
Abstract
Cancer remains a leading cause of death in industrialized countries despite advances in the detection and treatment of this disease (Heron et al., Natl Vital Stat Rep 57:1–134, 2009). Traditional models of cancer posit that neoplastic cells arise through the sequential accumulation of genetic mutations leading to independent and uninhibited replication, the evasion of apoptosis, sustained angiogenesis, and ultimately, invasion and metastasis. The latter is of particular clinical significance as metastasis is the leading cause of cancer related death (Pantel and Brakenhoff 2004; Colotta et al. 2009; Wu and Zhou 2009; Sica et al. 2008).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Esophageal Squamous Cell Carcinoma
- Foxo Protein
- Tumor Genesis
- Phosphorylated Tyrosine Residue
- Promote Cancer Cell Proliferation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Cancer remains a leading cause of death in industrialized countries despite advances in the detection and treatment of this disease (Heron et al. 2009). Traditional models of cancer posit that neoplastic cells arise through the sequential accumulation of genetic mutations leading to independent and uninhibited replication, the evasion of apoptosis , sustained angiogenesis, and ultimately, invasion and metastasis . The latter is of particular clinical significance as metastasis is the leading cause of cancer related death (Pantel and Brakenhoff 2004; Colotta et al. 2009; Wu and Zhou 2009; Sica et al. 2008).
In recent years, emerging evidence has challenged the previously held notion that metastasis is a late phenomenon in the natural history of malignant disease. The current understanding of the metastatic cascade represents a paradigm shift in which invasion and metastasis represent early occurrences in patients with cancer. Prior to the advent of the molecular characterization of neoplasia , aggressive phenotypes of primary tumors were observed histopathologically. For example, high grade, mitotically active cutaneous melanoma was shown to carry a worse prognosis than a less mitotically active tumor. Similar observations have been made across numerous malignancies including breast, thyroid and gastrointestinal tumors. The modern corollary to these observations is evident in the observation of a molecular metastatic phenotype evident in primary tumors whereby genetic expression profiles of metastatic lesions are mirrored in their primary counterparts (Pantel and Brakenhoff 2004; Colotta et al. 2009; Fidler 2003; Weigelt and Veer 2004).
Further analysis of the genes expressed in these metastatic signatures reveals that many are the same genes and proteins that have been the subject of laborious experimental analysis over the past decade and are not distinct. These genes and their translation products are the same ones implicated in the so called hallmarks of cancer, namely, evasion of apoptosis, self-sufficiency in growth signals, insensitivity to anti-growth signals, sustained angiogenesis, limitless replicative potential and tissue invasion and metastasis . In this chapter the basic pathways and some of the molecules involved in dysregulation of cell cycle control and self-sufficiency in growth signals will be reviewed (Weigelt and Veer 2004; Woelfle et al. 2003; Weigelt et al. 2003; Vijver et al. 2002; Ramaswamy et al. 2003).
1.1 Evidence for Metastasis as an Early Phenomenon
Emerging evidence supports the notion that metastasis is an inherent property of primary tumors and may arise early in the course of disease. Animal models have demonstrated distinct clonal populations within the primary tumor that display the propensity for metastasis to different sites (Weigelt et al. 2003). Similarly, the in vivo demonstration of circulating malignant cells (CMC’s) , whose poor prognostic significance has been demonstrated prospectively in a number of malignancies, have been observed in patients decades before the emergence of metastatic disease further supporting the notion of dissemination and metastasis as early events in neoplasia (Cools-Lartigue et al. 2008; Criscitiello et al. 2010; Fleitas et al. 2010). Additionally, the presence of small numbers of malignant cells within distant organs has been demonstrated in animal models prior to the development of clinically evident metastatic disease in several malignancies, including melanoma, breast, lung, and esophageal cancer (Cools-Lartigue et al. 2008; Minn et al. 2005; Lurje et al. 2010).
These observations support the hypothesis that malignancy is a systemic disease early in its evolution. This suggests that the ultimate emergence of clinically overt metastatic disease is the result of a complex interplay between the tumor and the host, which ultimately supports distant metastatic growth.
2 The Metastatic Phenotype
Recent evidence has challenged the traditional model of the metastatic cascade. The traditional model posits that within a primary tumor, individual cells undergo successive mutations, which confer a survival advantage, increasing their replicative success (Pantel and Brakenhoff 2004; Bernards and Weinberg 2002). This process continues in successive generations with the eventual emergence of rare cells capable of dissemination and metastasis . This theory implies therefore that the emergence of cells capable of metastasis is infrequent and a relatively late phenomenon in the tumor progression model (Pantel and Brakenhoff 2004; Bernards and Weinberg 2002).
Conceptually, this model has many flaws. Bernards and Weinberg suggest that a different conceptualization of tumor genesis is necessary, given that the emergence of cells capable of metastasizing confers no survival advantage to the primary tumor. The frequency of emergence of cells with metastatic potential in this construct is estimated at one in ten million. Accordingly, the impetus favoring metastasis is difficult to conceptualize in this model. The authors therefore posit that the metastatic potential is inherent to the primary tumor, that the development of metastasis may be an early phenomenon in tumor progression, and the genes responsible for metastasis are not distinct (Bernards and Weinberg 2002).
