu, 24.99; calculated, %: C, 28.94; H, 3.86; N, 7.94; Lu, 24.80. ESI-MASS, m/z: 684 ([M–Na]−), 665 ([M–H2O–Na]−), 582 ([M–(CH2)3SiOONa]−). 1H NMR, δ (D2O): 0.71 t (2H, –CH2–Si, J = 8.02 Hz), 1.77 m (2H, –CH 2 –CH2-Si), 3.01 t (2H, –CH 2 –CH2–CH2–Si), 2.55 t (2H, N–CH2−, J = 6.28 Hz), 2.72 t (2H, N–CH2−, J = 5.78 Hz), 2.94 t (2H, N-CH2−, J = 6.78 Hz), 2.97 t (2H, N–CH2−, J = 5.77 Hz), 3.29–3.70 m (10H, –CH2–COO−, –CH 2 –CONH–).
The lanthanide-containing nanocomposites were synthesized using adapted Stöber method [46]. TEOS (5 mmol, 1.12 ml) was added to a mixture of absolute ethanol (7.2 ml) and distilled water (0.08 ml) and stirred for 30 min, followed by the addition of 0.1 mmol of the Ln(III) complex (Ln-EDTA-APTMS or Ln-DTPA-APTMS) in 5 ml of water (pH = 7.0–7.5). Then 2 ml of NH4OH (25 %) was added. Stirring was continued at the same temperature during 30 min. The resulting samples were isolated by adding 20 ml of acetone, centrifuging and washing with ethanol and water several times to remove the unreacted materials that were dried at reduced pressure at 95–100 °C for 2 weeks. The molar ratio of the components in the synthesis was TEOS : Ln(III) complex = 50:1.
The modification of aminopolycarboxylic acids and formation lanthanide (III) complexes were confirmed by ESI-mass spectrometry, 1H NMR and FT-IR-spectroscopy. In the ESI-mass spectrum of EDTA-APTMS peaks with m/z 453 [M], 393 [M–2CH3O–CH3] and 317 [M–CH2Si(OCH3)3] are present. The spectrum of DTPA-APTMS displays peaks with m/z 493 ([M–2CH3–OCH3]−), 447 ([M–Si(OCH3)3]) and 391 ([M–(CH2)3Si–(OCH3)3]−). In the spectra of Eu(III) and Lu(III) complexes the most intense peaks correspond to molecular ion without coordinated water molecules.
The presence of trimethoxysilyl-groups in EDTA- APTMS and DTPA-APTMS due to its unsymmetrical structure causes magnetical non-equivalence of some protons and complication of 1H NMR spectra as compared to free EDTA and DTPA [47]. In the spectrum of DTPA-APTMS, for instance, the signals of N(CH2)2 N-protons appear as two triplets at 2.97 and 3.90 ppm, signals of NCH2COO−fragments are two singlets at 3.33 and 3.49 ppm. There are three signals corresponding to three methylene groups of propyl fragment at 0.59, 1.67, and 2.90 ppm.
The formation complex of EDTA- APTMS and DTPA-APTMS with Lu(III) ions leads to similar changes in 1H NMR spectra due to limitation of the conformational rotation around single C–C and C–N bonds under the coordination of the metal. Such changes in the spectra are typical for aminopolycarboxylates. For example, in the spectrum of Lu-DTPA-APTMS there is a small low field shift (0.10–0.12 ppm) of propyl protons signals, four triplets at 2.55–2.97 ppm were observed instead of two triplets of ethylenediamine protons in DTPA-APTMS, multiplet at 3.25–3.50 ppm was registered instead of two singlets from N–CH2–COO-fragments.
