
Abstract
Inflammasomes are a family of protein complexes that recognize diverse microbial and endogenous danger signals to promote innate immune responses, tissue inflammation and injury, or cell death via pyroptosis. Inflammasome activation results in the recruitment and activation of caspase-1, which is required for the production of the proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 that can modulate both adaptive and innate immune responses through effects on leukocyte survival, proliferation, differentiation, and migration. Recent studies suggest that inflammasome activity may also play important roles in several nonmicrobial disease states associated with chronic inflammation. For example, NLRP3 inflammasome expression and IL-1β production are both increased in obesity, and recent work has implicated NLRP3 inflammasome activation in a variety of obesity-linked conditions including gout, type 2 diabetes mellitus, metabolic liver disease, atherosclerosis, Alzheimer’s disease, cancer and rheumatoid arthritis. Further, many of the factors associated with these conditions, including elevated plasma glucose, fatty acids, uric acid and cholesterol crystals, and β-amyloid, have been shown to be elevated during obesity and to stimulate NLRP3 inflammasome expression or activation. Since chronic NLRP3 activation appears to play important roles in several common disease states, better understanding of inflammasome regulation and function may lead to better therapeutic approaches. Several agents that attenuate NLRP3 inflammasome activity or inhibit its primary effector, IL-1β, are currently under development or in early clinical trials as therapeutics to treat these common disease conditions. This chapter will review new research on inflammasome activation, its role in obesity and other chronic inflammatory states, and the status of approaches to attenuate NLRP3 inflammasome activity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Familial Mediterranean Fever
- NLRP3 Inflammasome
- Inflammasome Activation
- NLRP3 Inflammasome Activation
- Inflammasome Complex
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Inflammasomes Are Key Mediators of the Innate Immune System
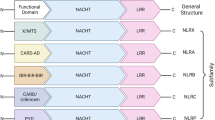
The innate immune system serves as a first line of defense against invading microbes and other harmful agents by sensing pathogen-associated molecular patterns (PAMPs), such as microbial nucleic acid, lipoproteins, and lipopolysaccharides (LPSs), or danger-associated molecular patterns (DAMPs) released during cell stress, including ATP, uric acid, heat shock proteins, and other cellular components [1]. The initiation and regulation of innate immune responses to these factors are orchestrated by several classes of germline-encoded pattern-recognition receptors (PRRs), including toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), and AIM2 (absent in melanoma 2)-like receptors (ALRs) [2, 3]. NLRs comprise a large protein family of intracellular sensors, the members of which share a conserved central nucleotide-binding, oligomerization domain (NOD), a leucine-rich repeat (LRR) region, and a variable N-terminal effector domain. NLRs can be classified as receptors (Nod1 and Nod2) or by their different central proteins and their diverse functions: negative regulators include NLRX1, NLRC5, and NLRP4 and inflammasome activators include NLRP1, NLRC4, NLRP3, and NLRC5 [4–7]. Several NLRs and AIM2 can form large multi-protein “inflammasome” complexes upon stimulation by PAMP or DAMP signals.
Following activation, NLRs oligomerize and recruit pro-caspases through direct interaction with the NLR caspase recruitment and activation domain (CARD) and through interaction with the pyrin domain (PYD) of the ASC (apoptosis-associated speck-like protein containing a CARD) adaptor protein. ASC resides in the nucleus, but its movement from the nucleus to the cytosol is required for inflammasome assembly [8]. Caspase zymogen recruitment into an inflammasome complex induces a conformational change leading to caspase autoactivation.
IL-1 and IL-18, generated by capase-1 maturation of pro-IL-1 and pro-IL-18 peptides, are the major functional effectors of inflammasome activation, inducing multiple responses to infectious agents. IL-1β induces fever, T cell survival, B cell proliferation, and leukocyte migration and promotes polarization of CD4+ T helper 1 (Th1) cells [9], while IL-18 synergizes with IL-12 to promote Th1 polarization and IFNγ production and facilitates proinflammatory Th17 responses [10]. Caspase-1 activity is also required for pyroptosis, a proinflammatory cell death mechanism for self-clearance of infected macrophages and dendritic cells thought necessary to prevent intracellular replication of pathogenic microorganisms (Fig. 2.1 from Zitvogel et al. [11]). Both IL-1 and IL-18 production and pyroptosis have implications for many diseases including infections, arthritis, metabolic syndrome, cancer, and inflammatory bowel disease. Caspase-1 also contributes to host defense through additional mechanisms, including secretion of leaderless peptides, cleavage of glycolytic pathway enzymes, restriction of bacterial replication, and augmentation of cell repair through control of lipid metabolism (summarized in Lamkanfi and Dixit [12]).

Pyroptosis as a possible outcome of caspase-1 activation. Activation of caspase-1 (casp-1) mediated by the inflammasome or by the supramolecular assembly of ASC dimers known as the “pyroptosome” can derive from the proteolytic cleavage of pro-caspase-1 (pro-casp-1) or follow less well-understood (perhaps conformational) mechanisms. Both examples of caspase-1 activation can lead to the proteolytic maturation of caspase-7, followed by the cleavage of several intracellular substrates and pyroptotic cell death. Nevertheless, cleaved caspase-1 is required for the processing of pro-IL-1β and pro-IL-18, as well as for the release of mature IL-1β and IL-18 into the microenvironment (reprinted with permission from Zitvogel et al. [11])
These cytokine- and caspase-1-regulated processes must be tightly controlled balancing the ability to curb infection and develop T and B cell memory to pathogen exposure vs. host injury and damage. Some invading microbes evade various steps in IL-1 and IL-18 production to overcome this host defense. However, uncontrolled activation of inflammasomes contributes to autoimmune diseases such as familial Mediterranean fever and Muckle-Wells syndrome (reviewed in Ting et al. [13]). Several distinct mechanisms can attenuate inflammasome-mediated responses [14]. The anti-inflammatory cytokine IL-10 can inhibit pro-IL-1 synthesis via the STAT3 signaling pathway, while activation of the STAT1 pathway signaling can inhibit IL-1 maturation by blocking caspase-1 processing through an unknown mechanism specific for NLRP3 and NLRP1 inflammasomes. IFNγ produced by activated CD4+ Th1 cells and CD8+ T cells can also transiently inhibit IL-1 production [15], which has been suggested to serve as negative feedback inhibition upon activation of the adaptive immune system. Effector and memory T cells can also inhibit inflammasome activity through the CD40L, OX40L, and RANKL signaling pathways [16].
2.2 The NLRP3 Inflammasome is a Major Mediator of Human Disease
NLRP3 inflammasomes are among the most highly studied, since Nlrp3 gene mutations have been linked to the autoinflammatory Muckle-Wells syndrome, familial cold auto-inflammatory syndrome, and neonatal-onset multisystem inflammation [17–19]. Most of these Nlrp3 genetic mutations produce constitutively active NLRP3 proteins that promote IL-1β secretion to elicit inflammatory responses [13]. Inhibition of the IL-1β pathway using an IL-1 receptor antagonist, anakinra, successfully ameliorates the severity of these inflammation-induced diseases, corroborating the essential role of NLRP3-induced IL-1β their inflammatory pathologies [20, 21]. The NLRP3 inflammasome is composed of the NLRP3 protein, the adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC), and the cysteine protease caspase-1. Human, but not mouse, NLRP3 inflammasome complexes also contain the Cardinal protein, whose function remains unclear [22].
Similar to other inflammasomes, the NLRP3 inflammasome activates caspase-1 to cleave pro-IL-1β and pro-IL-18 into IL-1β and IL-18. NLRP3 inflammasome activation can also induce pyroptosis and modulate immune responses by regulating the secretion of more than 20 leaderless cytokines and growth factors, including HMGB1, IL-1α, fibroblast growth factor 2 (FGF2), galectin-1, and galectin-3 [23]. NLPR3 inflammasome activity thus plays critical roles in inflammation, host defense, and other related activities.
2.2.1 NLRP3 Inflammasome-Priming Signals
IL-1β, unlike most other cytokines, is regulated by both gene transcription and posttranslational modification. Pro-IL-1β expression in myeloid cells, such as macrophages and dendritic cells (DCs), is induced upon exposure to LPS, a bacterial-derived TLR4 ligand, which activates the NF-κB signaling pathway. Many NF-κB pathway activators, including TNF-α, CpG, Pam3CSK4, poly(I:C), R848, imiquimod, and IL-1β itself, have also been shown to increase pro-IL-1β expression [24–28]. However, while NF-κB-activating stimuli are required to induce pro-IL-1β expression in vitro, it is unclear whether such priming agents are also required to increase in vivo pro-IL-1β expression. For example, intraperitoneal injection with alum or monosodium urate (MSU) crystals triggers IL-1β secretion without an exogenous priming step to induce NF-κB activation [29, 30]. Differences between in vitro and in vivo inflammasome responses could be explained, at least in part, by differences in the sensitivity of the responding cell types and their microenvironment.
Most NF-κB-dependent “priming” signals induce pro-IL-1β and increase NLRP3 protein levels, an essential step for inflammasome formation [24]. NLRP3 expression in bone marrow-derived dendritic cells (BMDCs) and macrophages is markedly increased upon TLR stimulation [24, 28], and signaling through the TLR adaptor proteins MyD88 and TRIF increases NLRP3 expression [24, 31]. In contrast, the inflammasome complex proteins caspase-1 and ASC are widely expressed in most cell types and are not further upregulated upon NF-κB activation.
2.2.2 The Assembly of the NLRP3 Inflammasome
NLRP3 is highly expressed in primary mouse neutrophils, peripheral blood mononuclear cells (PBMCs), and BMDCs and moderately expressed in established Th2 and macrophage cell lines [28, 32]. NLRP3 protein is sequestered in an auto-inhibitory conformation by heat shock protein 90 (HSP90) and suppressor of G2 allele of SKP1 (SGT1) in resting cells [33], but NLRP3 can recognize stimulatory signals through its LRR domain and undergo a conformational change, leading to ATP-dependent self-oligomerization via its NOD domain. NLRP3 oligomers then recruit the adaptor protein ASC via PYD-PYD domain interactions, which then recruits caspases-1 via CARD-CARD domain interactions to generate the final ~700 kDa NLRP3 inflammasome complex [34, 35]. This multi-protein complex assembly activates caspase-1, which cleaves pro-IL-1β and pro-IL-18 to generate mature IL-1β and IL-18 cytokines for secretion.