Experimental evidence supports this hypothesis. Ramsawamy et al. analyzed the gene expression profile of metastatic adenocarcinoma from several tumors, including lung, breast, prostate, colorectal and ovarian, and compared them to primary tumors of the same subtypes (Ramaswamy et al. 2003). The authors demonstrated distinct gene expression patterns in the two groups as would be expected according to the traditional model of metastasis. However, in a subset of tumors, an overlap in the expression profile of primary and metastatic lesions was observed, initially leading the authors to misclassify the primary tumors as metastases. Furthermore, the authors demonstrated that patients exhibiting this metastatic genetic profile in their primary tumors had significantly shorter overall survival compared to patients that did not. Of particular significance, the authors were able to refine this metastatic profile to a group of 17 genes of which 8 were up regulated and 9 were downregulated. This metastatic profile was able to predict outcome across the different malignancies examined in this study. Thus, the authors concluded that this signature is not specific to a particular neoplasm but representative of the processes governing metastasis as a whole.
Weigelt et al. performed a similar study in which the genetic profile of primary breast tumors was compared to samples from metastatic tumors. The authors demonstrated that in 6 of the 8-primary/metastatic pairs analyzed, the gene expression profile of the primary was closer to its metastatic counterpart than to the other primary tumors (Weigelt and veer 2004; Weigelt et al. 2003).
Van de Vijveer et al. demonstrated the presence of a poor outcome expression profile derived from estrogen receptor (ER) positive primary breast tumors in patients with advanced age. These patients demonstrated a different genetic profile than younger patients with ER positivity and a significantly increased propensity to die of metastatic disease than younger patients. This suggests that ER positivity in older patients is a distinct clinical entity, replete with a genetic predisposition for metastasis that is not mirrored in younger individuals with ER positive disease (Vijver et al. 2002).
Bhattacharjee et al. developed a molecular classification of carcinoma of the lung and were able to delineate genetic subtypes, which correlated with overall prognosis and metastatic potential. Collectively, these results support the hypothesis that metastatic potential may be inherent to the primary tumor (Bhattacharjee et al. 2001).
Data also supports the notion that features inherent to the primary tumor govern the site of metastasis. Woelfle et al. examined the gene expression profiles of primary breast tumors. In addition, patient lymph nodes and bone marrow (BM) were analyzed for the presence of metastases (micro- or macroscopic). The authors then compared the gene expression profiles of patients with BM+ and BM− disease, and patients with LN+ and LN− disease. The authors demonstrated that the gene expression profiles in patients with BM+ disease were distinct from those with BM− disease and did not overlap with the gene expression profiles in patients with LN+ disease (Woelfle et al. 2003).
Minn et al. demonstrated differential tissue tropism among metastases in a mouse model of metastatic breast carcinoma. The authors used cells derived from a primary tumor with a known poor prognosis profile as described by Woelfle et al. Tumor cells were injected systemically into immune deficient mice and single cell progenies were derived after harvesting metastases from different sites, including bone, lung and adrenals. The authors subsequently demonstrated homogeneous expression of the poor prognosis genotype amongst the various metastases. However, the authors also identified a subset of genes unique to progeny cells isolated from different metastatic sites, with cells with a metastatic tropism for bone displaying a distinct metastatic profile, which was not expressed in cells displaying other tissue tropisms. Furthermore, this metastatic genetic profile was distinct from the initial poor prognosis profile. Subsequent genomic analysis of tissue-specific metastatic phenotypes demonstrated that they were preserved in the metastasis of primary tumors that had demonstrated spread to their corresponding tissues. As a whole, this data further supports the postulate that the mechanisms required for dissemination and ultimately metastasis are present in the primary tumor (Woelfle et al. 2003; Minn et al. 2005).
2.1 The Genes Involved in Metastasis are Not Novel
Further analysis of the metastatic phenotype identified by Ramsawamy et al. demonstrated a cluster of 17 genes that could reliably predict survival in their cohort of patients. None of the identified genes were individually involved in metastasis and the authors concluded that the signature as a whole was predictive of metastasis. Of these 17 genes, 8 were up regulated and 9 were down regulated. Four of the 8 up regulated genes were members of the protein translational apparatus; one, securin, is involved in sister chromatid separation during the metaphase-anaphase transition, and the remaining three were cytoskeletal and extracellular matrix proteins. Of the 9 down-regulated genes, again, a large number of extracellular matrix proteins and cytoskeletal proteins were identified. These findings suggest that the mediators of metastasis are not novel, but play an already established role in tumor genesis (Ramaswamy et al. 2003).
Along these lines, Hong et al. demonstrated a metastatic signature in patients with left sided colon cancer, which was found to be predictive of metastasis-related mortality. The authors observed that the genes involved in predicting metastasis are indicative of “cell-wide” perturbations in basic cellular functions (Hong et al. 2010).
3 Growth Factor Systems in Metastasis
Taken together the above data suggests that the genes mediating metastasis play a role in basic cellular processes. Along these lines, a variety of growth factors have been identified and are known to play key roles in promoting progression of cancer cells through the cell cycle , thus promoting accelerated growth and progression. One of the hallmarks of cancer is the notion of self-sufficiency in growth factor signals (Colotta et al. 2009). Cancer cells must survive within foreign host tissues sometimes under harsh conditions. The metastatic microenvironment must be groomed to offer a fertile soil for cancer cells to thrive. Recent investigations have revealed that growth factor signals arise from numerous sources in the metastatic microenvironment. Contributions arise from numerous mechanisms and cell types in this highly orchestrated and complex process. Additionally, each metastatic signature mentioned implicates conserved pathways with multiple downstream effectors involved in cellular growth, replication and cell cycle regulation. Furthermore, these pathways have been the subject of cancer related study for decades and do not represent exclusively novel metastatic mediators.