The comparison of the FTIR spectra of the modified ligands, complexes, and hybrid materials was made. The FTIR spectra of DTPA-APTMS, its Eu(III) complex and hybrid material are shown as an example in Fig. 21.2. In the spectrum of DTPA-APTMS a broad band at 1052–1144 cm−1 and the band at 911 cm−1 correspond to the asymmetric and symmetric vibrations of the Si–O–C bonds, respectively. Peak at 480 cm−1 corresponds to the deformation vibrations O–Si–O, and the peak at 694 cm−1 probably caused by the stretching vibrations Si–C. Peaks at 1634 cm−1 and 1385–1398 cm−1 were attributed to νas (COO−) and νs (COO−), respectively. A shoulder at 1733 cm−1 corresponds to nonionized carboxyl group. Band of deformation vibrations of amino-group of APTMS at 1577 cm−1 [48] is absent in the spectrum of DTPA-APTMS, confirming the formation of amide bond. The direct observation of amide bond vibrations is complicated, probably, due to its overlay with the vibrations of carboxylic groups. The band at 2548 cm−1corresponds to stretching vibrations of R3N+–H fragments. Peaks at 2936, 2972, 3026 cm−1 were attributed to stretching vibrations of –C–H groups located near nitrogen atom.

FTIR spectra of DTPA-APTMS (a) Eu-DTPA-APTMS (b) and Eu-DTPA-APTMS-SiO2 (c) in KBr pellets
In the FTIR spectrum of the complex Eu-DTPA-APTMS (Fig. 21.2b) a shift of νas (COO−) (Δν = 40 cm−1) is observed, which confirms the coordination with Eu(III). The vibrations of R3N+–H fragment disappears in spectrum of complex. Stretching vibration of-CH2– groups are at 2855, 2927 and 2981 cm−1 and bending vibration are at 1328 and 1446 cm−1. νs(COO−) are manifested in the form of signals at 1385 and 1410 cm−1. The bands at 1094 and 1119 cm−1 are assigned to the stretching vibrations of Si–OH, which arise as a result of hydrolysis. The IR spectra also confirm the absence of polycondenzation of hydrolyzed fragments: peak corresponding to bending vibrations Si–O–Si at 799 cm−1 is absent. The bands at 679, 715 cm−1 correspond to oscillation of C–Si bond.
The FTIR spectra of sol–gel materials represent a superposition of spectra of SiO2 [49] and Ln-DTPA-APTMS (or Ln-EDTA-APTMS). The peaks at 468 and 799 cm−1 were attributed to O–Si–O and Si–O-Si bending vibrations, respectively; bands of symmetric and asymmetric stretching vibrations of Si–O–Si are located at 970 and 1088 cm−1. Some hydroxyl groups remaining in structure of materials give the band at 1639 cm−1 (bending vibrations of Si–OH groups) and a broad band at 3459 cm−1 (stretching vibrations of O–H groups). It is worth noting that position of the main absorption bands of the complex does not change, confirming the preservation of its structure in synthesized hybrid.
The calix[4]arene-derived hybrid materials were also synthesized in several steps. The lower rim of the TBC ligand was modified by reaction with 3-(triethoxysilyl)propyl isocyanate (TESPIC), wherein active isocyanine group reacts with hydroxyl group of calix[4]arene on the mechanism of nucleophilic addition, according to similar method described in [50, 51]. p-Tert-butylcalix[4]arene (0.324 g, 0.5 mmol) was dissolved in 20 ml of pyridine, and 3-(ethoxysilyl)propyl isocyanate (0.124 g, 0.5 mmol) in 5 ml of pyridine was added dropwise with stirring. The reaction mixture was kept under stirring at 60–65 °C for 8–10 h. After the end of reaction, the solvent was removed. The obtained product was recrystallized from toluene. Elemental analysis, calculated for C54H77NSiO8: C, 72.4, H, 8.6; found C, 72.6, H, 8.4. 1H NMR (CDCl3) δ, ppm: 0.63 (t, 2H, CH2Si), 1.10 (t, 9H, CH 3 CH2), 1.22 (s, 9H, C(CH3)3), 1.27 (s, 27H, C(CH3)3), 1.62–1.68 (m, 6H, CH3 CH 2 ), 3.15–3.21 (m, 2H, CH 2 CH2CH2), 3.51 (d, 4H, ArCH2Ar), 3.75–3.81 (m, 2H, CH 2 CH2CH2) 4.12 (d, 2H, ArCH2Ar), 4.25 (d, 2H, ArCH2Ar), 4.55 (s, 1H, NH), 7.06 (s, 6H, ArH), 7.23 (s, 2H, ArH), 10.35 (s, 1H, OH).