It remains unclear how NLRP3 recognizes the diverse range of stimuli that can activate NLRP3 inflammasome assembly and activation. It is plausible that NLRP3 is not a direct sensor of all the external stimuli and cellular “danger signals” it recognizes. Recent results have identified the non-NLR/ALR protein guanylate-binding protein 5 (GBP5) as a sensor for some NLRP3 inflammasome activators, stimulating NLRP3 inflammasome activation in response to pathogenic bacteria and soluble, but not crystalline, inflammasome-priming agents [36]. Interestingly, GBP5-deficient mice demonstrate impairments in both host defense and NLRP3-dependent inflammatory responses, indicating that proteins other than NLRP3, ASC, and caspase-1 are involved in sensing pathogenic “danger signals” that activate the NLRP3 inflammasome. Moreover, the restricted range of NLRP3 inflammasome stimuli recognized by GBP5 suggests that more than one sensor is involved in this process. These results, however, do not fully preclude the possibility of different sensor proteins giving rise to a common signaling intermediate that directly regulates the NLRP3 inflammasome complex. Given the ability of the NLRP3 inflammasome to regulate critical innate and adaptive immune responses to induce both protective and pathogenic responses, future studies are required to characterize signaling small molecules and proteins that regulate NLRP3 inflammasome activation.
2.3 The Diverse Regulation of NLRP3 Inflammasome Activation
Notably, the NLRP3 inflammasome is responsive to a wide array of stimuli, ranging from microorganisms of both endogenous and exogenous origin to inorganic agents such as asbestos, silica, and alum (Table 2.1). Specific activators have been identified for the NLRP1, NLRC4, and AIM2 inflammasomes, but much less is known about what triggers the assembly and activation of the NLRP3 inflammasome [37]. Studies indicate that reactive oxygen species (ROS), potassium efflux, and lysosomal damage are involved in NLRP3 inflammasome activation. Indeed, regulation of NLRP3 inflammasome activity by many stimuli can be explained by these three mechanisms; however, no single mechanism accounts for inflammasome activation by all known stimuli. The following sections summarize and discuss these models and the potential metabolic signals that can induce NLRP3 inflammasome responses.
2.3.1 Reactive Oxygen Species
Many NLRP3 agonists induce ROS formation, and reactive oxygen compounds such as hydrogen peroxide can induce inflammasome formation [38]. NADPH oxidase was initially postulated as the cellular ROS source that activated the NLRP3 inflammasome [39–41], since many factors that induce inflammasome activity also induce NADPH oxidase. However, studies performed with cells lacking various NADPH oxidase components indicate that NADPH oxidase activity is not required for NLRP3 inflammasome activation [42–44].
More recent studies have implicated mitochondrial ROS in inflammasome activation. Mitochondria are the main source of cellular ROS and markedly increase mitochondrial ROS production under various stress conditions [45]. Mitochondria also produce ROS during normal respiration, which can eventually alter respiratory chain function and lead to increased mitochondrial ROS production [46]. Such damaged mitochondria are continuously removed by a specialized form of autophagy, known as mitophagy, to avoid cellular damage from excess ROS exposure. Chemical and genetic manipulations that increase mitochondrial dysfunction and ROS production activate NLRP3 inflammasomes [47, 48], as does pharmacologic or genetic inhibition of mitophagy leading to the accumulation of damaged, ROS-producing mitochondria [46, 47, 49]. Both NLRP3 and ASC relocate to the perinuclear space upon inflammasome activation to colocalize with endoplasmic reticulum mitochondria, further implying a role for mitochondrial ROS in inflammasome activation [48].
However, while ROS have been repeatedly implicated in NLRP3 inflammasome activation, it is still not clear precisely how ROS alter the inflammasome complex to activate capase-1 activity. ROS are toxic, but can also serve as signals for various physiologic processes. One recent study found that thioredoxin-interacting protein (TXNIP), a binding partner of the antioxidant protein thioredoxin, can directly bind NLRP3 and regulate inflammasome activation in a ROS-inducible manner [50], and similar results were found in a second study, albeit with minor differences [51]. It is possible that NLRP3 is influenced directly by ROS, although there is currently no experimental evidence to support such a mechanism.
2.3.2 Cytoplasmic Potassium
Intracellular potassium is a common initiator of inflammasome activity, as inhibition of potassium efflux by hyperosmotic potassium can block NLRP3 inflammasome activation [39, 52, 53]. Cellular potassium concentration can serve as an indicator of membrane integrity, due to its high cellular and low extracellular levels. NLRP3 inflammasomes are activated by disruption of cell membrane integrity, but extracellular potassium as low as 20 mM is sufficient to block NLRP3 inflammasome formation [54]. NLRP3 inflammasome activity is thus likely regulated at least in part by potassium efflux during loss of membrane integrity, but many stimuli markedly induce inflammasome activity without significantly destabilizing the cell membrane. Inflammasome responses to several such stimuli, including PAMPs, DAMPs, and crystals, can be markedly attenuated by the potassium channel inhibitor glibenclamide [55], however, suggesting that potassium channels mediate many of these responses by increasing intracellular potassium efflux.
Changes in cell volume in response to low environmental osmolarity can also regulate NLRP3 inflammasome activation [56], at least in part by decreasing intracellular potassium concentrations. However, low intracellular potassium is required but not sufficient to induce NLRP3 inflammasome activation during cell swelling. Cell swelling was also found to alter transient receptor potential (TRP) channel activity to regulate an intracellular calcium increase that was found to stimulate transforming growth factor β-activated kinase 1 (TAK1) to activate the NLRP3 inflammasome.
2.3.3 Phagocytosis and Lysosomal Damage
Phagocytosis of crystalline or particulate structures, such as MSU, silica, asbestos, amyloid-β, and alum, can lead to the release of proteolytic lysosomal contents into the cytosol, resulting in NLRP3 inflammasome activation [42, 57]. Leakage of the lysosomal cysteine protease cathepsin B into the cytoplasm has been proposed to result in the cleavage of a putative factor regulating NLRP3 complex activity, since specific inhibition or genetic ablation of cathepsin B activity impairs inflammasome activation induced by phagocytosis of various particulates [42, 58, 59]. However, either cathepsin B or cathepsin L deficiency attenuated NLRP3 inflammasome activation in one of these studies, and repression was less pronounced at higher doses [58], implying functional redundancy or alternative protease activities. Further, not all studies found cathepsin B deficiency attenuated NLRP3 inflammasome activation by crystalline agonists [60].
2.3.4 The NLRP3 Inflammasome is Negatively Regulated by IFNγ and T Cell Responses
Emerging evidence indicates that cytokine signaling events can inhibit inflammasome activation and IL-1β production [15]. For example, IFNγ transiently inhibits LPS-stimulated IL-1β production in mouse macrophages and dendritic cells, although not in human cells [61], but is limited by LPS-induced SOCS1 expression, a negative regulator of the IFNγ signaling cascade. Endogenous IFNγ production in M. tuberculosis-infected mice also selectively inhibits IL-1α and IL-1β production by monocyte-macrophages and DCs [15]. IFNγ is a signature cytokine of CD4+ Th1 and CD8+ T cells [62, 63], suggesting that T cell IFNγ secretion during adaptive immune responses may act to attenuate inflammasome responses. Similarly, both effector and memory T cells can directly attenuate NLRP3 and NLRP1 inflammasome activity in macrophages and DCs through a cell-to-cell contact-dependent mechanism apparently mediated by TNF superfamily ligand-receptor interactions [16].
2.4 NLRP3 Inflammasome Is a Metabolic Regulator
Excess weight gain is associated with the dysregulation of multiple metabolic factors that increase inflammasome activation.
2.4.1 Fatty Acids
Elevated plasma free fatty acids (FFA) are strongly associated with insulin resistance and type 2 diabetes [64]. Saturated FFAs can activate toll-like receptor 4 (TLR4) signaling to induce proinflammatory responses in adipocytes, macrophages, and pancreatic β-cells to increase insulin resistance in target tissues and reduce pancreatic β-cell function [65, 66]. TLR4-NFκB pathway signaling induces transcription of multiple proinflammatory factors, including IL-1β [67]. Recent work indicates that the saturated FFA palmitate can induce NLRP3-inflammasome activity by suppressing AMP-activated protein kinase activity, resulting in decreased autophagy, accumulation of damaged mitochondria, and increased production of mitochondrial-derived ROS [68]. Thus high FFA levels can provide both the transcription and inflammasome activation signals required for IL-1β production.
Cellular palmitate uptake also induces ceramide accumulation, which can function as a second messenger in several key signaling pathways [69]. Plasma ceramides are increased in obese patients with type 2 diabetes and positively correlate with insulin resistance, implying a potential role for ceramide in obesity-induced insulin resistance [70]. Ceramide has been shown to potently activate NLRP3 inflammasomes in isolated macrophages and adipose tissue explants [71], suggesting a new mechanism for ceramide-mediated insulin resistance in target tissues.
2.4.2 Hyperglycemia
Hyperglycemia induces IL-1β production in multiple cell types, including endothelial cells, monocytes, and pancreatic islet β-cells [72–74], and has been shown to activate the PKCα-NFκB signaling pathway to stimulate IL-1β gene transcription [75]. More recent work suggests that TXNIP plays an important role in hyperglycemia-induced pancreatic β-cells IL-1β secretion by directly interacting with the NLRP3-inflammasome in a ROS-dependent manner [50]. TXNIP expression is consistently higher in β-cells of subjects with type 2 diabetes [76], and its expression is robustly induced in response to high glucose [77]. Similar results are found in human adipocytes, where high glucose markedly induces TXNIP expression, caspase-1 activation, and IL-1β secretion [78], and TXNIP knockdown reduces hyperglycemia-induced IL-1β production, albeit primarily through IL-1β transcription [78]. Thus, both FFA and hyperglycemia appear to act through the NF-κB signaling pathway to stimulate IL-1β gene transcription, but use alternate mechanisms to provide the second signal required for inflammasome activation, IL-1β processing, and release.
2.4.3 Uric Acid and MSU Crystals
Multiple studies have indicated that uric acid or MSU crystals can stimulate inflammasome activity. For example, mice intraperitoneally injected with MSU crystals demonstrate NLRP3 inflammasome activation and develop IL-1β-dependent peritonitis [79]. MSU crystals are frequently found in patients with gout, but uric acid has also been implicated in inflammasome activation in other acute and chronic proinflammatory disease conditions. For example, during bleomycin-induced acute lung injury leading to pulmonary inflammation and fibrosis, uric acid released from injured cells can activate the NLRP3 inflammasome to induce IL-1β production [80]. Both adipose tissue uric acid levels [81] and inflammasome activity [82] are elevated in obesity, suggesting that uric acid may also promote NLRP3 inflammasome activation and IL-1β production in adipocytes, although there is no direct evidence for such a mechanism. Similarly, since plasma uric acid concentrations were one of the original diagnostic criteria for metabolic syndrome, it is tempting to speculate on the role of uric acid on systemic complications of obesity. Several recent studies have indicated that elevated plasma uric acid confers increased risk for multiple disease states associated with metabolic syndrome, including obesity, insulin resistance, hypertension, and cardiovascular disease [83].