3.1 Transforming Growth Factor Beta (TGF-β)
Original reports on the effects of TGF-β signaling on cancer cells suggested that it inhibited cell cycle progression and induced apoptosis. However, the tumor suppressive aspects of TGF-β signaling were countered by studies where complete abrogation of TGF-β in cancer cells could also promote metastasis in vivo (Bierie and Moses 2006). TGF-β ligands reside in the extracellular matrix and require activation via a number of mechanisms to carry out their signal. Integrins, a number of proteases such as elastase or matrix metalloproteinase 9 and glycoproteins such as thrombospondin can all interact with TGF-β ligands, resulting in activation and eventual intracellular signaling. Indeed many of these ‘activators’ of TGF-β signaling are known to be involved in the metastatic process and have frequently been described as essential components of the metastatic tumor microenvironment (Bierie and Moses 2009).
TGF-β signaling is a complex affair. Ligation of its receptors TβRI and TβRII leads to transactivation, resulting in signaling through the SMAD pathway . SMAD signaling leads to transcriptional control. In addition, TGF-β can signal through numerous SMAD independent networks (Shi and Massague 2003). The net effect of the TGF-β pathway is highly dependent on the co-activation of parallel pathways. As mentioned, these effects can include such dichotomous anti-tumorigenic and pro metastatic phenotypes as induction of apoptosis and increased motility and invasion . These diametrically opposed effects have been reconciled by recent studies that delineate how inflammatory cells of the tumor microenvironment can modulate the effects of TGF-β signaling (Bierie and Moses 2009). Although TGF-β may suppress entry into the cell cycle , it also has been shown to suppress chemokine production that is largely responsible for the attraction of myeloid-derived suppressor cells (MDSC) to the tumor microenvironment. This host-tumor interaction is increasingly recognized as an essential component of tumor progression. The immune suppression that is effected locally by MDSC has been reported to promote metastatic growth. These findings highlight the importance of studying signaling events within the context of a heterogeneous and complex tumor microenvironment.
3.2 Epidermal Growth Factor Receptor
Epidermal growth factor receptor is over-expressed in most epithelial malignancies (Kalyankrishna and Grandis 2006). The downstream effects of EGFR signaling have been extensively studied and its role in cancer progression has been described at many levels (Hynes and Lane 2005). Indeed, EGFR is involved in the pathogenesis of some cancers and is also responsible for some aspects of progression to metastasis. Elevated expression of EGFR correlates with poor prognosis. EGFR can signal through the ERK pathway to promote proliferation in response to TGF-alpha binding (Albanell et al. 2001). Furthermore, signaling through the STAT3 pathway has been shown to promote tumor growth (Grandis et al. 1998; Thomas et al. 2003). Finally, by activating PLC-γ, EGFR activation can promote tumor cell survival (Thomas et al. 2003). Together, these characteristics of the EGFR pathway contribute to a pro-metastatic phenotype in cancer cells that over express this receptor and that are exposed to its ligand.
While EGFR signaling can be triggered by a number of ligands, it can also be triggered by transactivation via receptors like Insulin Growth Factor 1 Receptor (IGF-1R) (Adams et al. 2004). Such a pathway is of particular clinical significance. Trials that have incorporated EGFR inhibitors have yielded inconsistent results. Although such inhibitors may have potent action on the ligand mediated effects of EGFR signaling, transactivation of the pathway via a receptor like IGF-1R may detract from the efficacy of such an inhibitor. The complexity of these systems is at the root of the inefficacy of certain growth factor inhibitors like the EGFR blockers. Similarly to the impact of the tumor microenvironment on cancer cell proliferation, the cross talk between various growth factor pathways is key to robust oncogenic progression. Thus, therapies must be targeted broadly to the cancer cell, the microenvironment and the multiple facets via which growth factors affect the cancer cell cycle .
3.3 Inflammatory Cytokines and the Cell Cycle
Perhaps one of the most striking discoveries in recent years for the field of cancer metastasis is the pivotal role played by supporting inflammatory cells (Coussens and Werb 2002). Over a century and half ago, Virchow first drew the parallel between cancer progression and the inflammation present in healing wounds by noting the presence of abundant leukocytes within tumors (Wu and Zhou 2009). Later, cancer was likened to a non-healing wound. Wounds require trophic factors to bridge an epithelial gap. Multiple growth factors are required to induce proliferation of epithelial cells and to promote their migration across the wound. The wound microenvironment is rich with inflammatory cells that orchestrate and drive the process. Many seminal studies over the past 10 years have shown that cells like macrophages, neutrophils and MDSC are very active players in the metastatic process (Sica et al. 2008; Bunt et al. 2006; McDonald et al. 2009). One key aspect of their function is to promote cancer cell proliferation via the production of inflammatory cytokines .
One of the gateways to the production of inflammatory cytokines is NFκB activation. Activation of NFκB in cancer cells has been shown to suppress apoptosis, thus promoting survival and progression (Karin 2006). The result of NFκB activation is the production of inflammatory cytokines like TNF-α, IL-1 and IL-6 to name a few. Production of cytokines like IL-6 in the tumor microenvironment has been shown to cause STAT3 phosphorylation, which is a key signaling step related to metastatic progression (Groner et al. 2008). Indeed, STAT3 activation leads to protection from apoptosis and progression through the cell cycle . Sources of IL-6 can arise from the cancer cell or from the microenvironment. Multiple reports have shown that cancer cells are capable of producing inflammatory cytokines and thus may support their own growth via this mechanism. However, the overwhelming source of inflammatory mediators within the tumor microenvironment appears to be derived from inflammatory cells like tumor-associated macrophages. As alluded to earlier, these cells are key to metastatic progression and act as cytokine factories (Spicer et al. 2010). The effects of cytokines go well beyond cell cycle control as they promote angiogenesis and support stromal cells to create a milieu that is welcoming to cancer progression. Nevertheless, these inflammatory mediators have the net effect of stabilizing cancer cells and favoring their survival in an otherwise harsh metastatic environment.
