
Abstract
The recent trend towards Bayesian and adaptive study designs has led to a growth in the field of pharmacokintetics and pharmacodynamics (PK/PD). The mathematical models used for PK/PD analysis can be extremely computationally intensive and particularly sensitive to messy data and anomalous values. The techniques of paneling and creating polygon summaries help to bring clarity to potentially messy graphics. An understanding of the expected shape of the data, combined with the right choice of graphic can help identify unusual patterns of data.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Pharmacokinetics and pharmacodynamics (PK/PD) describe the manner in which a compound enters and exits the body and the effect the compound has on the body using mathematical–statistical models. The often complex mathematical models used in PK/PD analysis provide quantitative descriptions of compounds, which are critical for dose finding and safety assessments during drug development. With fewer drugs making it to market and pipelines seemingly drying up, the focus has turned towards Bayesian and adaptive study designs and towards PK/PD analysis as a means of accelerating the early phases of clinical drug development.
Specifically a typical pharmacokinetic (PK) analysis involves modeling the concentration of an administered drug over time using structural models (often compartmental models) and exploring covariate relationships using statistical techniques. Pharmacodynamic (PD) analysis attempts to describe the relationship between the concentration of a compound and some measurable clinical response that represents an improvement in patient well-being. Like PK data, PD data is often temporal with the pharmacodynamic effect measured at discrete time points.
The primary aim of the PK/PD exploratory data analysis (EDA) process is to establish the quality of the data and to confirm that the intended structural model for the system is appropriate. Here we describe some general principles for graphing longitudinal PK/PD data and present some custom graphs applied to PK/PD data. The intent is not only to present specific types of plots but also to demonstrate general principles of creativity for efficient EDA. The techniques are of particular interest for data involving multiple measurements made for each individual over time.
2 Datasets and Graphics
Graphics have been drawn using a combination of simulated datasets that have been created using the MSToolkit package (http://r-forge.r-project.org/projects/mstoolkit/) and a popular simulated dataset distributed with the Xpose package (Jonsson and Karlsson 1999). The MSToolkit package is a suite of R functions developed specifically in order to simulate trial data and manage the storage of the simulated output. The MSToolkit package has been selected due to the ease in which PK/PD data including covariate interactions can be generated. The Xpose package data has been selected due to a commonly occurring challenge with PK/PD data. It consists of measurements over time for numerous individuals (subjects), which can make trends difficult to spot due to the large amount of information available on the same data range.
All plots are created with either the lattice package (Deepayan Sarkar 2009) or with the ggplot2 package (Hadley Wickham 2009) in R (R Development Core Team 2008).
3 Repeated Measures Data
Clinical trials are typically planned such that for each subject the primary endpoint(s) of interest (and perhaps a number of other key endpoints) is collected at baseline and then again at key points throughout the study. A pharmacodynamic analysis seeks to understand the effect that dose/concentration has on the endpoint over time. A PK analysis is similarly concerned with repeated measures over time; however, the aim of the PK analysis is to understand the change in concentration over time. In either case, such repeated measures data are not independent; there is correlation between the measurements of each individual, and the statistical techniques used must reflect this.
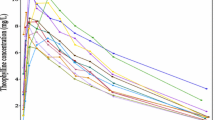
We focus first on PK data and start by plotting individual subject concentration-time profiles together in a single plot. Figure 6.1 shows the change over time in observed concentration for a number of simulated subjects. Usually this “spaghetti” plot serves only to confirm that our data is in range as any points not in range would skew the axes. For a large number of subjects the spaghetti plot can be difficult to read due to the number of intersecting lines on the plot and even if semi-transparent lines are used it is difficult to see how one subject compares with the rest of the population. As a solution to this we partition our plot into panels where each panel contains a single subject’s time profile. In Fig. 6.2 time profiles for the first four subjects in our dataset have been plotted in a 2 ×2 grid. This is sufficient to ascertain the shape of the profile but the free scaling of the axes provides little indication of scale or location. It is therefore difficult to compare subject profiles.