2.4.4 Cholesterol
Plasma cholesterol levels are strongly linked to atherosclerosis, and cholesterol crystals are a recognized hallmark of advanced atherosclerotic lesions [84]. Cholesterol crystals can induce robust, dose-responsive, and caspase-1-dependent macrophage IL-1β secretion by inducing lysosomal damage [58]. Bone marrow NLRP3, ASC, or IL-1α/β deficiency markedly decreases atherosclerosis and inflammasome-dependent IL-18 levels, suggesting that NLRP3 inflammasomes play major roles in atherosclerosis [58]. Recently, autophagosomes were identified in atherosclerotic plaques, but autophagy was found to be defective [85]. Complete deficiency of autophagy in the apolipoprotein E-deficient mouse led to accelerated atherosclerosis with increased IL-1β levels and ASC protein bodies indicative of inflammasome complex formation and activation. These results are consistent with observations suggesting that lysosomal cholesterol crystals alter autophagic processes [58, 86]. These changes, in addition to increased vascular ROS, contributed to inflammasome activation.
2.4.5 Amyloid
Islet amyloid polypeptide (IAPP), which is co-secreted with insulin, can form amyloid deposits in the pancreas during type 2 diabetes. IAPP aggregates have cytotoxic properties that are believed to play a key role in β-cell loss and type 2 diabetes progression [87]. Recent work now indicates that IAPP crystals can trigger pancreatic NLRP3 inflammasome activation and IL-1β secretion [51].
2.5 Obesity and the NLRP3 Inflammasome
Chronic inflammation and recruitment of macrophages has been recognized as a hallmark of obesity for decades. Recent work indicates that innate and adaptive immune processes in adipose tissue play central roles in the development of obesity-induced inflammatory responses and indicates potential roles for NLRP3 inflammasome activity in these processes.
2.5.1 Chronic Inflammation in Adipose Tissue
Adipose tissue is an important endocrine organ, which can undergo marked changes in both its cellular composition and secretory profile during the onset of obesity. Caloric excess alters adipocyte function and dramatically increases the relative abundance and proinflammatory phenotype of adipose-resident immune cells, resulting in increased serum levels of proinflammatory adipokines [88, 89], while reductions in fat mass strongly correlate with serum adipokine decreases [90], suggesting that adipose tissue plays a central role in the systemic proinflammatory milieu of obesity. Adipocytes are important endocrine cells in this process, and adipocyte secretion of proinflammatory or pathogenic adipokines, such as TNFα, leptin, plasminogen activator inhibitor-1, monocyte chemotactic protein-1, IL-6, resistin, angiotensinogen, and IL-1β, markedly increases with obesity [88, 89]. Obesity also increases the relative adipose abundance of proinflammatory CD4 T cells (Th1) and CD8 T cells, resulting in increased adipose expression of IFNγ [91, 92]. The polarization of adipose tissue macrophages (ATMs) from an anti-inflammatory (M2) to a proinflammatory (M1) phenotype likely results from this increase in adipose IFNγ [93]. However, other immune cells, including B cells [94], mast cells [95], eosinophils [96], and neutrophils [97], have also been reported to regulate obesity-induced inflammation. The trigger signals for immune cell accumulation and polarization are unclear, although adipocyte death, hypoxia, and adipokines secreted in response to metabolic overload may all direct inflammatory cells to adipose tissue [98].
NLRP3 inflammasome components are highly expressed in ATMs, whose abundance can dramatically increase with the onset of obesity and contribute to the metabolic dysregulation and insulin resistance that give rise to obesity-associated complications. IL-1β directly inhibits adipocyte insulin signaling by decreasing insulin receptor substrate-1 [99], and both IL-1β and IL-18 can induce adipose-resident Th1 and CD8+ T cells [71, 100] to further increase adipose tissue inflammation. NLRP3 inflammasome activation in preadipocytes inhibits adipocyte differentiation and fat accumulation by reducing preadipocyte expression of PPARγ, a key regulator of adipogenesis, while caspase-1-deficient adipocytes are more metabolically active, suggesting that obesity-induced changes in NLRP3 inflammasome activity can negatively impact adipocyte function [82]. High-fat diet (HFD)-fed IL-1R knockout mice have less adipose inflammation and better insulin sensitivity than wild-type control mice [101]. Similarly, HFD-fed Nlrp3−/−, Asc−/−, and Casp1−/− mice demonstrate decreased weight gain and body fat, reduced inflammation, and improved insulin sensitivity [71, 82, 102], although both IL-1R and IL-18 deficiencies were subsequently found to induce hyperphagia resulting in delayed-onset obesity and insulin resistance in chow-fed mice [103–105].
2.5.2 NLRP3 Inflammasome Expression and Activity in Obese Mice
Several studies have investigated IL-1β/IL-18 production and inflammasome activation in mouse models of diet-induced obesity. Visceral adipose IL-1β and Nlrp3 mRNA expression is reported to positively correlate with body weight and adiposity in mice fed with standard chow diet and to markedly decrease upon caloric restriction [71], while adipose tissue IL-1β protein is increased in obese db/db and HFD-fed C57BL/6 mice relative to their lean controls [71, 82]. Similar to IL-1β, adipose tissue and circulating IL-18 protein levels are increased in HFD-fed or genetically obese mice [71, 82, 106], despite no IL-18 mRNA differences, suggestive of increased adipose inflammasome activity. Caspase-1 mRNA and activity are increased in multiple tissues of HFD-fed and genetically obese mice [71, 82]. NLRP3 ablation attenuates obesity-induced caspase-1 activity in adipose tissue and liver, but not kidney [71], implying that kidney caspase-1 activation is either NLRP3 independent or complemented by another inflammasome. NLRP3 inflammasome-dependent IL-1β secretion by the pancreas also increases during obesity and likely mediates chronic obesity-induced pancreatic damage [107].
2.5.3 NLRP3 Inflammasome Expression and Activity in Obese Patients
Circulating IL-1β concentrations are low in healthy human subjects. Elevated circulating IL-1β levels have been reported in some studies of obese patients [108, 109], but it is questionable whether these concentrations, typically <100 pg/mL, can induce biologically meaningful systemic effects. IL-1β protein concentrations in metabolic tissues, such as adipose tissue and liver, however, are clearly increased in obese human subjects, at levels likely to produce pathological effects, and weight loss reduces adipose and liver IL-1β mRNA expression [71, 110]. IL-1β secretion from human omental fat explants is correlated with the donor’s body mass index [111], while LPS-stimulated IL-1β production from primary monocytes of obese alcoholics is correlated with body mass index, percent body fat, abdominal circumference, and total histologic score [112]. Similar to IL-1β, circulating IL-18 concentrations are higher in overweight subjects [113] and are decreased by weight loss [114]. However, unlike IL-1β, weight loss significantly decreases IL-18 mRNA expression in liver, but not in adipose tissue [110].
Few studies have investigated NLRP3 inflammasome genes in obese human subjects. In one study, adipose tissue caspase-1 mRNA expression increased in obese subjects, while NLRP3, IL-1β, and IL-18 mRNA expressions were not significantly different in normal-weight and obese subjects [115]. Results from weight loss studies are contradictory, with weight loss reducing NLRP3 expression in one study [71] but having no effect in another [110], although gender, ethnicity, and adiposity differences between these cohorts may explain this discrepancy.
No comparable data is available for the effect of weight gain and loss on caspase-1 activity in human adipose tissue. However, a recent analysis of paired subcutaneous and visceral adipose tissue biopsies from ten overweight subjects found that IL-1β and IL-18 production as well as caspase-1 activity was dramatically higher in visceral than subcutaneous adipose tissue [100]. Interestingly, caspase-1 activity levels were positively correlated with CD8+ T cell numbers present in both tissues. These findings are consistent with well-established results that visceral adipose tissue is more inflammatory than subcutaneous adipose tissue, and suggest a possible role for caspase-1 activity in the infiltration of CD8+ T cells into human adipose tissue. Further, our recent microarray analysis and RT-PCR analyses of high-purity human and mouse adipocyte fractions from subcutaneous and visceral adipose tissue indicate that the expression of NLR pathway genes, including NLRP3 and ASC, is significantly upregulated during obesity (unpublished data), suggesting that obesity may induce adipocyte inflammasome activity.
2.5.4 Mechanisms of NLRP3 Inflammasome Activation in Adipose Tissue
Adipose tissue TLR4 signaling plays an important role in obesity-associated insulin resistance [66]. In obesity, increased FFA levels stimulate adipose tissue TLR4 signaling by increasing adipose expression of fetuin A, an endogenous TLR4 ligand [116]. Since the proinflammatory TLR4-NFκB signaling cascade regulates both NRLP3 and IL-1β transcription, increased TLR4 signaling may provide the priming signal for the increased NLRP3 inflammasome activity observed in obesity. However, while TLR4 signaling may prime the system, it is unclear what triggers NLRP3 inflammasome activation during obesity. Extracellular ATP can active the NLRP3 inflammasome via a P2X purinoceptor 7 (P2RX7)-dependent mechanism [117], but inflammasome activation and adipose and metabolic phenotypes are similar in HFD-fed P2RX7-deficient and wild-type mice [118], excluding it from a role in this process. Obesity-associated metabolic danger signals, such as elevated FFA and ceramide, hyperglycemia, and mitochondrial dysfunction, may trigger NLRP3 inflammasome activation, as discussed above. Due to the central role adipose inflammation plays in obesity-related complications, identification of the responsible signal(s) regulating adipose tissue inflammasome activity is an important challenge for future investigation.
2.6 The NLRP3 Inflammasome in Metabolic Diseases Associated with Obesity
The prevalence of obesity has led to the increased incidence of obesity-associated metabolic diseases and become a serious threat to public health in the developed world. Overweight and obesity are associated with increased risk of atherosclerosis, type 2 diabetes, nonalcoholic fatty liver disease (NAFLD), neurodegenerative disease, and cancer [119]. Chronic inflammation, a key feature of obesity, plays an important role in the development of many metabolic diseases. As discussed above, the NLRP3 inflammasome has been recognized to mediate inflammatory reactions to several metabolic danger signals that are increased in obesity, implying that these stimuli induce NLRP3-driven inflammation to produce tissue injury in obesity. Indeed, the major effectors of inflammasome activation, IL-1β and IL-18, have been linked to inflammatory responses in various metabolic diseases [9, 120]. However, new research has just begun to explain the specific stimuli and molecular mechanisms acting on the NLRP3 inflammasome for individual metabolic diseases.