3.4 The PI3-Akt Pathway
This family of lipid kinases has been intensely studied since its discovery in the 1980s and its regulation in diverse cellular processes such as survival, proliferation, and differentiation has been demonstrated. Not surprisingly, dysregulated functioning of this pathway has been linked to many of the processes at play in malignancy, which have been collectively referred to as the hallmarks of cancer (Colotta et al. 2009).
The PI3-Akt pathway is a conserved pathway, which transduces signals from various cytokines and growth factors via the association of the PI3 lipid kinase and activated receptor tyrosine kinases (RTK), G-protein coupled receptors (GPCR) and oncogenes such as RAS (Fig. 7.1) (Vivanco and Sawyers 2002). The PI3 lipid kinase is a heterodimer composed of a regulatory p85 subunit and a catalytic p110 subunit. The p85/p110 complex is capable of participating in multiple protein-protein interactions via its association with phosphorylated tyrosine residues. In the context of RTK initiated signaling, binding of a ligand to its receptor initiates recruitment of PI3k to the plasma membrane via the association of the p85 regulatory subunit with phosphorylated tyrosine residues. The now active p110 subunit subsequently catalyzes the phosphorylation of phosphatidyl inositol 4,5, bisphosphate (PtdIns(4,5)P2) to PtdIns(3,4,5)P3 which, among other processes, recruits the serine threonine kinase Akt to the cell membrane via the association of its pleckstrin homology (PH) domain to PtdIns (3,4,5)P3. This association induces a conformational change in Akt permitting its phosphorylation, and concomitant activation by 3-phosphoinositide dependent kinases (PDK). The major regulatory protein, phosphatase and tensin homologue (PTEN), acts to limit the availability of PtdIns(3,4,5)P3 and active PDK through the removal of phosphate groups. Activated Akt participates in a broad range of downstream signaling events (Adams et al. 2004; Kelly-Spratt et al. 2009). In the context of this chapter, its role in cell cycle regulation will be discussed (Fig. 7.2, 7.3).

The PI3-Akt Pathway. The PI3 lipid kinase is a heterodimer composed of a regulatory p85 subunit and a catalytic p110 subunit. The p85/p110 complex is capable of participating in multiple protein-protein interactions via its association with phosphorylated tyrosine residues. Binding of a ligand to its receptor initiates recruitment of PI3k to the plasma membrane via the association of p85 with phosphorylated tyrosine residues. Active p110 subsequently catalyzes the phosphorylation PtdIns(4,5)P2 to PtdIns(3,4,5)P3 which recruits the serine threonine kinase Akt to the cell membrane via the association of its PH domain to PtdIns (3,4,5)P3. This association induces a conformational change in Akt permitting its phosphorylation, and concomitant activation by 3-phosphoinositide dependent kinases (PDK). The major regulatory protein, PTEN, acts to limit the availability of PtdIns(3,4,5)P3 and active PDK through the removal of phosphate groups. Activated Akt participates in a broad range of downstream signaling events including regulation of the cell cycle. Figure adapted from Vivanco et al.(2002)

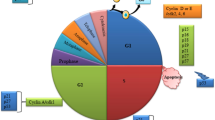
The cell cycle. The progression from G1 to S and through the S phase is a multi-step process mediated by the association of D, E and A type cyclins with CDK4 or 6, and CDK2, respectively. The expression of the D-type cyclins occurs early in G1; as such, the D type cyclins are commonly regarded as nuclear relays of extracellular signaling

Postranslational regulation of Cyclin D1. Cyclin D1 normally shuttles between the nucleus and the cytoplasm. Within the nucleus, it is able to complex with CDK4/6 and drive cell cycle progression. Its phosphorylation at Threonine 286 by GSK beta renders it amenable to nuclear export in a CRM-1 dependent manner. Within the cytoplasm, it is vulnerable to ubiquitination by the SCF ubiquitin ligase and proteosomal degradation. Figure adapted from Witzle et al. (2010)
3.4.1 The PI3-Akt Pathway and Proliferation
The insulin growth factor 1 receptor (IGF-1R) can control transition through the cell cycle via multiple signaling pathways . However, the summative evidence would suggest that it causes progression at the G1-S interface via ERK and PI-3K/AKT signaling (Samani et al. 2007). Increased expression of IGF-1R and its ligands IGF-1 and IGF-2 are elevated in a number of malignancies. The coincidental presence of high levels of ligand and receptor in the tumor microenvironment suggest that the IGF axis can function both in an autocrine and paracrine fashion. Indeed, studies that genetically engineered liver metastasizing H-59 Lewis lung carcinoma cells to express the soluble form of IGF-1R had significantly fewer metastases, suggesting that the paracrine effect of IGF-1 liver expression is tumor promoting (Brodt et al. 2001). Confirming this hypothesis was a series of experiments where the size of primary tumor and liver metastases were increased by the addition of exogenous IGF-1 to liver-specific IGF-1 deficient mice in an orthotopic model of colon carcinoma metastasis (Wu et al. 2002). The role of IGF-1 was further delineated in a model of bone metastasis where prostate cancer cells expressed urokinase plasminogen activator (uPA) to degrade IGF-1 binding protein. As a result the bioavailability of IGF-1 in the metastatic microenvironment was bolstered (Koutsilieris and Polychronakos 1992).