Observed concentration versus time after dose by subject

Observed concentration versus time after dose by subject
In Fig. 6.2, each subject profile is drawn a free scale which is automatically generated using data ranges applicable only to that particular subject. As a by-product of this, 2 profiles appearing to be almost identical at first glance, for example the profiles for subjects 2 and 3, may actually be very different when closer attention is paid to the axes. In fact the maximum concentration for subject 2 is approximately 8 times higher than the maximum concentration for subject 3. By default, most software packages fix the scaling such that the data ranges for the full population are used when constructing the panels. This example highlights the danger of overriding this default. It should be noted, however, that for non-homogeneous data, for example for data in which the subjects took different doses, fixing the scales can hide data characteristics such as the shape of the concentration-time curve for subjects taking a low dose.
In Fig. 6.2 only the first 4 subjects from our simulated pharmacokinetic dataset have been selected. It is natural to plot these four profiles in a 2 ×2 grid in order to fill the available space. For an EDA for which the primary intention is to establish the quality of the data by comparing an individual subject profile to the rest of the data, this approach usually suffices. However, when the primary aim is to make comparisons between panels the layout is far more important. For example, in Fig. 6.2 the primary variable of interest is concentration, which is shown on the y-axis. It is much easier to compare concentrations between two subjects when the y-axes are aligned. That is to say when the two plots are located side by side as opposed to vertically stacked. In order to make comparisons between panels, the panels should be stacked in the direction that best facilitates the comparison of interest.
For an exploratory analysis it is useful to compare individual profiles to the remaining population so that we can ascertain the relative location of the profile and identify outlying profiles more quickly. One technique that will enable such a comparison is to plot the overall data as a background “shadow.” An illustration of this technique is given in Fig. 6.3. As in Fig. 6.2, the first 4 subjects from our simulated data have been plotted in separate panels. In each panel the subject of interest has been drawn in a thick, black line, with the remaining subjects from the simulated dataset plotted in gray. For each subject we see instantly how their profile compares to the rest of the population. Note also that plotting the four panels side by side also now facilitates comparison between these four subjects.

Observed concentration versus time after dose by subject (other subjects shown in gray)
These techniques form the foundation for an exploratory repeated measures analysis. Later we see how they can be further extended in order to investigate dose and other covariates.
4 Data Quality
Many data quality issues are simple to spot, particularly incorrectly formatted values and data values that are significantly out of range. However, incorrect values that are still within the overall data range (such as values that have been transpose/swapped) are more difficult to identify. In this case, we must rely on the structural information we anticipate within the data in order to spot outlying values.
If an incorrectly coded value is still within the anticipated data range, a basic univariate analysis is not able to identify the issue. In this case, some errors may be impossible to spot without quality control (QC) steps against the data source. We can, however, use basic graphical methods to spot observations that do not seem to follow similar trends. These observations may be erroneous or may be highly informative.
In this section we concentrate on 2 key variables: the observed concentration variable to be modeled and “time after dose,” taken as the primary independent variable.
In the field of PK, we typically understand the ways in which the direction of trend of the concentration curve should change, based on the dosing mechanism employed. For example, after a drug is orally administered the concentration typically increases sharply to a maximum concentration, then smoothly decreases as the drug is metabolized and eliminated.
As such, we can often spot erroneous data in a dependent versus independent relationship by simply analyzing the sign of the gradient at each step within a graph of observed concentration versus time after dose. The gradient is the slope of the line connecting two observations, so a switch in sign indicates a switch in the direction.
Figure 6.4 graphs observed concentration versus time after dose, split by the number of changes in the sign of the gradient during the time period studied. In this plot, an upward facing green triangle is used for a value that is larger than the previous value and downward facing red triangle for a value that is less than the previous value. A change in the direction occurs at locations where the plotting symbol changes.

Observed concentration versus time after dose, split by the number of times the sign of the gradient changes direction
As you can see here, the majority of the data has at most one change in the sign of the gradient. Selecting subjects where there are more shifts in gradient sign often exposes data groups which may require further analysis. Figure 6.5 reproduces the above graphic for selected subjects with a high number of gradient sign changes. We expect that these short-term increases are due to measurement or reporting inaccuracies.

Observed concentration versus time after dose, split by the number of times the sign of the gradients changes direction (including only selected subjects)
Another way to look for potentially erroneous data that could be contained within the acceptable range of the data is to scale the data against a model-agnostic smoother (such as a loess smoother). In order to perform this scaling, the following steps are used:
-
Log the concentration data
-
Fit a smooth line to the logged data
-
Calculate differences from this smooth line
-
Calculate the mean “difference from smooth line” for each subject, and subtract this from the differences (effectively “centering” each subject’s data)
-
For each time point, subtract the mean of the data and divide by the standard deviation
-
Plot the calculated values vs. time and look for outlying values
Figure 6.6 presents the scaled data generated versus “time after dose.” In this graphic, the subject identifiers are shown in dark blue, with subject groups linked by light gray lines. The change of color here focuses the reading on the subject values themselves (darker color) as opposed to the “trend” of each subject (lighter color). Horizontal grid lines have been added to allow ease of reading.