2.6.1 Type 1 and Type 2 Diabetes
Diabetes is characterized by uncontrolled blood glucose elevations resulting from inadequate insulin production due to pancreatic β-cell loss (type 1 diabetes) or insulin resistance in skeletal muscle and other metabolic tissues associated that is with a slower decline in pancreatic β-cell function (type 2 diabetes). Mounting evidence now indicates that inflammasome activity plays critical roles in the dysregulation of both insulin production and insulin signaling.
IL-1β production by both pancreatic β-cells and infiltrating immune cells is known to regulate β-cell viability and insulin secretion. IL-1β treatment of both rodent and human pancreatic islets potently inhibits pancreatic β-cell-specific transcription factors and insulin secretion [121], while IL-1β secretion from invading immune cells induces pancreatic β-cell death during the development of autoimmune type 1 diabetes [122], partially through increased β-cell iNOS expression and NO production [123, 124]. Chronic hyperglycemia can also stimulate pancreatic β-cells to produce IL-1β, further impairing their viability and insulin secretion [73], through ROS-mediated activation of the NLRP3 inflammasome [50]. Finally, during type 2 diabetes high insulin secretion can result in the formation of cytotoxic IAPP aggregates that induce NLRP3 inflammasome activation and IL-1β secretion to stimulate β-cell apoptosis [51].
2.6.2 Metabolic Liver Disease
NAFLD is strongly associated with abdominal adiposity, and proinflammatory liver gene expression increases with adiposity [125]. NAFLD affects more than one-third of the Western world population, but only about 25 % of cases develop nonalcoholic steatohepatitis (NASH), which is characterized by chronic inflammation. NASH is a major cause of cirrhosis and liver transplantation, but the mechanism(s) underlying NAFLD progression to NASH remains elusive. Liver expression of NLRP3 inflammasome genes, caspase-1 activity, and mature IL-1β is, however, dramatically increased in mouse models of NASH [126]. NAFLD, the precursor to NASH, is also characterized by a marked increase in hepatocyte lipid accumulation (steatosis), which is attenuated by genetic ablation of NLRP3 inflammasome components [71, 82]. Saturated fatty acid accumulation in NAFLD may play an important role in hepatic inflammation, since the saturated fatty acid palmitate can induce inflammasome activation in cultured hepatocytes [126]. Hepatic inflammasome activation appears to require TLR2 signaling, which is induced by saturated fatty acids, since hepatic caspase-1 activation and serum IL-1β levels are suppressed in TLR2−/− mice fed with a NASH-inducing choline-deficient diet [127]. Finally, a gene regulating the elongation of C12- to C16-length saturated and monounsaturated fatty acids, Elovl6, has also been reported to regulate NASH development, hepatic NLRP3 inflammasome activation, and IL-1β release [128]. Taken together these results suggest that fatty acid accumulation directly or indirectly stimulates NLRP3 inflammasome activity. Genetic or pharmaceutical blockade of IL-1R signaling also attenuates NASH progression in mice [129, 130]. However, NAFLD/NASH phenotypes in these studies could result from non-hepatic inflammasome effects on insulin resistance and inflammation. Liver-specific NLRP3 inflammasome knockouts are required to address this argument.
2.6.3 Atherosclerosis
Chronic inflammation is a well-known contributor to atherosclerosis. Macrophage phagocytosis of modified low-density lipoprotein cholesterol present in the vasculature results in the accumulation of cholesterol-laden foam cells, increased oxidative stress, and vascular inflammation, which can ultimately lead to plaque destabilization and thrombosis. Moreover, IL-1β has been shown to participate in both the development and destabilization of atherosclerotic lesions. For example, genetic ablation of IL-1β or the IL-1 receptor in apolipoprotein E-deficient (Apoe−/−) mice, a standard mouse model of atherosclerosis, significantly decreased atherosclerosis development [131, 132], as did treatment with an IL-1 receptor antagonist [133, 134]. Results from low-density lipoprotein receptor-deficient (Ldlr−/−) mice fed with a high-cholesterol diet also indicated that macrophage-derived NLRP3 and IL-1 were essential for cholesterol-driven atherosclerosis, since these mice revealed significantly less atherosclerosis after receiving bone marrow transplants from NLRP3- or IL-1α/IL-1β-deficient mice [58]. Surprisingly, however, NLRP3 inflammasome activity was not required for normal atherosclerosis progression in Apoe−/− mice, since mice deficient in NLRP3, ASC, or caspase-1 expression revealed no decrease in atherosclerosis [135].
It is not clear why these results differ from those of Apoe−/− mice lacking IL-1β or IL-1 receptor expression or Ldlr−/− mice with NLRP3- or IL-1α/IL-1β-deficient bone marrow. One possible explanation, however, may be that Apoe−/− mice, unlike Ldlr−/− mice fed with a high-cholesterol diet, represent a nonobese mouse model of atherosclerosis and may therefore lack a metabolic stimulus found in obese mice. Furthermore, while this data indicates that inflammasome activity can play an important role in the pathogenesis of atherosclerosis, the agents that trigger NLRP3 inflammasome activation remain unclear. Duewell et al. have reported that cholesterol crystals, detected in early atherosclerotic lesions, induce NLRP3 inflammasome-dependent IL-1β production in murine macrophages [58], with similar results observed for human macrophages [86]. However, several other factors associated with increased atherosclerotic risk or the atherosclerotic microenvironment (hyperglycemia, ROS, uric acid) may also impact inflammasome activation.
2.6.4 Gout
Gout is the accumulation of MSU crystals in the joints, often leading to an inflammatory arthritis associated with elevated plasma uric acid. MSU crystals have long been identified as the causative agent of gout [136], but only recently has progress been made regarding the mechanism underlying their recognition as a proinflammatory danger signal. MSU crystals induce the production of inflammatory cytokines from monocytes and macrophages, particularly IL-1β [137–139], but do not induce IL-1β production in macrophages with ablations of various NRLP3 inflammasome components [79], indicating that inflammasome activity plays an essential role in MSU recognition. Elevated plasma uric acid was proposed as one of the original criteria for diagnosis of obesity-associated metabolic syndrome [140], and obese patients are more than twice as likely to develop gout [141] and develop it about 11 years earlier than their normal-weight counterparts [142].
2.6.5 Alzheimer’s Disease
Cerebral accumulation of amyloid-β plaques is one of the main pathologic features of Alzheimer’s disease (AD), the most common form of dementia, and this is believed to be a critical stimulus for many proinflammatory components of AD that induce neuronal death and memory loss. Although the role of IL-1β in the pathogenesis of AD is controversial, numerous studies suggest that IL-1β can induce neuronal death and recruit inflammatory cells into the central nervous system [143]. Amyloid fibrils have been reported to trigger IL-1β release from microglia and monocytes [144, 145], suggesting a potential role for inflammasome activity in AD pathology. In keeping with this hypothesis, microglial NLRP3 inflammasome activity and IL-1β secretion are activated by phagocytosis of amyloid-β protein [57], while microglia with genetic ablation of caspase-1 or inflammasome components revealed lack of attenuated or defective inflammatory responses to amyloid-β exposure. Taken together, this data suggests that the inflammasome may represent a novel therapeutic target for the treatment of AD. Small molecule caspase-1 inhibitors such as those currently in clinical trials (see below) appear to be the best candidates for AD therapeutics, due to the difficulty expected in delivering recombinant protein therapeutics across the blood–brain barrier.
2.7 Cancer, Obesity and Inflammation
Mounting evidence suggests that inflammation promotes cancer development. Inflammatory responses are associated with 15–20 % of all cancer deaths worldwide, and inflammatory cells and factors are present in the microenvironment of most tumors, where they are often associated with metastasis and poor prognosis [146]. Proinflammatory cytokines, including IL-1β, IL-6, and TNFα, implicated to link inflammation and increased cancer risk, are expressed by adipose tissue and demonstrate increased systemic expression in overweight and obese human subjects [90]. Body fat is convincingly associated with colorectal, kidney, esophageal, pancreatic, endometrial, and postmenopausal breast cancer, and a recent meta-analysis of prospective cancer studies has expanded this list to include thyroid cancer, leukemia, multiple myeloma, and non-Hodgkin lymphoma in both men and women and increased risk of malignant melanoma in men [147]. Body weight is also associated with increased cancer mortality, since a prospective analysis of more than 900,000 US adults proposed that overweight and obesity may account for 14 % and 20 % of cancer deaths in men and women ≥50 years of age, respectively [148]. Based on the growing worldwide obesity epidemic, weight gain may represent the largest avoidable cause of cancer in nonsmokers.
2.7.1 Cell Death and Inflammasome Activity
An important hallmark of cancer is the ability of cancer cells to escape immunosurveillance [149]. Cancer cells are often resistant to various types of programmed cell death including apoptosis, programmed necrosis, and mitotic catastrophe [150]. Similarly inflammasome-mediated cell death, or pyroptosis, also appears to be involved in oncogenesis, since dysregulation of pyroptosis is tightly associated with tumor progression [151, 152]. Pyroptosis is stimulated by caspase-1 activity and plays key roles in anti-pathogen inflammatory responses, since bacterially infected macrophages and dendritic cells are usually eliminated through pyroptosis [152]. Caspase-1 activity can be activated by several mechanisms during pyroptosis. First, caspase-1 activity can be induced by classical inflammasomes, which usually contain NLRP3 or the cytoplasmic DNA sensor protein AIM2 [153]. Second, ASC can be recruited to a single subcellular location and aggregate into a polymer called an “ASC focus,” “ASC speck,” or “pyroptosome” that can then recruit and activate caspase-1 [154–156]. Finally, the NLRC4 inflammasome can also activate caspase-1 via a nonclassical, ASC-independent mechanism [157].
In pyroptosis, activated caspase-1 catalyzes the proteolytic activation of caspase-7, but not caspase-3, caspase-8, or caspase-9, to initiate programmed cell death [158]. However, while pyroptosis shares certain features with classical programmed cell death mediated via apoptosis, recent studies have identified characteristics that distinguish pyroptosis from apoptosis. First, apoptotic and pyroptotic cells demonstrate distinct DNA damage patterns. Both apoptotic and pyroptotic DNA damage can be detected by TUNEL staining [159, 160], but pyroptotic cells have a distinct nuclear morphology and usually lack the DNA laddering pattern characteristic of apoptotic DNA damage [159, 161]. Second, these processes have differential effects on cell membrane composition and integrity. For example, apoptosis triggers phosphatidylserine translocation from the inner to the outer plasma membrane, resulting in a cell surface annexin V-staining pattern. By contrast, during pyroptosis the formation of plasma membrane pores allows annexin V to enter the cell to produce an inner cell membrane staining pattern [162–164]. Finally, apoptotic and pyroptotic cell debris demonstrate different fates. During apoptosis, dying cells are cleaved into spherical membrane-bound structures known as apoptotic bodies that are ultimately cleared by phagocytosis [165], while the cytosolic contents of pyroptotic cells are released into extracellular space [159].