Ln-TBC-containing hybrid materials were prepared without isolation of Ln(III) complex. To a solution of TBC-TESPIC (0.0216 g, 0.024 mmol) in 5 ml of DMF an equimolar amount of lanthanide chloride was added. The reaction mixture was stirred for 3 h, tetraethoxysilane (TEOS) was added (0.250 g, 1.2 mmol) and stirring was continued for another 4 h. Then 0.02 ml of concentrated hydrochloric acid and 30 ml of distilled water was added to the resulting solution under careful stirring. The final hybrids were isolated by adding 20 mL of acetone, centrifuging, and washing with ethanol and water several times to remove the unreacted materials. The molar ratio of the components in the synthesis was TEOS:Ln(III) complex = 50:1.
The presence of TBC ligand covalently bonded to a silica polymeric host was identified by means of FTIR spectroscopy. In the spectrum of TBC-TESPIC stretching vibration bands of the hydroxyl groups is shifted to higher frequencies compared to TBC (ν(OH) = 3163 cm−1) and overlaps with the absorption band of the NH-vibrations forming a broad band with a peak at 3338–3345 cm−1. Increasing the intensity of absorption bands with maxima at 2964 and 2913 cm−1 is more than twofold due to the superposition of vibration bands of substituent CH2-groups and signals of methylene bridge of calixarene macrocycle. The vibrations of N=C=O groups originating from TESPIC were not obserfed at 2270–2280 cm−1, indicating the modification of TBC. In contrast to the initial calix[4]arene, there are also bands of stretching vibrations of the carbonyl group (ν(c=o) = 1658 cm−1) and ethoxysilylic fragment (ν(Si–O) = 1112 cm−1, ν(Si–C) = 1210 cm−1).
As in the case of aminopolycarboxylates the FTIR spectra of calix[4]arene-containing hybrids consist of bands of Ln(III)–TBC complex [52, 53] superimposed on absorption bands of SiO2. The spectra reveal a red shift of the band of phenolic groups bending vibrations (Δδc-O-H = 18–20 cm−1) and the shift in the low-frequency of bands of stretching vibrations of carbonyl fragments (ν(c=o) = 1635 cм−1), these changes indicate the coordination of the se groups with lanthanide ion. Reduction of the frequency of Si–O-bonds oscillations (ν(Si–O) = 1040 cm−1) caused due to the hydrolysis of the fragments and the formation of SiO2-skeleton matrix.
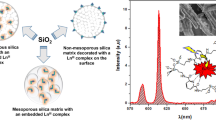
Thus sol–gel organic–inorganic hybrid materials Ln-EDTA-APTMS/SiO2, Ln-DTPA-APTMS/SiO2, and Ln-tBC-TESPIC/SiO2 (Ln(III) = Nd, Eu, Tb, Yb, Er, Lu) were synthesized (Fig. 21.3). The scanning electron microphotographs demonstrate the microstructure of hybrids. The morphology of the Ln(III) aminopolycarboxylate-containing hybrids Ln-DTPA-APTMS/SiO2 is rough and the intensive tendency of growing into inorganic polymer is observed with reservation of the coordinated positions in corresponding bulk materials. In the case of calix[4]arene-based hybrid the configuration of the organosilane is mixed up and it is difficult to form an organized structure under the weak interactions such as π–π-staking and because of the coordination between organic groups in TBC-TESPIC and lanthanide ions.

Schematic structure of synthesized organic–inorganic hybrids (a) Ln-EDTA-APTMS/SiO2 (b) Ln-DTPA-APTMS/SiO2 (c) Ln-TBC-TESPIC/SiO2 (d) and SEM-images of DTPA (e) and TBC-based samples (f)
The microstructure of sol–gel materials significantly depends on conditions of the synthesis and the formation and the growth rates of the sol particles during the stage of sol formation [53]. Thus, at pH < 7 the rate of particles formation is greater than the rate of growth that leads to a large number of little particles (less than 10 nm) which form a three-dimensional structure. At pH = 7–10 the growth rate of particles dominates and their size can reach about 1000 nm, but further increase in size may lead to their deposition from solutions. On the photo the formation of spherical nanoparticles could not be observed that suggests their aggregation with forming of a three-dimensional matrix on the early stages of growth (up to 5 nm).