These studies highlight the contribution of the host metastatic organ in terms of trophic factors that can promote cancer cell proliferation. Such environmental particularities contribute to the site specificity of certain malignancies. In addition, cancer cells benefit from being equipped with an armament of growth factor receptors and proteolytic enzymes to match the growth factor production line of the metastatic site where they attempt to implant. Metastatic inefficiency is in part attributed to this frequent mismatch. Cells capable of expressing high levels of growth factor in addition to high levels of the corresponding receptor may be more aggressive and metastasize more widely. Cancer cells with a poor match for the eventual metastatic microenvironment may not survive or may require the arrival of supporting inflammatory cells to engage further growth.
Activated Akt appears to promote progression through the cell cycle and thus cellular replication. This is achieved in two general ways. First, activated Akt inhibits the function of the forkhead family of transcription factors , whose production is associated with cell cycle arrest in G1 and at the G2-M transition (Medema et al. 2000). Similarly, Akt mediates the inhibition of glyceraldehyde synthase kinase 3 beta (GSK3b), which is similarly implicated in cell cycle arrest at the G1-S interface via its inhibitory role on cyclin dependent signaling (Burgering and Kops 2002).
4 Cell Cycle Control
Dysregulation of the Cell Cycle in Malignancy
The cell cycle is a tightly controlled process and its dysregulation is implicated in tumor genesis. Transition from G1-S, progression through the S-phase, G2-M transition and completion of mitosis are processes that are regulated through differential expression of cyclin dependent kinases (CDK) and their binding partners, cyclins. While the expression of the CDK is relatively stable throughout the cell cycle, the cyclins are a diverse group of proteins, which exhibit periodic expression at key phases. These proteins function as allosteric modulators of the CDK (Slingerland and Pagano 2000).
The transition from G1 to S has demonstrated aberrant function in nearly all human malignancies. Under normal conditions, the progression from G1 to S and through the S phase, induced, for example, by exposure to mitogens, is a multi-step process mediated by the association of D, E and A type cyclins with CDK4 or 6, and CDK2, respectively. The expression of the D-type cyclins occurs early in G1; as such, the D type cyclins are commonly regarded as nuclear relays of extracellular signaling (Slingerland and Pagano 2000).
Progression Through the Cell Cycle and Its Regulation
In response to mitogen stimulated increased transcription, translation and inhibited proteolysis of Cyclin D takes place via both MAPK and PI3-Akt dependent pathways respectively. Consequently, intra-nuclear accumulation of cyclin D, with its resultant association with CDK4 or 6, results in the phosphorylation of the retinoblastoma (RB) tumor suppressor gene which in turn results in increased E2F mediated transcription. This leads to enhanced transcription and translation of E type cyclins, which, in association with CDK2 promote ongoing E2F mediated transcription and are responsible for progression through the G to S restriction point. Finally, A type cyclins, in conjunction with CDK1 or 2 are responsible for progression through the S phase (Slingerland and Pagano 2000; Witzel et al. 2010).
Regulation of cyclin:CDK complexes is under the control of 2 families of CDK inhibitors; the INK4 and Cip/Kip families. INK4 proteins (p16ink41, p15ink4b, p18ink4c, p19ink4d) bind and inhibit CDK4 and 6 while the Cip/Kip proteins (p21cip1/waf1/sdil, p27kip1, p57kip2) inhibit CDK2/cyclin E and CDK1/2:cylin A. As levels of CDK4/6/Cyclin D rise throughout early G1, these complexes sequester Cip/Kip family CDK inhibitors facilitating CDK2/Cyclin E complex formation and progression from G1-S. Finally, the cyclins are under posttranslational control at the level of sub-cellular localization and proteolysis, all of which have been implicated in malignancy (Witzel et al. 2010; Alt et al. 2000).
4.1 D type Cyclins in Malignancy
D-type Cyclins in Human Cancers Demonstrate Translocations, Amplifications, and Subtle Polymorphisms
Not surprisingly, increased expression of D type cyclins has been demonstrated in human cancers including, breast, bladder, head and neck, lung and esophageal cancer. A variety of mechanisms for the aberrant expression of Cyclin D have been observed including chromosomal translocations, and amplifications as well as single nucleotide polymorphisms that alter the sensitivity of cyclin D to proteolysis.
In mantle cell lymphoma , the demonstration of the t(11;14)(q13;q32) translocation, which places the cyclin D1 gene CCND1 under the control of the immunoglobulin heavy chain (IgH) promoter has been observed in over 90 % of cases. The postulate here is that cyclin D1 expression is constitutive, thereby driving cellular proliferation (Schmitz et al. 2005).
Cyclin D1 overexpression has also been demonstrated through amplification of the CCND1 locus at 11q13 in multiple human cancers including esophageal squamous cell carcinoma. The study by Shinozaki et al. demonstrated over 3-fold amplification of 11q13 in 23 % of 122 primary esophageal squamous cell carcinomas examined. The authors also demonstrated a statistically significant decrease in overall survival as well as an increase in the presence of distant metastasis in patients harboring this genetic abnormality. Thus, the over expression of cyclin D1 in these patients is of clinical significance (Shinozaki et al. 1996).