Similar to apoptosis, pyroptosis is thought to contribute to cell-autonomous tumor suppression. In support of this hypothesis, caspase-1 is downregulated in most human prostrate cancers, and caspase-1 overexpression in cultured prostate cancer cells enhances their sensitivity to radiation-induced killing [166]. One study has also reported that NLRP3-deficient mice are more susceptible to colitis-associated colon cancer [167], while another has reported that NLRC4-deficient mice, but not NLRP3-deficient mice, have more tumor load than wild-type mice after exposure to azoxymethane-dextran sulfate sodium (AOM-DSS) [168]. Thus, while the function of specific inflammasomes in tumor suppression remains a matter of debate, these results appear to support the hypothesis that pyroptosis plays an important regulatory role in cancer development.
2.7.2 The Inflammasomes and Carcinogenesis
Chronic inflammation is often linked with increased cancer risk, and proinflammatory cytokines, such as IL-1β, TNF-α, and IL-6, are frequently associated with tumor progression [169]. However, the effect of inflammasomes on specific proinflammatory processes associated with increased cancer risk, such as colitis and colitis-associated cancer, remains controversial. For example, capase-1 or NLRP3 deficiency is reported to reduce colitis severity in DSS-treated mice [170, 171]. Consistent with these results, high levels of IL-1 in the tumor microenvironment usually correlate with a poor prognosis [172]. However, other authors have reported that mice with NLRP3, ASC, caspase-1, NLRP6, or NLRP12 deficiencies are more susceptible to DSS-induced colitis and death [167, 173–177]. In one case, an IL-18 deficit following AOM-DSS-induced intestinal damage in caspase-1 and ASC-deficient mice caused a systemic spread of commensal bacteria due to the inability of these mice to repair the mucosal barrier [174]. Colitis-associated cancer has also been reported to be significantly increased upon genetic deletion of components of the NLRP3 inflammasome (ASC, NLRP3, or caspase-1) [167, 168, 177]. These contradictory observations for NLRP3 inflammasome effects on AOM-DSS-induced colitis may be due to animal facility-dependent differences in gut microflora [178].
Despite contradictory reports on inflammasome effects on colitis, in vivo experiments suggest that IL-1β and IL-18 play important roles in promoting gastric, hepatic, and breast cancer progression [179–181]. Further, gastric-specific IL-1β expression in transgenic mice induces gastric tumorigenesis [182], while IL-1β-deficient mice have reduced and retarded subcutaneous tumor development in response to transdermal 3-methylcholanthrene [183], suggesting that local IL-1β-induced inflammation is strongly associated with carcinogenesis.
Based on this link, approaches that inhibit NLRP3, caspase-1, IL-1β, and IL-18 have been used to develop novel therapies to that have been assessed in a variety of experimental cancer systems. Small compounds targeting inflammasomes or NLRP3 have not been successful due to off-target effects [184–187]. However, reagents targeting IL-1β and IL-18, including monoclonal antibodies and recombinant derivatives [188, 189], have been more successful. For example, in patients with smoldering or indolent multiple myeloma, treatment with anakinra, a recombinant non-glycosylated form of the human IL-1 receptor antagonist (IL-1ra), was found to decrease myeloma proliferation rates and high-sensitivity C-reactive protein (hsCRP) levels, leading to a chronic disease state with improved progression-free survival [190]. IL-1 inhibitors have also been shown to reduce the side effects of anticancer therapy [191, 192]. Based on the ability of IL-1R antagonism to reduce tumor burden and metastasis and the relatively low risk of in vivo IL-1 inhibition, future preclinical and clinical trials are needed to examine the effect of IL-1 inhibition on cancer outcomes [193].
2.7.3 Inflammasome-Dependent Anticancer Responses
While the inflammasome has often been linked to innate immunity, chronic inflammation, and carcinogenesis, mounting evidence suggests that the inflammasome is also involved in anticancer responses. Inflammasomes are hypothesized to inhibit tumor progression through several mechanisms, including (1) triggering innate immune reactions against potentially carcinogenic microbiota; (2) inducing the pyroptotic demise of premalignant, infected cells; and (3) facilitating antitumor adaptive immune responses [11].
IL-1β plays an essential role in stimulating adaptive immune responses and facilitating anticancer immunosurveillance [194]. Anticancer chemotherapies primarily eliminate tumors by inducing immunogenic cell death [195]. The immunogenic signals secreted by dying tumor cells not only attract innate immune effector cells into the tumor bed but also stimulate P2RX7 receptor-mediated activation of the NLRP3 inflammasome to produce IL-1β [196]. IL-1β, in conjunction with IL-23, then induces IL-17 secretion by γδ T cells [197, 198] and the polarization of CD8+ αβ T cell responses toward increased IFN-γ secretion [199]. Mice lacking P2RX7, NLRP3, ASC, caspase-1, or IL-1R1 do not respond to chemotherapies that elicit immunogenic cell death signals in wild-type mice [196]. Similarly, blockade of IL-1β signaling significantly impairs the growth-inhibitory effects of tumor chemotherapies in a variety of mouse models [197, 200]. Finally, P2RX7-mediated activation of the NLRP3 inflammasome is also required for the efficacy of anticancer therapies in patients [196, 201–203]. Taken together, these results indicate that inflammasome activity contributes to antitumor adaptive immune responses induced during chemotherapy. The study of the precise molecular mechanisms through which inflammasomes modulate anticancer immune responses may therefore yield insight into better anticancer therapeutics.
2.8 Pharmaceutical Interventions Targeting the Inflammasome
Several treatments designed to attenuate complications of metabolic disorders have now been developed using approaches that target NLRP3 inflammasome activity. Most of these strategies have focused on the development of therapeutic agents to attenuate IL-1β activity, although other agents that directly target inflammasome function and caspase-1 activity are also under development and may provide alternative therapeutic approaches with distinct advantages and disadvantages. One major concern of all these approaches, however, is the potential for reduced tissue repair and immune surveillance, as a result of excessive suppression of NLRP3 inflammasome responses.
2.8.1 Therapeutics Targeting IL-1β Activity
IL-1β can reduce tissue insulin sensitivity and inhibit insulin production by pancreatic β-cell [204, 205], while antagonism of IL-1β signaling, by receptor blockade or cytokine neutralization approaches, has been shown to ameliorate several proinflammatory conditions, including type 2 diabetes. For example, short-term treatment of a small cohort of type 2 diabetic patients with anakinra, a recombinant non-glycosylated form of the human IL-1 receptor antagonist (IL-1ra) that is FDA approved for treatment of rheumatoid arthritis, was found to significantly improve glycemia and β-cell function, while decreasing the plasma level of two surrogate markers of systemic inflammation, hsCRP and IL-6 [206]. These effects occurred within 4 weeks of treatment and were sustained at 13 weeks of treatment. However, no significant improvements were observed in insulin sensitivity, insulin-regulated skeletal muscle gene expression, or serum adipokine levels, indicating anakinra treatment effects were due primarily to improvements in β-cell function rather than enhanced glucose disposal. Similar results were found in a second study performed with a cohort of insulin-resistant but nondiabetic obese human subjects, where anakinra also failed to improve insulin sensitivity [207]. Anakinra-mediated IL-1β antagonism does not, therefore, appear to significantly impact skeletal muscle insulin sensitivity, although the reason for this failure is unclear. Adverse events associated with anakinra usage in these studies were relatively mild, primarily consisting of transient injection-site reactions that most likely result from the solution used to dissolve the recombinant protein, although this is aggravated by a need for daily subcutaneous injections due to the short half-life (<1 h) of anakinra [208].
IL-1 neutralizing agents provide an alternate means to attenuate the deleterious effects of inflammasome activation. Rilonacept, a recombinant therapeutic agent used to neutralize free IL-1α and IL-1β, has been shown to significantly reduce pain scores and plasma hsCRP levels in a small study of ten patients with chronic gouty arthritis, refractive to standard treatment approaches, and to prevent acute gout flares during the initiation of urate-lowering therapy [209, 210]. Rilonacept, however, attenuates both IL-1α and IL-1β activity and thus cannot differentiate between inflammasome-mediated IL-1β effects and those of IL-1α. More recent approaches to specifically inhibit IL-1β bioactivity in the treatment of human inflammatory disorders have resulted in the production of new agents such as canakinumab, a fully human monoclonal antibody that neutralizes IL-1β bioactivity. Canakinumab can be administered intravenously or subcutaneously and is FDA approved for the treatment of cryopyrin-associated periodic syndromes (CAPS), including familial cold auto-inflammatory syndrome and Muckle-Wells syndrome [211], which are associated with mutations in NLRP3 [13]. Canakinumab is also now being used in clinical trials for the treatment of a number of inflammation-related disorders such as type 2 diabetes and its derived complications and chronic obstructive pulmonary disease, with the CANTOS trial (cardiovascular risk reduction in type 2 diabetes) representing the largest trial of any anti-cytokine drug to date [212].
In addition to these three FDA-approved drugs, a number of agents targeting the IL-1 pathway are under preclinical or clinical development. The majority of these agents are neutralizing antibodies, such as gevokizumab (XOMA-052) and LY2189102, that reduce IL-1β bioavailability [212], although IL-1 receptor blocking antibodies, such as MEDI-8968 and AMG-108 (aka MEDI-78998), are also under study as novel therapeutics for treatment of chronic inflammatory diseases. XOMA-052 has been granted orphan drug status by the FDA for the treatment of Behçet’s disease, a rare condition where chronic immune-mediated vascular inflammation can result in severe neurological, pulmonary, gastrointestinal, and cardiovascular complications [213]. XOMA-052, LY2189102, MEDI-8968, and AMG-108 are all now under investigation in phase II clinical trials: XOMA-052 and LY2189102 for the treatment of type 2 diabetes, MDI-8968 for the treatment of chronic obstructive pulmonary disease [212], and AMG-108 for the treatment of rheumatoid arthritis. Both MEDI-8968 and AMG-108 are fully human monoclonal antibodies that selectively bind IL-1R to inhibit the binding and subsequent signaling of both IL-1α and IL-1β, resulting in inhibition that is not restricted to inflammasome-mediated IL-1β responses. However, a clinical trial of AMG-108 effects on patients with rheumatoid arthritis or osteoarthritis of the knee found moderate disease improvement coupled with an excellent safety profile [214, 215], suggesting that an “off-target” suppression of IL-1α may not produce severe side effects. Finally, induction of endogenous antibodies by therapeutic vaccines has proven to be a safe and effective means of attenuating other disease conditions [216]. Recently, this approach has also been extended to cytokine-induced disease conditions, and a vaccine targeting IL-1β (CYT-013) is currently in phase I clinical trial in patients with type 2 diabetes [212].