The emission spectra of silica-modified ligands were studied to estimate the position of singlet and triplet levels from the fluorescence and phosphorescence spectra and the possibility of the excitation energy transfer to Ln(III) ion. The emission spectra of Lu-EDTA-APTMS/SiO2 (Fig. 21.4a) and Lu-DTPA-APTMS/SiO2 at 298 K show fluorescence in the range of 400–600 nm with maximum at 421 and 413 nm, respectively. At 77 K the maxima of this band shift to 446 nm and 483 nm for EDTA-and DTPA-derived samples, respectively. In the case of Lu-TBC-TESPIC/SiO2 the sample exhibits fluorescence at 406 nm and phosphorescence at 419 nm in the form of broad bands in the range 380–520 nm (Fig. 21.4b). The maxima of these spectra are shift hypsochromically at 3 and 5 nm, respectively, compared with lutetium complexes with TBC and the same for samples in the solid state and in solution. Therefore, the energy of the singlet states of Lu-EDTA-APTMS/SiO2, Lu–DTPA-APTMS/SiO2, and Lu-TBC-TESPIC/SiO2 amounts to 23,750 cm−1, 24,210 cm−1, and 24,630 cm−1, respectively, and the energy of the triplet states—22,420 cm−1, 21,900 cm−1, and 23,870 cm−1, respectively.

Fluorescence (solid line) and phosphorescence (dashed line) spectra of Lu-EDTAAPTMS/SiO2 (a) and Lu-TBC-TESPIC/SiO2 (b)
The values of the triplet levels are suitable to realize the intramolecular excitation energy transfer to Ln(III) ions and to observe 4f-luminescence. The position of the energy levels of aminopolycarboxylate-derived organic–inorganic materials is more suitable for energy transfer to Eu(III) than Tb(III) ions. The energy gap between triplet state and the emissive 5D0-level of Eu(III) (17280 cm−1) is in the range of 4600–5100 cm−1, while for 5D4-level of Tb(III) (20500 cm−1)—1400–1920 cm−1 and the energy back transfer processes are expected in Tb-EDTA-APTMS/SiO2 and Tb-DTPA-APTMS/SiO2. The difference between triplet level of TBC-based material and Eu(III) and Tb(III) ions is at about 6600 and 3370 cm−1, respectively, indicating more efficient energy transfer to Tb(III) ion in contrast to aminopolycarboxylates. The large energy gap for Eu-TBC-TESPIC/SiO2 assumes the greater contribution of radiationless losses of the excitation energy on O–H-, C–H-vibrations in quenching of 4f-luminescence.
The excitation spectra of Eu-EDTA-APTMS/SiO2 and Eu-DTPA-APTMS/SiO2 (Fig. 21.5a) consist of symmetric and broad band ranging from 300 to 400 nm. As can be seen, the matrix—the organic ligand within silica polymeric host is able to transfer much more energy to europium ion than it absorbs at 394 nm (f–f-transition 7F0 → 5L6). The corresponding emission spectrum contains 5D0 → 7FJ (J = 0, 1, 2, 3, 4) transition lines of Eu(III) with the red emission 5D0 → 7F2 as the most intensive one. The observed transitions are mainly of an electric dipole nature, except the 5D0 → 7F1 line which has a predominant magnetic dipole contribution. It is worth noting that the fluorescence of matrix in the 390–450 nm region is quenched more than 75 % as compared to Lu(III) complexes. The emission spectrum of Eu-TBC-TESPIC (Fig. 21.5b) represents the band of matrix fluorescence and very poor 4f-luminescent signal of Eu(III), probably because of the efficient energy back transfer processes.

Excitation a and emission b spectra of Eu-DTPA-APTMS/SiO2 (solid line) and Eu-EDTA-APTMS/SiO2 (dashed line)
The 4f-luminescence intensity of obtained xerogels was compared with the corresponding molecular complexes (Eu-EDTA, Eu-DTPA) (Table 21.1).