Finally, cyclin D1 over expression has been linked to more subtle genetic alterations. More than 100 polymorphisms have been identified at the CCND1 locus. One such polymorphism, the G/A substitution at position 870, has been extensively studied. This polymorphism generates a truncated protein known as cyclin D1b, which retains the capacity to associate with CDK4/6 but lacks the GSK b phosphorylation site and PEST sequence necessary for nuclear export and ubiquitin mediated degradation, respectively. This polymorphism has been demonstrated in approximately 5 % of Mantle cell lymphomas and approximately 60 % of human bladder cancers (Krieger et al. 2006; Kim et al. 2009).
Mechanisms of Cyclin D oncogenesis
Cyclin D overexpression alone is insufficient to drive neoplastic transformation
The mechanism by which Cyclin D influences tumor progression remains incompletely understood. Its observed over expression among multiple malignancies has led to the postulate that it is a proto-oncogene. However, the ability of cyclin D to transform cells alone has not been supported by experimental data. Over expression of cyclin D1 in 3T3 fibroblasts by itself is unable to induce transformation. Over expression of cyclin D1 alone in murine lymphocytes is similarly unable to elicit transformation unless it is co-transfected with c-myc. A similar observation was made by Rodriguez et al. wherein over expression of cyclin D1 and ras results in the formation of skin tumors in mice (Rodriguez-Puebla et al. 1999).
Along these lines, Opitz et al. demonstrated that over expression of Cyclin D1 alone is insufficient to generate oral squamous cell carcinoma (OSCC) in mice. The authors, knowing that cyclin D was over expressed and p53 was under expressed in human OSCC, generated mice with differential expression of these proteins. The authors bred mice which over expressed cyclin D1 (L2D1+) and either demonstrated wild type p53 expression (p53+ / +), were heterozygous for p53 expression (p53+ / −) or lacked p53 expression altogether (p53−/−). The authors demonstrated tumor formation in only L2D1 + p53 + / − or p53 −/−. No L2D1 p53 + / + mice exhibited tumor formation (Opitz et al. 2002).
Thus, experimental data suggests that isolated over expression of cyclin D1 is generally insufficient to drive neoplastic transformation. Instead data suggests that it can potentiate ras and myc oncogenesis in vitro and can induce tumors efficiently in vivo in the absence of additional regulatory mechanisms as evidenced by the generation of OSCC in only those mice lacking appropriate p53 expression. Given the extent to which cyclin D over expression is observed in human cancers, additional mechanisms explaining its oncogenic activity were sought.
Post-Translational Regulation of Cyclin D and Enhanced Oncogenic Potential
Experimental evidence supports the hypothesis that the subcellular localization of cyclins influences their role in tumor formation. Cyclin D1 normally shuttles between the nucleus and the cytoplasm. Within the nucleus, it is able to complex with CDK4/6 and drive cell cycle progression. Its phosphorylation at Threonine 286 by GSK beta renders it amenable to nuclear export in a CRM-1 dependent manner. Within the cytoplasm, it is vulnerable to ubiquitination by the SCF ubiquitin ligase and proteosomal degradation. Data supports the hypothesis that excessive intranuclear CDK/cyclinD1 is involved in tumor genesis (Kelly-Spratt et al. 2009; Gladden et al. 2006).
Chul Jang Kim et al. demonstrated increased invasiveness and anchorage independent growth of bladder cancer cell lines transfected with cyclin D1b, a variant not amenable to nuclear export, in vitro. Furthermore, this effect was abrogated by siRNA specific for cyclin D1b. The results of their study suggest that the malignant phenotype of urothelial carcinoma is enhanced by the expression of cyclin D1b and may be related to its nuclear localization (Kim et al. 2009).
Experimental data supports this hypothesis. Alt et al. demonstrated that 3T3 fibroblasts transfected with cyclin D T286A, a mutant not amenable to GSK phosphorylation and CRM-1 mediated nuclear export, exhibited contact independent growth and were immortalized. By contrast, 3T3 fibroblasts transfected with wild type Cyclin D constructs exhibited only a shortened G1 phase, consistent with previous reports that over expresion of cyclin D1 alone is not sufficient to effect transformation (Alt et al. 2000).
This phenomenon was demonstrated in vivo by Gladden et al. who generated transgenic mice with constitutive expression of cyclin D T286A. These mice exhibited a significantly shorter lifespan compared to controls attributable to disseminated B-cell lymphoma . Of particular interest, however, was the observation that a large proportion of B-cells that were driven to proliferate by the mutant cyclin D1 demonstrated apoptosis, suggesting that S-phase entry in these cells is countered by increased apoptosis (Gladden et al. 2006). Thus, lymphomatous cells that escape apoptosis must acquire a second hit rendering them resistant to apoptosis. Indeed aberrations in the p19ARF-MDM2-p53 pathway were observed in these malignant cells and corroborate the experimental observation that cyclin D over expression in conjunction with p53 under expression is sufficient to drive tumor development.
The above data suggests that cell cycle control is aberrant across a heterogeneous group of malignancies. This deregulation is mediated in part through proliferation driven by increased cyclin D1 activity, which is mediated by multiple mechanisms including amplifications, translocations, and mutations rendering nuclear export and proteolysis less efficient.
4.2 Forkhead Transcription Factors in Cell Cycle Control and Malignancy
Cell Cycle Arrest via Forkhead Transcription Factors
Forkhead transcription factors, also known as Foxo proteins, are collectively implicated in cell cycle arrest and apoptosis . They belong to a family of transcription factors related through homology in their DNA binding domain. They are important downstream targets of PI3-Akt signaling. Phosphorylation of Foxo proteins by Akt increases their cytoplasmic localization as a result of inhibited interaction with nuclear DNA binding proteins such as 14-3-3. Within the cytoplasm, they are vulnerable to degradation. In the absence of mitogenic stimulation, Foxo proteins are retained within the nucleus and drive transcription of their targets (Medema et al. 2000; Burgering and Kops 2002).