2.8.2 Therapeutic Approaches Acting on the Inflammasome or Caspase-1
Inflammasomes play important roles in immune surveillance and tissue repair. Thus, approaches designed to directly target pathological inflammasome activity need to maintain a careful balance between attenuating deleterious inflammatory activity and maintaining necessary host defense and tissue repair responses. Relatively little is known about the exact mechanisms that regulate inflammasome assembly and caspase-1 activation, however, and these may differ according to the stimulus and NLR subtype of the inflammasome complex. Caspase-1 blockade thus appears to be a far more attractive target for treatment of inflammasome-related disorders due to the greater knowledge base available for caspase-1 inhibition.
Two small molecule caspase-1 inhibitors, VX-765 and VX-740 (pralnacasan), have been tested in clinical trials for the treatment of chronic plaque psoriasis, rheumatoid arthritis, and psoriasis [217]. VX-765 has also been tested on six patients with Muckle-Wells syndrome, resulting from mutations in NLRP3, where it was found to significantly reduce inflammatory markers, recurrent fevers, and arthritis [212]. The rheumatoid arthritis clinical trial using VX-740 was discontinued, however, due to liver abnormalities in animal toxicology studies [217]. No caspase-1 inhibitors are currently approved for clinical use, but other approved drugs with known anti-inflammatory activity may function in part by attenuating inflammasome complex activity. For example, treatment with glyburide, a sulfonylurea drug frequently used for the treatment of type 2 diabetes [51, 55], has been shown to attenuate inflammation under diabetic conditions by blocking IAPP-stimulated NLRP3 inflammasome activation and IL-1β secretion [51]. Glyburide has also been shown to prevent NLRP3 inflammasome activation in response to pathogen-associated and DAMP signals and biological crystals [55]. Glyburide inhibits ATP-sensitive potassium channels (KATP) in pancreatic β-cells to induce insulin secretion, but NLRP3 activation and glyburide-mediated inhibition were preserved in macrophages lacking KATP subunits and ATP-binding cassette transporter proteins, indicating that glyburide does not inhibit NLRP3 inflammasome activity through attenuating potassium efflux. Moreover, glyburide had no effect on NLRP3 ATPase activity, strongly suggesting that glyburide acts upstream of the NLRP3 inflammasome [55].
2.8.3 Other Therapeutic Targets to Attenuate Inflammasome Activity
Improved knowledge of the signaling networks involved in inflammasome activation has led to the discovery and testing of new therapeutic targets, such as the P2X purinoceptor 7 (P2RX7), an ATP receptor that regulates potassium efflux to induce inflammasome-mediated caspase-1 activation [117]. However, while several studies have now been performed with P2RX7 antagonists, none have shown significant action to reduce inflammation. AZD9056, an oral P2RX7 antagonist, has been evaluated in a phase IIa and subsequent phase IIb clinical trial for treatment of rheumatoid arthritis, but failed to demonstrate any significant efficacy [218]. The P2RX7 antagonist CE-224,535 also failed to reveal efficacy in patients with active rheumatoid arthritis and an inadequate response to methotrexate [219]. Finally, the P2RX7 modulator GSK1482160 was also recently investigated for single-dose safety, tolerability, pharmacokinetics, and pharmacodynamics, in healthy human subjects, but simulations lead to the conclusion that it was not possible to achieve the desired level of pharmacology, resulting in the termination of GSK1482160 development for chronic inflammatory pain [220].
2.9 Summary
Increasing evidence indicates multiple metabolic and cellular factors contribute to NLRP3 inflammasome activation, which plays a key role in adipose tissue inflammation and resultant obesity-associated tissue injury and metabolic derangement. These observations have widespread implications for a variety of diseases that are increased in obesity including diabetes, atherosclerosis, NASH, Alzheimer’s disease, and cancer. Better understanding of the roles of NLRP3, IL-1β and IL-18 in mediating specific tissue injuries coupled with new therapeutics that target inflammasome activity may permit the development of novel and more precise interventions to prevent or treat these important disease conditions. Therapeutic control of the inflammasome is in our grasp.
References
Strowig T, Henao-Mejia J, Elinav E, Flavell R (2012) Inflammasomes in health and disease. Nature 481:278–286
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140:805–820
Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ et al (2010) NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 141:483–496
Meylan E, Tschopp J, Karin M (2006) Intracellular pattern recognition receptors in the host response. Nature 442:39–44
Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z et al (2008) NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 451:573–577
Xia X, Cui J, Wang HY, Zhu L, Matsueda S, Wang Q, Yang X, Hong J, Songyang Z, Chen ZJ et al (2011) NLRX1 negatively regulates TLR-induced NF-kappaB signaling by targeting TRAF6 and IKK. Immunity 34:843–853
Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C (2009) Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182:3173–3182
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6:1123–1132
Zitvogel L, Kepp O, Galluzzi L, Kroemer G (2012) Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol 13:343–351
Lamkanfi M, Dixit VM (2012) Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 28:137–161
Ting JP, Kastner DL, Hoffman HM (2006) CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev 6:183–195
Rathinam VA, Vanaja SK, Fitzgerald KA (2012) Regulation of inflammasome signaling. Nat Immunol 13:332–333
Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A (2011) Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35:1023–1034
Guarda G, Dostert C, Staehli F, Cabalzar K, Castillo R, Tardivel A, Schneider P, Tschopp J (2009) T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature 460:269–273
Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, Meyrier A, Watts RA, Scott DG, Nicholls A et al (2002) New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet 70:1498–1506
Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, de Saint Basile G (2002) Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 71:198–203
Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29:301–305
Hawkins PN, Lachmann HJ, McDermott MF (2003) Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. N Engl J Med 348:2583–2584
Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, Anderson JP, Wanderer AA, Firestein GS (2004) Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 364:1779–1785
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J (2004) NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20:319–325
Nickel W, Rabouille C (2009) Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol 10:148–155
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA et al (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183:787–791
Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, Nunez G (2007) Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of toll-like receptor signaling. Immunity 26:433–443
Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P et al (2006) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440:233–236
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228–232
Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galan JE, Askenase PW et al (2006) Critical role for NALP3/CIAS1/cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24:317–327
Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL (2006) MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 116:2262–2271
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P et al (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223
Guarda G, Zenger M, Yazdi AS, Schroder K, Ferrero I, Menu P, Tardivel A, Mattmann C, Tschopp J (2011) Differential expression of NLRP3 among hematopoietic cells. J Immunol 186:2529–2534
Anderson JP, Mueller JL, Rosengren S, Boyle DL, Schaner P, Cannon SB, Goodyear CS, Hoffman HM (2004) Structural, expression, and evolutionary analysis of mouse CIAS1. Gene 338:25–34
Mayor A, Martinon F, De Smedt T, Petrilli V, Tschopp J (2007) A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat Immunol 8:497–503
Jin C, Flavell RA (2010) Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol 30:628–631
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832
Shenoy AR, Wellington DA, Kumar P, Kassa H, Booth CJ, Cresswell P, MacMicking JD (2012) GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 336:481–485
Latz E (2010) The inflammasomes: mechanisms of activation and function. Curr Opin Immunol 22:28–33
Martinon F (2010) Signaling by ROS drives inflammasome activation. Eur J Immunol 40:616–619
Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J (2008) Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320:674–677
Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V et al (2009) Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459:433–436
Underhill DM, Rossnagle E, Lowell CA, Simmons RM (2005) Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 106:2543–2550
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E (2008) Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9:847–856
Meissner F, Molawi K, Zychlinsky A (2008) Superoxide dismutase 1 regulates caspase-1 and endotoxic shock. Nat Immunol 9:866–872
Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A (2010) Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 116:1570–1573
Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS (2004) Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol 287:C817–C833
Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B (2010) A reaction–diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol 6:e1000657
Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12:222–230
Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225
Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M et al (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456:264–268
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11:136–140
Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z et al (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol 11:897–904
Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS (2008) The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci U S A 105:9035–9040
Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA (2008) Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453:1122–1126
Franchi L, Kanneganti TD, Dubyak GR, Nunez G (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282:18810–18818
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM (2009) Glyburide inhibits the cryopyrin/Nalp3 inflammasome. J Cell Biol 187:61–70
Compan V, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Martinez CM, Angosto D, Montero MT, Herranz AS, Bazan E, Reimers D et al (2012) Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37:487–500
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9:857–865
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M et al (2010) NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464:1357–1361
Sharp FA, Ruane D, Claass B, Creagh E, Harris J, Malyala P, Singh M, O’Hagan DT, Petrilli V, Tschopp J et al (2009) Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Natl Acad Sci U S A 106:870–875
Dostert C, Guarda G, Romero JF, Menu P, Gross O, Tardivel A, Suva ML, Stehle JC, Kopf M, Stamenkovic I et al (2009) Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4:e6510
Masters SL, Mielke LA, Cornish AL, Sutton CE, O’Donnell J, Cengia LH, Roberts AW, Wicks IP, Mills KH, Croker BA (2010) Regulation of interleukin-1beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep 11:640–646
O’Rourke RW, White AE, Metcalf MD, Winters BR, Diggs BS, Zhu X, Marks DL (2012) Systemic inflammation and insulin sensitivity in obese IFN-gamma knockout mice. Metabolism 61:1152–1161
Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, Libby P (2008) Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res 103:467–476
Boden G, Shulman GI (2002) Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur J Clin Invest 32(suppl 3):14–23
Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K et al (2012) Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab 15:518–533