The luminescence intensity of Eu-EDTA-APTMS/SiO2 is about 63 % from Eu-EDTA intensity, and intensity of Eu-DTPA-APTMS/SiO2 is 72 % from Eu-DTPA. When assessing these values the concentration of the ion-emitter in the sample should also be taken into account. The content of Eu(III) in Eu-DTPA-APTMS/SiO2, for instance, is 6.7 times lower in comparison with the Eu-DTPA. The 4f-luminescence intensity of Eu-DTPA-APTMS/SiO2 is 1.5 times higher than of Eu-EDTA-APTMS/SiO2. A slight increase of Eu(III) excited state lifetime is observed in hybrids: from 329 μs in Eu-EDTA to 343 μs in Eu-EDTA-APTMS/SiO2 and from 625 μs in Eu-DTPA to 672 μs in Eu-DTPA-APTMS/SiO2.
The energy transfer from the hybrid matrix to Tb(III) is not as efficient as ion in Eu(III)-containing materials. The luminescence spectra of Tb-EDTA-APTMS/SiO2 and Tb-DTPA-APTMS/SiO2 (Fig. 21.6a) registered upon excitation wavelength at 350 nm show wide intensive band of fluorescence at 420 nm and series of peaks, corresponding to Tb(III) transitions 5D4 → 7FJ (J = 3, 4, 5, 6). The intensity of the matrix fluorescence is two times larger than the 4f-luminescence. Thus, excitation at 350 nm leads both to luminescence of Tb(III) and fluorescence of hybrid matrix, which excitation energy is not completely transferred to the lanthanide ion. The reason is probably in the value of the energy levels of the matrix and Tb(III) ion, that causes energy back transfer. In contrast to aminoplolycarboxylates, the emission spectrum of Tb-TBC-TESPIC/SiO2 shows very intensive 4f-luminescence (Fig. 21.6b),

Emission spectra of Tb-DTPA-APTMS/SiO2 (dashed line) and Tb-TBC-TESPIC/SiO2 (solid line)
In silica materials doped with lanthanide complexes with TBC 4f-luminescence of Nd(III), Er(III), and Yb(III) is realized in the IR-region. For erbium-containing compound 4f-luminescence is observed with the maximum at 1530–1532 nm (6540 cm−1) corresponding to the transition 4I13/2 → 4I15/2 (Fig. 21.7a). It was found that in the silicate particles the intensity of Er(III) luminescence is 3.2–3.6 times higher than in the solution of the initial complex Er-TBC.

Emission spectra of a Er-TBC-TESPIC/SiO2 and b YB-TBC-TESPIC/SiO2
Upon excitation in the macrocycle absorption band (312–320 nm) particles containing complex Nd-TBC exhibit characteristic 4f-luminescence: bands with maxima at 889–895 nm (11,173–11,250 cm−1 transition 4F3/2 → 4I9/2), 1065–1069 nm (9350–9400 cm−1) and 1073–1076 nm (9250–9300 cm−1) corresponding to the splitting transition 4F3/2 → 4I11/2.
The spectrum of 4f-luminescence of Yb-TBC-TESPIC/SiO2 represents a band, corresponding to the band split to three peaks with maxima at 976, 1006 and 1022 nm (Fig. 21.7b). Such splitting is a consequence of reducing the symmetry of the coordination polyhedron of the ytterbium complex in silica matrix.
In a series of synthesized compounds EDTA - and DTPA-APTMS are more suitable precursors for obtaining Eu(III) complexes and hybrids with effective 4f-luminescence in visible and TBC-TESPIC—for Tb(III) and Lanthanides emitting in near IR-range. Despite the inefficient energy transfer in Tb(III) aminopolycarboxylate-containing hybrids they could be useful objects for research as wide-range dual-wave emitters due to simultaneous fluorescence and 4f-luminescence. It may be concluded that sol–gel organic–inorganic hybrids with covalently bonded Ln-EDTA, Ln-DTPA and Ln-TBC complexes are promising materials for further research in practical application.