Scaled standard deviations from time point centers versus time after dose, with subject identified
From this analysis, we can identify subjects that may warrant further analysis based on large absolute differences from 0. For example, the following subjects have at least 1 value that is more than 3 standard deviations from the mean time point center in the above plot: 10, 25, 36, 39, and 141. Creating a plot of observed concentration versus time after dose for these subjects produces the graph shown in Fig. 6.7.

Observed concentration vs. time after dose including only those subjects flagged in the above analysis, with blue shaded areas representing the range and interquartile range for the overall data and the overall median drawn in black
In this instance, we want to illustrate how these subjects compare to the general trend of the data. If we were to include all subjects in this graph, the visualization might look too “busy,” and the focal message could be confused. As such, in order to represent the “general trend” of the data, we have instead included polygon “shadows” of color representing the 0, 25, 75, and 100 percentiles of the total data set on each plot; these are represented as light and dark blue polygons (0th and 100th percentiles drawn in light color, overlapped with dark polygons representing the 25th to 75th percentiles). In addition, the “center” of the data is included as a (black) median line. To contrast against this background, the individual subject data has been graphed as bright red points linked by a line.
From this analysis, subjects 10 and 36 certainly warrant further investigation. In particular, we should look at the observation at time point 3 for subject 10 (which appears to be comparatively low) and the reasons that the concentrations peak comparatively late for subject 36 when compared with the general trend of the data (since most concentrations peak at 2 h).
5 Baseline Effects
The techniques discussed thus far are applicable to both PK and PD repeated measures data.
Baseline is almost always a highly important covariate when modeling a PD endpoint. If the baseline distribution of the dependent variable differs between dose groups, it can often be seen to undermine the findings of a study even when the baseline effects have been accounted for in our model. This is particularly true of smaller early phase studies for which the more extreme baseline values have a greater influence on population summaries.
For the EDA, the effect of baseline can blur the effects of other covariates and unless we are interested investigating interactions with baseline we should look to try and remove the baseline effect in order to focus on the covariate of interest. For longitudinal data, this usually means plotting the change from baseline over time such that all profiles begin from the same place. In Fig. 6.8 we first plot our dependent variable over time and then the change from baseline in the dependent variable over time. In the left-hand plot the change in the response variable appears random over time. In the right-hand plot however, it is apparent that there is increased variability over time, as would be expected for a controlled trial in which subjects must meet certain baseline criteria in order to be randomized to the study.

Improved clarity after adjusting for baseline, subject profiles are plotted using semi-transparent lines
When plotting change from baseline we highlight the challenge of choosing an appropriate y-axis range. It is often easy to neglect this choice since almost all software packages automatically generate a y-axis based on the observed range of our data, occasionally adding a small amount of “white space” to this range in an attempt to aesthetically enhance the default plots. It is not uncommon for the observed bounds of our data to closely coincide with this default range and so this default range is usually sufficient for our needs. When we plot change from baseline data however, the possible range is often much larger than the observed range. Therefore if we adopt the default range, then a small change in the profile of our data might appear to be much larger than it actually is, potentially leading us down the wrong path. Often the range we choose lies somewhere between a change of potential clinical importance and a plausible range.
6 Investigating Covariates
Once we are satisfied with the quality of our data, the next step is to investigate dose and any other covariates that we are considering for inclusion in our model. Any covariate that we choose to investigate (including dose) falls into one of the two categories, continuous and discrete. We look at each in turn, beginning with discrete covariates.
When plotting by dose or any other discrete covariate we have a choice to make. We can either use grouping techniques to distinguish between the levels within a single plot or partition the plot into panels. The advantage of grouping within a single plot is that all profiles are on the same axis, and thus it is theoretically easier to compare the profiles. For a spaghetti plot of individual subject profiles, color or line type can be used to differentiate between the groups. However, with a large number of subjects or when the data is highly variable it can be difficult to extract any differences between groups using this method. For this reason it is common to plot average data profiles for each group, for example median values. When plotting using a graphics device that allows semi-transparent coloring we may also decide to plot “ribbons” or error regions to give an idea of the range of the data. However the use of error bars or ribbons can crowd the plot, particularly when the covariate has many levels. Even with just three levels a plot can look crowded if the differences between the groups are small. For that reason we are usually limited to plotting a single summary statistic for each subgroup over time. Figure 6.9 shows the change over time in a simulated response variable by country. In this particular dataset there are 9 levels of the country variable. A simple plot of the median values for each country highlights a sudden rise in response between weeks 8 and 10 for China in contrast to the majority of countries for whom the response tends to decrease between weeks 8 and 10.

Change in response over time by Country
In the previous example plotting median values helped highlight an interesting feature of our data. However without any indication of variability we cannot be sure whether this is truly interesting or simply random fluctuation. Figure 6.10 plots the change from baseline over time in the dependent variable by dose group. The blue regions within each panel represent the range and interquartile range for the dose group with the median represented by a black line. In addition, the range of response values across all doses is shown in gray. The position of the blue area in relation to the shadow helps to identify a dose effect with increased response in the higher dose group.