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015–3025
Haversen L, Danielsson KN, Fogelstrand L, Wiklund O (2009) Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis 202:382–393
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, Ting JP (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12:408–415
Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148:852–871
Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, Kirwan JP (2009) Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58:337–343
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17:179–188
Asakawa H, Miyagawa J, Hanafusa T, Kuwajima M, Matsuzawa Y (1997) High glucose and hyperosmolarity increase secretion of interleukin-1 beta in cultured human aortic endothelial cells. J Diabetes Complications 11:176–179
Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110:851–860
Shanmugam N, Reddy MA, Guha M, Natarajan R (2003) High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 52:1256–1264
Dasu MR, Devaraj S, Jialal I (2007) High glucose induces IL-1beta expression in human monocytes: mechanistic insights. Am J Physiol Endocrinol Metab 293:E337–E346
Parikh H, Carlsson E, Chutkow WA, Johansson LE, Storgaard H, Poulsen P, Saxena R, Ladd C, Schulze PC, Mazzini MJ et al (2007) TXNIP regulates peripheral glucose metabolism in humans. PLoS Med 4:e158
Stoltzman CA, Peterson CW, Breen KT, Muoio DM, Billin AN, Ayer DE (2008) Glucose sensing by MondoA:Mlx complexes: a role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc Natl Acad Sci U S A 105:6912–6917
Koenen TB, Stienstra R, van Tits LJ, de Graaf J, Stalenhoef AF, Joosten LA, Tack CJ, Netea MG (2011) Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes 60:517–524
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J (2006) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237–241
Gasse P, Riteau N, Charron S, Girre S, Fick L, Petrilli V, Tschopp J, Lagente V, Quesniaux VF, Ryffel B et al (2009) Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med 179:903–913
Cheung KJ, Tzameli I, Pissios P, Rovira I, Gavrilova O, Ohtsubo T, Chen Z, Finkel T, Flier JS, Friedman JM (2007) Xanthine oxidoreductase is a regulator of adipogenesis and PPARgamma activity. Cell Metab 5:115–128
Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A et al (2010) The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 12:593–605
Lyngdoh T, Marques-Vidal P, Paccaud F, Preisig M, Waeber G, Bochud M, Vollenweider P (2011) Elevated serum uric acid is associated with high circulating inflammatory cytokines in the population-based Colaus study. PLoS One 6:e19901
Small DM (1988) George Lyman Duff memorial lecture. Progression and regression of atherosclerotic lesions. Insights from lipid physical biochemistry. Arteriosclerosis 8:103–129
Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF (2012) Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15:534–544
Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK (2010) Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One 5:e11765
Westermark P, Andersson A, Westermark GT (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev 91:795–826
Clément K (2010) Adipokines, inflammation, and obesity. In: Leff TAG, James G (eds) Adipose tissue in health and disease. Wiley-VCH, Weinheim
Trayhurn P, Wood IS (2005) Signalling role of adipose tissue: adipokines and inflammation in obesity. Biochem Soc Trans 33:1078–1081
Lyon CJ, Law RE, Hsueh WA (2003) Minireview: adiposity, inflammation, and atherogenesis. Endocrinology 144:2195–2200
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S et al (2009) CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15:914–920
Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F et al (2009) Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15:921–929
Lumeng CN, Bodzin JL, Saltiel AR (2007) Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117:175–184
Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, Tsui H, Wu P, Davidson MG, Alonso MN et al (2011) B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17:610–617
Liu J, Divoux A, Sun J, Zhang J, Clement K, Glickman JN, Sukhova GK, Wolters PJ, Du J, Gorgun CZ et al (2009) Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med 15:940–945
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, Chawla A, Locksley RM (2011) Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332:243–247
Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y et al (2012) Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18(9):1407–1412
Lumeng CN, Maillard I, Saltiel AR (2009) T-ing up inflammation in fat. Nat Med 15:846–847
Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF (2007) Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 148:241–251
Koenen TB, Stienstra R, van Tits LJ, Joosten LA, van Velzen JF, Hijmans A, Pol JA, van der Vliet JA, Netea MG, Tack CJ et al (2011) The inflammasome and caspase-1 activation: a new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology 152:3769–3778
McGillicuddy FC, Harford KA, Reynolds CM, Oliver E, Claessens M, Mills KH, Roche HM (2011) Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes 60:1688–1698
Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, Neale GA, Hooiveld GJ, Hijmans A, Vroegrijk I et al (2011) Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A 108:15324–15329
Garcia MC, Wernstedt I, Berndtsson A, Enge M, Bell M, Hultgren O, Horn M, Ahren B, Enerback S, Ohlsson C et al (2006) Mature-onset obesity in interleukin-1 receptor I knockout mice. Diabetes 55:1205–1213
Netea MG, Joosten LA, Lewis E, Jensen DR, Voshol PJ, Kullberg BJ, Tack CJ, van Krieken H, Kim SH, Stalenhoef AF et al (2006) Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med 12:650–656
Zorrilla EP, Sanchez-Alavez M, Sugama S, Brennan M, Fernandez R, Bartfai T, Conti B (2007) Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc Natl Acad Sci U S A 104:11097–11102
Membrez M, Ammon-Zufferey C, Philippe D, Aprikian O, Monnard I, Mace K, Darimont C (2009) Interleukin-18 protein level is upregulated in adipose tissue of obese mice. Obesity (Silver Spring) 17:393–395
Youm YH, Adijiang A, Vandanmagsar B, Burk D, Ravussin A, Dixit VD (2011) Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 152:4039–4045
Aygun AD, Gungor S, Ustundag B, Gurgoze MK, Sen Y (2005) Proinflammatory cytokines and leptin are increased in serum of prepubertal obese children. Mediators Inflamm 2005:180–183
Garanty-Bogacka B, Syrenicz M, Syrenicz A, Gebala A, Lulka D, Walczak M (2005) Serum markers of inflammation and endothelial activation in children with obesity-related hypertension. Neuro Endocrinol Lett 26:242–246
Moschen AR, Molnar C, Enrich B, Geiger S, Ebenbichler CF, Tilg H (2011) Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol Med 17:840–845
Nov O, Kohl A, Lewis EC, Bashan N, Dvir I, Ben-Shlomo S, Fishman S, Wueest S, Konrad D, Rudich A (2010) Interleukin-1beta may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology 151:4247–4256
Bunout D, Munoz C, Lopez M, de la Maza MP, Schlesinger L, Hirsch S, Pettermann M (1996) Interleukin 1 and tumor necrosis factor in obese alcoholics compared with normal-weight patients. Am J Clin Nutr 63:373–376
Jung C, Gerdes N, Fritzenwanger M, Figulla HR (2009) Circulating levels of interleukin-1 family cytokines in overweight adolescents. Mediators Inflamm 2010:958403
Esposito K, Pontillo A, Di Palo C, Giugliano G, Masella M, Marfella R, Giugliano D (2003) Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA 289:1799–1804
Goossens GH, Blaak EE, Theunissen R, Duijvestijn AM, Clement K, Tervaert JW, Thewissen MM (2012) Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Mol Immunol 50:142–149
Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, Ray S, Majumdar SS, Bhattacharya S (2012) Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med 18(8):1279–1285
Di Virgilio F (2007) Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol Sci 28:465–472
Sun S, Xia S, Ji Y, Kersten S, Qi L (2012) The ATP-P2X7 signaling axis is dispensable for obesity-associated inflammasome activation in adipose tissue. Diabetes 61:1471–1478
Lumeng CN, Saltiel AR (2011) Inflammatory links between obesity and metabolic disease. J Clin Invest 121:2111–2117
Feve B, Bastard JP (2009) The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol 5:305–311
Dinarello CA, Donath MY, Mandrup-Poulsen T (2010) Role of IL-1beta in type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 17:314–321
Mandrup-Poulsen T (1996) The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia 39:1005–1029
Corbett JA, McDaniel ML (1992) Does nitric oxide mediate autoimmune destruction of beta-cells? Possible therapeutic interventions in IDDM. Diabetes 41:897–903
Corbett JA, McDaniel ML (1995) Intraislet release of interleukin 1 inhibits beta cell function by inducing beta cell expression of inducible nitric oxide synthase. J Exp Med 181:559–568
Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11:183–190
Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G (2011) Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54:133–144
Miura K, Yang L, van Rooijen N, Brenner DA, Ohnishi H, Seki E (2013) TLR2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation. Hepatology 57(2):577–589
Matsuzaka T, Atsumi A, Matsumori R, Nie T, Shinozaki H, Suzuki-Kemuriyama N, Kuba M, Nakagawa Y, Ishii K, Shimada M et al (2012) Elovl6 promotes nonalcoholic steatohepatitis in mice and humans. Hepatology 56(6):2199–2208
Isoda K, Sawada S, Ayaori M, Matsuki T, Horai R, Kagata Y, Miyazaki K, Kusuhara M, Okazaki M, Matsubara O et al (2005) Deficiency of interleukin-1 receptor antagonist deteriorates fatty liver and cholesterol metabolism in hypercholesterolemic mice. J Biol Chem 280:7002–7009
Miura K, Kodama Y, Inokuchi S, Schnabl B, Aoyama T, Ohnishi H, Olefsky JM, Brenner DA, Seki E (2010) Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 139(323–334):e327
Chi H, Messas E, Levine RA, Graves DT, Amar S (2004) Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apolipoprotein E heterozygote model: pharmacotherapeutic implications. Circulation 110:1678–1685
Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M (2003) Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 23:656–660
Elhage R, Maret A, Pieraggi MT, Thiers JC, Arnal JF, Bayard F (1998) Differential effects of interleukin-1 receptor antagonist and tumor necrosis factor binding protein on fatty-streak formation in apolipoprotein E-deficient mice. Circulation 97:242–244
Isoda K, Ohsuzu F (2006) The effect of interleukin-1 receptor antagonist on arteries and cholesterol metabolism. J Atheroscler Thromb 13:21–30
Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J (2011) Atherosclerosis in ApoE-deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis 2:e137
Shi Y, Mucsi AD, Ng G (2010) Monosodium urate crystals in inflammation and immunity. Immunol Rev 233:203–217
Di Giovine FS, Malawista SE, Nuki G, Duff GW (1987) Interleukin 1 (IL 1) as a mediator of crystal arthritis. Stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL1. J Immunol 138:3213–3218
Landis RC, Yagnik DR, Florey O, Philippidis P, Emons V, Mason JC, Haskard DO (2002) Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages. Arthritis Rheum 46:3026–3033
Yagnik DR, Hillyer P, Marshall D, Smythe CD, Krausz T, Haskard DO, Landis RC (2000) Noninflammatory phagocytosis of monosodium urate monohydrate crystals by mouse macrophages. Implications for the control of joint inflammation in gout. Arthritis Rheum 43:1779–1789
Reaven GM (1988) Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37:1595–1607
Juraschek SP, Miller ER III, Gelber AC (2013) Body mass index, obesity, and prevalent gout in the United States in 1988–1994 and 2007–2010: body mass index, obesity, and gout. Arthritis Care Res (Hoboken) 65(1):127–132
DeMarco MA, Maynard JW, Huizinga MM, Baer AN, Kottgen A, Gelber AC, Coresh J (2011) Obesity and younger age at gout onset in a community-based cohort. Arthritis Care Res (Hoboken) 63:1108–1114
Shaftel SS, Griffin WS, O’Banion MK (2008) The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation 5:7
Lorton D, Kocsis JM, King L, Madden K, Brunden KR (1996) Beta-amyloid induces increased release of interleukin-1 beta from lipopolysaccharide-activated human monocytes. J Neuroimmunol 67:21–29
Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR (2000) Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J Neurochem 74:1017–1025
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357:539–545