Abbreviations
- APTMS:
-
(3-aminopropyl)trimethoxysilane
- DMF:
-
N,N-Dimethylformamide
- DTPA:
-
Diethylenetriaminepentaacetic acid
- EDTA:
-
Ethylenediaminetetraacetic acid
- ESI:
-
Electrospray ionization
- ET:
-
Energy transfer
- FTIR:
-
Fourier transform infrared spectroscopy
- IR:
-
Infrared
- ISC:
-
Intersystem crossing
- Ln(III):
-
Lanthanide(III)
- NMR:
-
Nuclear magnetic resonance
- OLED:
-
Organic light-emitting diode
- TBC:
-
25,26,27,28-tetrahydroxy-p-tert-butylcalix[4]arene
- TEOS:
-
Tetraethyl orthosilicate
- TESPIC:
-
3-(triethoxysilyl)propyl isocyanate
- THF:
-
Tetrahydrofuran
- UV:
-
Ultraviolet
- m:
-
Multiplet (NMR-spectra)
- s:
-
Singlet
- t:
-
Triplet
- I4f :
-
4f-Luminescence intensity
- S0 :
-
Ground singlet state
- S1 :
-
First excited singlet state
- T1 :
-
First excited triplet state
- λex :
-
Excitation wavelength
- λem :
-
Emission wavelength
- τ:
-
Excited state lifetime
References
Albrecht M, Osetska O, Klankermayer J et al. (2007) Enhancement of near-IR emission by bromine substitution in lanthanide complexes with 2-carboxamide-8-hydroxyquinoline Chem Commun 18:1834–1836
Glover P, Basett A, Nockemann P et al (2007) Fully fluorinated imidodiphosphinate shells for visible- and NIR-emitting lanthanides: hitherto unexpected effects of sensitizer fluorination on lanthanide emission properties. Chem Eur J 13:6308–6320
Eliseeva S, Bünzli J-C (2010) Lanthanide luminescence for functional materials and bio-sciences. Chem Soc Rev 39:189–227
Weissman S (1942) Intramolecular energy transfer the fluorescence of complexes of europium. J Chem Phys 10:214–217
Crosby G, Whan R, Alire R (1961) Intramolecular energy transfer in rare earth chelates. Role Triplet State J Chem Phys 34:743–748
Crosby G, Whan R, Freeman J (1962) Spectroscopic studies of rare earths chelates. J Phys Chem 66:2493–2499
Muller G, Kean S, Parker D et al (2002) Temperature and pressure dependence of excitation spectra as a probe of the solution structure and equilibrium thermodynamics of a Eu(III) complex containing a modified dota ligand. J Phys Chem A 106:12349–12355
Frey S, Horrocks W Jr (1995) On correlating the frequency of the 7F0 → 5D0 transition in Eu3+ complexes with the sum of ‘nephelauxetic parameters’ for all of the coordinating atoms Inorg Chim Acta 229: 383–390
Bünzli J-C, Klein B, Wessner D (1982) Crystal structure and emission spectrum of tris(nitrato)-1,4,7,10-tetraozacyclododecane-Europium(III) Inorg Chim Acta 59:269–274
Bünzli J-C (2010) Lanthanide luminescence for biomedical analyses and imaging. Chem Rev 110:2729–2755
Samuel A, Xu J, Raymond K (2009) Predicting efficient antenna ligands for Tb(III) emission. Inorg Chem 48:687–698
Archer R, Chen H, Thompson L (1998) Synthesis, characterization, and luminescence of europium(III) schiff base complexes. Inorg Chem 37:2089–2095
Latva M, Takalo H, Mukkala V et al (1997) Correlation between the lowest triplet state energy level of the ligand and lanthanide(III) luminescence quantum yield J. Luminescence 75:149–169
Dadabhoy A, Faulkner S, Sammes P (2002) Long wavelength sensitizers for europium(III) luminescence based on acridone derivatives. J Chem Soc Perkin Trans 2:348–357
Dang S, Yu JB, Wang X et al (2010) A study on the NIR-luminescence emitted from ternary lanthanide [Er(III), Nd(III) and Yb(III)] complexes containing fluorinated-ligand and 4,5-diazafluoren-9-one. J Photochem Photobiol A Chem 214:152–160
Monguzzi A, Tubino R, Meinardi F et al (2009) Er3+ perfluorinated complexes for broadband sensitized near infrared emission. Chem Mater 21:128–135
dos Santos E, Freire R, da Costa N et al (2010) Theoretical and experimental spectroscopic approach of fluorinated Ln3+-diketonate complexes. J Phys Chem A 114:7928–7936