Change in median response variable over time by dose group with blue shaded areas representing the range and interquartile range for each dose group and the overall data range included as a gray shadow on the plot
The approach is one that is easily extended into higher dimensions. In Fig. 6.11 we look at two discrete covariates, the sex of the subjects and whether or not they smoke. Again, the 0th, 25th, 50th, 75th, and 100th percentiles are displayed for each covariate group and as with the last graphic, the 0–100 range is in light blue, 25–75 in darker blue, and a black line shows the median. Figure 6.11 shows higher concentrations for female subjects, and a later peak in concentrations for male subjects. The graph also suggests a slight increase in concentration values for smokers. The consistency across the plots plot would also seem to indicate a lack of any interaction between sex and smoking habit. Note that to facilitate comparison of the observed concentration values, the plots are aligned from left to right as opposed to a 2 ×2 grid layout.

Observed concentration vs. time after dose plots split by sex/smoking Indicator, with blue shaded areas representing the range and interquartile range for the subgroup and the overall data range included as a grey shadow on the plot
When the covariate of interest is continuous we no longer have the option of paneling available to us since to panel by a variable implies that it is discrete. We can of course split the continuous variable into discrete categories of interest and apply the same techniques already discussed for categorical variables. In a modeling scenario we generally wish to avoid partitioning our data in this way as we create artificial boundaries between data points. However for an EDA this actually may help us to spot an effect, and if we do intend to categorize a continuous variable, then we can use paneling as a way of selecting the breakpoints. Of course it is vital that the natural order is retained when a discrete variable has been formed from a continuous one.
If we decide not to cut our continuous covariate up into a discrete variable, then we face a dimensionality challenge; we are effectively already treating both the response variable and time as continuous variables and the within-subject correlation adds further dimensionality. When faced with this challenge, it can be tempting to move to a three-dimensional coordinate system. Given that we only have 2 dimensions available to us, this approach should be adopted with caution. In particular the angle of rotation has a strong effect on our interpretation of the graphic and interesting features of the data can easily be hidden. An alternative approach is to treat time as a discrete variable. This is possible for clinical trial data since the response information is collected at scheduled (discrete) times. We can then investigate the response variable against our covariate of interest via a sequence of scatter plots. If a relationship exists between the two variables, we would expect to see a pattern develop as we move from left to right across the panels. As with all ordinal categorical variables it is vital that when we panel by time we maintain the order and arrange the panels from left to right.
In Fig. 6.12 we have paneled by time in order to investigate how the relationship between Y and X changes over time. Varying the plotting symbol and color by dose, we see how the dose groups begin to separate over time.

Comparison of response against weight by dose
This method is not without its drawbacks. The length of time between the discrete data collection points is usually wider towards the end of a subject’s participation in a study. Care must therefore be taken when paneling by the discrete time points to preserve the distance between time points. In Fig. 6.12 therefore the first time point has been removed so that the difference between each time point is equal. Alternatively, spacing could be used to indicate larger time intervals.
7 Conclusion
For the PK/PD modeler, the EDA is the first step in the model building process. Exploring the data graphically presents many challenges due to the within-subject correlation that results from repeated measures data. An understanding of this structure can help us to break the data down and spot troublesome with-range anomalies. The aim of the EDA is to attain a better understanding of the data prior to commencing a model building process. It is therefore important to focus the exploratory work on identifying patterns within the data that may influence this process. Plot change from baseline data when applicable and consider the comparison of subgroups with the population as a whole in addition to with each other.
References
Cleveland WS, Grosse E, Shyu WM (1992) Local regression models. In: Chambers JM, Hastie TJ (eds) Statistical models in S, Chap. 8. Wadsworth and Brooks/Cole, Pacific Grove, CA
Jonsson EN, Karlsson MO (1999) Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Meth Programs Biomed 58(1):51–64. (Available from http://xpose.sourceforge.net/)
R Development Core Team (2008) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Sarkar D (2009) Lattice: multivariate data visualization with R. Springer, New York
Wickham H (2009) ggplot2: Elegant graphics for data analysis. Springer, New York
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, New York
About this chapter
Cite this chapter
Roosen, C., Pugh, R., Nicholls, A. (2012). Exploring Pharmacokinetic and Pharmacodynamic Data. In: Krause, A., O'Connell, M. (eds) A Picture is Worth a Thousand Tables. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-5329-1_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5329-1_6
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-5328-4
Online ISBN: 978-1-4614-5329-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)