Renehan AG, Tyson M, Egger M, Heller RF, Zwahlen M (2008) Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 371:569–578
Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003) Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348:1625–1638
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S et al (2011) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19:107–120
Kepp O, Galluzzi L, Zitvogel L, Kroemer G (2010) Pyroptosis—a cell death modality of its kind? Eur J Immunol 40:627–630
Miao EA, Rajan JV, Aderem A (2011) Caspase-1-induced pyroptotic cell death. Immunol Rev 243:206–214
Gross O, Thomas CJ, Guarda G, Tschopp J (2011) The inflammasome: an integrated view. Immunol Rev 243:136–151
Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM (2010) Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med 207:1745–1755
Broz P, von Moltke J, Jones JW, Vance RE, Monack DM (2010) Differential requirement for caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8:471–483
Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J (1999) ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem 274:33835–33838
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430:213–218
Lamkanfi M, Kanneganti TD, Van Damme P, Vanden Berghe T, Vanoverberghe I, Vandekerckhove J, Vandenabeele P, Gevaert K, Nunez G (2008) Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol Cell Proteomics 7:2350–2363
Brennan MA, Cookson BT (2000) Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol 38:31–40
Chen LM, Kaniga K, Galan JE (1996) Salmonella spp. are cytotoxic for cultured macrophages. Mol Microbiol 21:1101–1115
Watson PR, Gautier AV, Paulin SM, Bland AP, Jones PW, Wallis TS (2000) Salmonella enterica serovars Typhimurium and Dublin can lyse macrophages by a mechanism distinct from apoptosis. Infect Immun 68:3744–3747
Fink SL, Cookson BT (2006) Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8:1812–1825
Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH (1994) Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84:1415–1420
Silveira TN, Zamboni DS (2010) Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect Immun 78:1403–1413
Majno G, Joris I (1995) Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146:3–15
Winter RN, Kramer A, Borkowski A, Kyprianou N (2001) Loss of caspase-1 and caspase-3 protein expression in human prostate cancer. Cancer Res 61:1227–1232
Allen IC, TeKippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, Herfarth HH, Jobin C, Ting JP (2010) The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207:1045–1056
Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, Eisenbarth SC, Flavell RA (2010) Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A 107:21635–21640
Karin M, Lawrence T, Nizet V (2006) Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124:823–835
Bauer C, Duewell P, Mayer C, Lehr HA, Fitzgerald KA, Dauer M, Tschopp J, Endres S, Latz E, Schnurr M (2010) Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59:1192–1199
Siegmund B, Lehr HA, Fantuzzi G, Dinarello CA (2001) IL-1 beta-converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A 98:13249–13254
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R (2004) Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118:229–241
Chen GY, Liu M, Wang F, Bertin J, Nunez G (2011) A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186:7187–7194
Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KS, McIntire CR, LeBlanc PM, Meunier C, Turbide C, Gros P, Beauchemin N et al (2010) Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32:367–378
Normand S, Delanoye-Crespin A, Bressenot A, Huot L, Grandjean T, Peyrin-Biroulet L, Lemoine Y, Hot D, Chamaillard M (2011) Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A 108:9601–9606
Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32:379–391
Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti TD (2010) IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol 185:4912–4920
Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI et al (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145:745–757
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899
Reed JR, Leon RP, Hall MK, Schwertfeger KL (2009) Interleukin-1beta and fibroblast growth factor receptor 1 cooperate to induce cyclooxygenase-2 during early mammary tumourigenesis. Breast Cancer Res 11:R21
Schafer M, Werner S (2008) Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol 9:628–638
Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG et al (2008) Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14:408–419
Krelin Y, Voronov E, Dotan S, Elkabets M, Reich E, Fogel M, Huszar M, Iwakura Y, Segal S, Dinarello CA et al (2007) Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res 67:1062–1071
Green DR, Kroemer G (2005) Pharmacological manipulation of cell death: clinical applications in sight? J Clin Invest 115:2610–2617
Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H (2010) Identification of a primary target of thalidomide teratogenicity. Science 327:1345–1350
Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, Meng R, Quong AA, Latz E, Scott CP et al (2010) Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem 285:9792–9802
Keller M, Sollberger G, Beer HD (2009) Thalidomide inhibits activation of caspase-1. J Immunol 183:5593–5599
Dinarello CA (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117:3720–3732
Srivastava S, Salim N, Robertson MJ (2010) Interleukin-18: biology and role in the immunotherapy of cancer. Curr Med Chem 17:3353–3357
Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ, Buadi FK et al (2009) Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc 84:114–122
Wu ZQ, Han XD, Wang Y, Yuan KL, Jin ZM, Di JZ, Yan J, Pan Y, Zhang P, Huang XY et al (2011) Interleukin-1 receptor antagonist reduced apoptosis and attenuated intestinal mucositis in a 5-fluorouracil chemotherapy model in mice. Cancer Chemother Pharmacol 68:87–96
Zhu J, Zhang J, Xiang D, Zhang Z, Zhang L, Wu M, Zhu S, Zhang R, Han W (2010) Recombinant human interleukin-1 receptor antagonist protects mice against acute doxorubicin-induced cardiotoxicity. Eur J Pharmacol 643:247–253
Dinarello CA (2010) Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev 29:317–329
Eisenbarth SC, Flavell RA (2009) Innate instruction of adaptive immunity revisited: the inflammasome. EMBO Mol Med 1:92–98
Zitvogel L, Kepp O, Kroemer G (2011) Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol 8:151–160
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E et al (2009) Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15:1170–1178
Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, Boucontet L, Apetoh L, Ghiringhelli F, Casares N et al (2011) Contribution of IL-17-producing gamma delta T cells to the efficacy of anticancer chemotherapy. J Exp Med 208:491–503
Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009) Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31:331–341
Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G et al (2011) Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 334:1573–1577
Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ (2011) Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 71:4809–4820
Kunzli BM, Bernlochner MI, Rath S, Kaser S, Csizmadia E, Enjyoji K, Cowan P, d’Apice A, Dwyer K, Rosenberg R et al (2011) Impact of CD39 and purinergic signalling on the growth and metastasis of colorectal cancer. Purinergic Signal 7:231–241
Sluyter R, Dalitz JG, Wiley JS (2004) P2X7 receptor polymorphism impairs extracellular adenosine 5′-triphosphate-induced interleukin-18 release from human monocytes. Genes Immun 5:588–591
Stagg J, Smyth MJ (2010) Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 29:5346–5358
Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, Svenson M (1986) Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science 232:1545–1547
Mandrup-Poulsen T, Bendtzen K, Nerup J, Dinarello CA, Svenson M, Nielsen JH (1986) Affinity-purified human interleukin I is cytotoxic to isolated islets of Langerhans. Diabetologia 29:63–67
Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356:1517–1526
van Asseldonk EJ, Stienstra R, Koenen TB, Joosten LA, Netea MG, Tack CJ (2011) Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab 96:2119–2126
Tack CJ, Stienstra R, Joosten LA, Netea MG (2012) Inflammation links excess fat to insulin resistance: the role of the interleukin-1 family. Immunol Rev 249:239–252
Schumacher HR Jr, Sundy JS, Terkeltaub R, Knapp HR, Mellis SJ, Stahl N, Yancopoulos GD, Soo Y, King-Davis S, Weinstein SP et al (2012) Rilonacept (interleukin-1 trap) in the prevention of acute gout flares during initiation of urate-lowering therapy: results of a phase II randomized, double-blind, placebo-controlled trial. Arthritis Rheum 64:876–884
Terkeltaub R, Sundy JS, Schumacher HR, Murphy F, Bookbinder S, Biedermann S, Wu R, Mellis S, Radin A (2009) The interleukin 1 inhibitor rilonacept in treatment of chronic gouty arthritis: results of a placebo-controlled, monosequence crossover, non-randomised, single-blind pilot study. Ann Rheum Dis 68:1613–1617
Dhimolea E (2010) Canakinumab. MAbs 2:3–13
Dinarello CA, Simon A, van der Meer JW (2012) Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov 11:633–652
Gul A, Tugal-Tutkun I, Dinarello CA, Reznikov L, Esen BA, Mirza A, Scannon P, Solinger A (2011) Interleukin-1beta-regulating antibody XOMA 052 (gevokizumab) in the treatment of acute exacerbations of resistant uveitis of Behcet’s disease: an open-label pilot study. Ann Rheum Dis 71:563–566
Cardiel MH, Tak PP, Bensen W, Burch FX, Forejtova S, Badurski JE, Kakkar T, Bevirt T, Ni L, McCroskery E et al (2010) A phase 2 randomized, double-blind study of AMG 108, a fully human monoclonal antibody to IL-1R, in patients with rheumatoid arthritis. Arthritis Res Ther 12:R192
Cohen SB, Proudman S, Kivitz AJ, Burch FX, Donohue JP, Burstein D, Sun YN, Banfield C, Vincent MS, Ni L et al (2011) A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res Ther 13:R125
Weiner LM, Surana R, Murray J (2010) Vaccine prevention of cancer: can endogenous antigens be targeted? Cancer Prev Res (Phila) 3:410–415
Leemans JC, Cassel SL, Sutterwala FS (2011) Sensing damage by the NLRP3 inflammasome. Immunol Rev 243:152–162
Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB (2012) Clinical evaluation of the efficacy of the P2X7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine. Ann Rheum Dis 71:1630–1635
Stock TC, Bloom BJ, Wei N, Ishaq S, Park W, Wang X, Gupta P, Mebus CA (2012) Efficacy and safety of CE-224,535, an antagonist of P2X7 receptor, in treatment of patients with rheumatoid arthritis inadequately controlled by methotrexate. J Rheumatol 39:720–727
Ali Z, Laurijssens B, Ostenfeld T, McHugh S, Stylianou A, Scott-Stevens P, Hosking L, Dewit O, Richardson JC, Chen C (2013) Pharmacokinetic and pharmacodynamic profiling of P2X7 receptor allosteric modulator GSK1482160 in healthy human subjects. Br J Clin Pharmacol 75(1):197–207
Martinon F, Tschopp J (2004) Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117:561–574
Willingham SB, Bergstralh DT, O’Connor W, Morrison AC, Taxman DJ, Duncan JA, Barnoy S, Venkatesan MM, Flavell RA, Deshmukh M et al (2007) Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2:147–159
Kanneganti TD, Body-Malapel M, Amer A, Park JH, Whitfield J, Franchi L, Taraporewala ZF, Miller D, Patton JT, Inohara N et al (2006) Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J Biol Chem 281:36560–36568
Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A (2009) Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med 206:79–87
Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J (2008) The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452:103–107
Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG (2006) Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126:1135–1145
Li H, Ambade A, Re F (2009) Cutting edge: necrosis activates the NLRP3 inflammasome. J Immunol 183:1528–1532
Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14:1583–1589
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Deng, T. et al. (2013). Inflammasomes and Obesity. In: Dannenberg, A., Berger, N. (eds) Obesity, Inflammation and Cancer. Energy Balance and Cancer, vol 7. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-6819-6_2
Download citation
DOI: https://doi.org/10.1007/978-1-4614-6819-6_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-6818-9
Online ISBN: 978-1-4614-6819-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)