Hasegawa Y, Murakoshi K, Wada Y et al (1996) Enhancement of luminescence of Nd3+ complexes with deuterated hexafluoroacetylacetonato ligands in organic solvent. Chem Phys Lett 248:8–12
Hasegawa Y, Murakoshi K, Wada Y, et al. (1996) Characteristic emission of β-diketonato Nd3+ complexes dressed with perfluoroalkyl groups in DMSO-d 6 Chem Phys Lett, 260: 173–177
Bischof C, Wahsner J, Scholten J et al (2010) Quantification of C − H quenching in near-IR luminescent ytterbium and neodymium cryptates. J Am Chem Soc 132:14334–14335
Glover P, Bassett A, Nockemann P et al. (2007) Fully fluorinated imidodiphosphinate shells for visible- and NIR-emitting lanthanides: hitherto unexpected effects of sensitizer fluorination on lanthanide emission properties. Chem–Eur J, 13: 6308–6320
Iwamuro M, Adachi T, Wada Y, et al. (2000) Photosensitized Luminescence of Neodymium(III) Coordinated with 8-Quinolinolates in DMSO-d6 Bull Chem Soc Japan 73: 1359–1363
Binnemans K, Gnrller-Walrand C (2002) Lanthanide containing liquid crystals and surfactants. Chem Rev 102:2303–2345
Peter S, Panigrahi B, Viswanathan K et al. (1992) Fluorescence enhancement of dysprosium, europium and terbium using sodium benzoate-trioctylphosphine oxide-triton X-100 Anal Chim Acta 260: 135–141
Wenlian L, Weili L, Gui Y, et al. (1993) Enhanced luminescence and energy transfer of Eu(III) and Tb(III) in chelates in micelle solutions J Alloys and Compounds 191: 107–110
Ermolaev V, Gruzdev V (1984) Novel spectral-kinetic methods for investigation of ligand exchange in labile metal complexes in solutions. Inorg Chim Acta 95:179–185
Asano-Someda M, Kaizu Y, (2001) Hot bands of (f,f*) emission from ytterbium(III) porphyrins in solution J Photochem Photoboil A: Chem 139: 161–165
Korovin Yu, Rusakova N (2001) Infrared 4f-luminescence of lanthanides in the complexes with macrocyclic ligands Rev. Inorg Chem 21:299–329
Beeby A, Dickins R, Fitzgerald S et al. (2000) Porphyrin sensitization of circularly polarised near-IR lanthanide luminescence: enhanced emission with nucleic acid binding J Chem Soc Chem Commun: 1183–1184
Steemers F, Verboom W, Reinhoudt D et al (1995) New sensitizer-modified calix[4]arenes enabling near-UV excitation of complexed luminescent lanthanide ions. J Am Chem Soc 117:9408–9414
Ramírez F, Charbonnière L, Muller G et al. (2001) A p-tert-butylcalix[4]arene functionalised at its lower rim with ether-amide pendant arms acts as an inorganic–organic receptor: structural and photophysical properties of its lanthanide complexes J Chem Soc, Dalton Trans 3205–3213
Kajiwara T, Katagiri K, Hasegawa M et al (2006) Conformation-controlled luminescent properties of lanthanide clusters containing p-tert-butylsulfonylcalix[4]arene. Inorg Chem 45:4880–4882
Skripacheva V, Mustafina A, Rusakova N et al (2008) Novel heterometallic Co(III)-Ln(III) (Ln = Gd, Tb, Dy) complexes on p-sulfonatothiacalix[4]arene platform exhibiting redox-switchable metal-to-metal energy transfer Eur J. Inorg Chem 2008:3957–3963
Eddaoudi M, Moler D, Li H et al (2001) Modular chemistry: secondary building units as a basis for the design of highly porous and robust metal—organic carboxylate frameworks. Acc Chem Res 34:319–330
Daiguebonne C, Kerbellec N, Bernot K et al (2006) Synthesis, crystal structure, and porosity estimation of hydrated erbium terephthalate coordination polymers. Inorg Chem 45:5399–5406
Misra V, Mishra H (2008) Photoinduced proton transfer coupled with energy transfer: Mechanism of sensitized luminescence of terbium ion by salicylic acid doped in polymer J Chem Phys 128: 244701-1-244701-8
Raj D, Francis B, Reddy M et al (2010) Highly luminescent poly(methyl methacrylate)-incorporated europium complex supported by a carbazole-based fluorinated β-diketonate ligand and a 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene oxide co-ligand. Inorg Chem 49:9055–9063