Foxo Proteins Inhibit Cell Cycle Progression via Upregulation of Cip/Kip Cdk Inhibitors
Foxo proteins are implicated in increased production of the Cip/Kip family cyclin dependent kinase inhibitors including p27kip1. In the study by Medena et al. the authors demonstrated that over expression of AFX, a forkhead transcription factor, induces G1 arrest in 3T3 fibroblasts. This effect was dependent on the inhibition of cyclin dependent signaling via increased levels of p27kip1, and was independent of downstream effectors of cyclin dependent signaling such as the retinoblastoma (Rb) tumor suppressor (McDonald et al. 2009).
4.3 Foxo Proteins As Tumor Suppressors
Abundant evidence suggests that Foxo proteins are bona fide tumor suppressors in mammals. Zou et al. demonstrated that FOXO 1 and 3 inhibit estrogen receptor (ER) mediated signaling in the estrogen-dependent human breast cancer cell line MCF 7, and this effect is associated with reduced proliferation in vitro. The authors generated MCF-7 breast cancer cell lines transfected with a FOXO expression vector. The decreased ER signaling observed was mediated through direct contact of FOXO proteins and ER and was associated with increased expression of cyclin dependent kinase (CDK) inhibitors including p27kip1 and reduced expression of cyclin D1. These findings were associated with a significant reduction in proliferation in the transfected MCF-7 cell lines compared to wild-type (Zou et al. 2008).
Furthermore, the authors demonstrated that inhibition of FOXO3 in MCF-7 cells promotes tumor genesis in vivo. The authors constructed an MCF-7 FOXO3 knockdown derivative and demonstrated the development of tumors in the mammary fat pad of nude mice in the absence of estrogen stimulation. This effect was not observed with wild-type MCF-7 cells in the absence of exogenous 17-b estradiol.
Further evidence supporting the role of the forkhead transcription factors as tumor suppressors stems from the study by Paik et al. Here, the authors demonstrated the development of a widespread cancer phenotype in mice with FOXO gene deletions. The authors also demonstrated that the tumors were cell lineage specific, with thymic FOXO deletions producing aggressive lymphomas while endothelial cell targeted mutations resulted in a widespread hamartomatous phenotype associated with premature death. With respect to lymphangiogenesis, the authors demonstrated that tumor genesis in this model was mediated in part by increased cell cycle progression, inferred from strong down regulation of p27kip1 in the context of the FOXO null mice (Paik et al. 2007).
Tothova et al. demonstrated similar results. The authors again generated conditional FOXO null mice. This was associated with the development of a non-fatal myeloproliferative phenotype as well as quantitative and qualitative abnormalities of lymphoid cells. These abnormalities were mediated in part by abnormal cell cycle regulation as demonstrated by a two-fold increase of hematopoetic stem cells in S/G2/M in FOXO null mice compared to wild type. This finding was associated with aberrant expression of FOXO target genes including down regulation of p27 and up regulation of cyclin D2 (Tothova et al. 2007).
Dysregulated FOXO Function is Mediated by Akt Dependent and Independent Mechanisms in Humans
Aberrant function of Foxo proteins, either through dysregulated PI3-Akt signaling or mutation, has an experimentally established role in tumor genesis. In fact, such a role has been demonstrated in humans as well. Chronic myelogenous leukemia (CML) is characterized by the BCR-ABL translocation, which results in strong Akt activation. This activity is inhibited by the RTK inhibitor imatinib mesylate and is associated with a significant survival benefit. In this model, Akt activity drives the nuclear export and degradation of FOXO proteins, supporting ongoing proliferation. In the presence of RTK inhibitors, nuclear localization of Foxo proteins is restored, driving cell cycle arrest and apoptosis (Naka et al. 2010).
Alveolar rhabdomyosarcoma demonstrates characteristic translocations t(2;13)(q35;q14) and t(1;13)(p36;q14) which generate fusion proteins between PAX and forkhead member transcription factors . These proteins are resistant to Akt mediated cytoplasmic redistribution and are collectively localized to the nucleus where the fusion proteins drive the transcription of genes involved in proliferation, apoptosis and motility (Sumegi et al. 2010).
Kornblau et al. demonstrated that high levels of phosphorylated FOXO3A are an independent marker of poor prognosis in AML. Similarly, Song et al. demonstrated that loss of FOXA1 and FOXA2 are critical steps in the epithelial to mesenchymal transition in pancreatic ductal carcinoma, an important step in tumor genesis and predicted precursory step to metastasis. Along these lines, forkhead family transcription factors have been implicated in invasion and metastasis in multiple other human malignancies including leukemia, breast, thyroid and esophageal cancer (Song et al. 2010; Kornblau et al. 2010).
5 CDK Inhibitors are Implicated in Tumor Genesis
5.1 P27 and Cell Cycle Progression
P27 is one of the downstream effectors of forkhead family transcription factors implicated in cell cycle control, tumor genesis and metastasis. As alluded to previously, this molecule is extensively involved in the regulation of cell cycle progression particularly in the G1-S transition through its association with cyclin E-CDK2, and its subsequent inhibition of ongoing RB phosphorylation and E2F mediated transcription of genes necessary to complete the G1-S transition. However, further studies implicate p27 in processes including aberrant cell cycle control and metastasis.