O’Riordan A, O’Connor E, Moynihan S et al (2005) Narrow bandwidth red electroluminescence from solution-processed lanthanide-doped polymer thin films. Thin Solid Films 491:264–269
Binnemans K (2009) Lanthanide-based luminescent hybrid materials Chem Rev 109:4283–4374
Feng J, Zhang H (2012) Hybrid materials based on lanthanide organic complexes. Rev Chem Soc Rev. doi:10.1039/C2CS35069F
Anderegg G, Arnaud-Neu F, Delgado R et al (2005) Critical evaluation of stability constants of metal complexes of complexones for biomedical and environmental applications. Pure Appl Chem 77:1445–1495
Alpha B, Lehn J-M, Mathis G (1987) Energy transfer luminescence of europium(III) and terbium(III) cryptates of macrobicyclic polypyridine ligands Angew Chem Int Ed 26: 266–267
Sabbatini N, Perathoner S, Balzani V, Alpha B, Lehn J-M (1987) Supramolecular photochemistry. Reidel, Dordrecht
Qiaoyu N, Jianxin M, Haiming W et al (2006) Novel terbium chelate doped fluorescent silica nanoparticles. J Rare Earths 24:193–196
Rieter W, Kim J, Taylor K et al (2007) Hybrid silica nanoparticles for multimodal imaging. Angew Chem Int Ed 46:3680–3682
Stöber W, Fink A (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26:62–69
Choppin G, Baisden P, Khan S (1979) Nuclear magnetic resonance studies of diamagnetic metal-diethylenetriaminepentaacetate complexes. Inorg Chem 18:1330–1332
Kim J, Seidler P, Wan L et al (2009) Formation, structure, and reactivity of amino-terminated organic films on silicon substrates. J Colloid Interface Sci 329:114–119
Yan B, Zhang H, Wang S et al (1997) Luminescence properties of the ternary rare earth complexes with β-diketones and 1,10-phenanthroline incorporated in silica matrix by a sol-gel method. Mater Chem Phys 51:92–96
Lu H-F, Yan B, Liu J-L (2009) Functionalization of calix[4]arene as a molecular bridge to assemble luminescent chemically bonded rare-earth hybrid systems. Inorg Chem 48:3966–3975
Li Y-J, Yan B, Wang L (2011) Rare earth(Eu3+, Tb3+) mesoporous hybrids with calix[4]arene derivative covalently linking MCM-41:Physical characterization and photoluminescence property J Solid State Chem 184: 2571–2579
Katsyuba S, Kovalenko V, Chernova x et al (2005) Vibrational spectra, co-operative intramolecular hydrogen bonding and conformations of calix[4]arene and thiacalix[4]arene molecules and their para-tert-butyl derivatives. Org Biomol Chem 3:2558–2565
Leyton P, Domingo C, Sanchez-Cortes S et al (2007) Reflection–absorption IR and surface-enhanced IR spectroscopy of tetracarboethoxy t-butyl-calix[4]arene, as a host molecule with potential applications in sensor devices. Vib Spectrosc 43:358–365
Brinker CJ, Scherer GW (1990) Sol-gel science: the physics and chemistry of sol-gel processing. Academic Press, New York
This work was supported by the National Academy of Sciences (DCNTP “Nanotechnology and nanomaterials”, project 6.22.7.43).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this paper
Cite this paper
Smola, S.S., Snurnikova, O.V., Fadeyev, E.N., Rusakova, N.V. (2013). Sol–Gel Organic–Inorganic Hybrid Materials Containing Lanthanide Complexes with Polydentate Acyclic and Cyclic Ligands: Synthesis and Spectral-Luminescent Properties. In: Fesenko, O., Yatsenko, L., Brodin, M. (eds) Nanomaterials Imaging Techniques, Surface Studies, and Applications. Springer Proceedings in Physics, vol 146. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-7675-7_21
Download citation
DOI: https://doi.org/10.1007/978-1-4614-7675-7_21
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-7674-0
Online ISBN: 978-1-4614-7675-7
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)