The regulation of p27 itself is complex and a full discussion is beyond the scope of this chapter. Briefly, however, early in G1, p27 plays a role in stabilizing cyclin D-cdk4/6 complexes within the cytoplasm, assisting in their localization to the nucleus and hence the phosphorylation of Rb. However, p27 also acts to inhibit cyclin E-CDK 2, thereby keeping levels of cyclin A low and preventing progression through S phase. It is only when levels of p27 drop in late G1, which is believed to be mediated at least in part by phosphorylation, nuclear export and ubiquitin mediated degradation of p27, that the activity of cyclin E-cdk2 increases and progression through the cell cycle occurs (Kelly-Spratt et al. 2009; Alt et al. 2000).
5.2 Post-Translational Mechanisms Mediate Aberrant P27 Function in Malignant Disease
This tightly regulated process has been shown to be dysfunctional in a variety of human cancers. Considering the importance of p27 in the regulation of the cell cycle, one would expect mutations of this allele to be common in human cancers. While loss of a single allele has been demonstrated among various malignancies, loss of both alleles has only been rarely observed. Instead, dysregulation appears to occur at the level of p27 protein, not gene transcription. Low levels of p27 protein are independent predictors of poor outcome in multiple malignancies including lung, colon, ovarian, breast, prostate and gastric carcinoma. In fact, it has been estimated that up to 50 % of human cancers lack normal p27 expression. Furthermore, in a variety of these cancers, the low protein levels observed appear to be related to increased ubiquitin mediated proteolytic degradation (Zhou et al. 2003).
Calvisi et al., in a model of hepatocellular carcinoma (HCC), demonstrated that increased ubiquitination of p27 among other cell cycle regulators was associated with increased cellular proliferation. In a separate study, these authors demonstrated that susceptibility to the development of HCC in a rat model was dependent on ubiquitin-mediated degradation of cell cycle regulatory proteins, including p27. The authors examined two rat strains of known divergent susceptibility to the development of HCC induced by exogenous dietary nitrosamine. Only 35 % of Norway Brown (BN) rats exposed to exogenous nitrosamines developed overt HCC in the study, compared to all F344 rats. The authors demonstrated that although no difference in p27 mRNA was observed between the two groups over the course of the study, levels of p27 protein were significantly higher in BN compared to F344 rats. In order to elucidate the posttranslational mechanism responsible for the observed disparity in p27, the authors demonstrated significantly lower levels of SCF ubiquitin ligase components (Skp-2, Cks-1) in the resistant compared to the susceptible rats. Furthermore an inverse correlation between increased levels of cell cycle regulatory proteins, including p27, and proliferation based on Ki67 protein levels was observed. Taken together, this data supports the role of ubiquitin-mediated degradation of p27 in disease progression of HCC (Calvisi et al. 2010).
In keeping with this theme, Liu et al. demonstrated that the anti-neoplastic effects of hinokitiol, which induces G1 cell cycle arrest in human FEM melanoma cell lines is associated with inhibition of ubiquitin driven degradation of p27. This effect was mediated both through decreased phosphorylation of p27 at threonine 187, which targets p27 for proteosomal degradation, and down regulation of skp2, a subunit of the SCF ubiquitin protein ligase. This finding further implicates increased proteolysis as one mechanism whereby p27 activity is reduced (Liu and Yamauchi 2009).
5.3 Cytoplasmic Sequestration of P27 is Involved in Tumor Progression and Metastasis
A subset of human cancers has failed to demonstrate markedly reduced levels of p27 protein, prompting the search for alternate modalities of p27 inactivation in these neoplasms. Additional mechanisms for aberrant p27 function and cell cycle regulation have accordingly been observed (Besson et al. 2008).
Akt Independent Mechanisms
Active p27 has been shown to be sequestered in cyclinD:CDK complexes in association with Myc mediated proliferation in vitro (Yamamoto et al. 2009; Wu et al. 2009). Consequently, p27 is unable to bind and inhibit cyclinE:CDK complexes leading to cell cycle progression (Zhou et al. 2003). In vivo cytoplasmic sequestration of p27 in cyclin-D1:CDK4 complexes has been observed in some human lymphomas (Qi et al. 2006). Furthermore, Her-2 over expression in breast cancer is associated with c-myc over expression, upregulation of D-type cyclins and p27 sequestration in cyclinD:Cdk4 complexes (Acosta et al. 2008).
Akt Dependent Mechanisms
Akt mediated cytoplasmic accumulation of p27 has been documented in a variety of human cancers including melanoma , breast, colon, thyroid and esophageal cancer. Liang et al. demonstrated impaired nuclear import and cytoplasmic accumulation of p27 in both human mammary epithelial cells (HMECS) and WM35 cell lines transfected with constitutively active Akt in vitro. Furthermore, the authors demonstrated an abrogated G1 arrest in response to exogenous TGF-beta in Akt over expressing cell lines. Finally, the authors demonstrated that impaired nuclear import of p27 was the result of Akt phosphorylation at threonine 157 on p27. Furthermore, this shift in p27 compartmentalization is associated with increased cdk2 activity and cell cycle progression. Accordingly, cytoplasmic sequestration of p27 was impaired in cells expressing a mutant p27T157A, not amenable to phosphorylation by Akt at this site. Thus the authors concluded that abnormal progression through G1 may be mediated by aberrant Akt signaling and consequent cytoplasmic sequestration of p27 (Liang et al. 2002). Additional in vitro support of this hypothesis is evident in the observation that the pharmacologic PI3K inhibitor LY294002 abrogates this effect, restoring nuclear localization (Motti et al. 2005).
In vivo evidence supporting cytoplasmic accumulation of p27 in tumor genesis has also been demonstrated. Viglietto et al. demonstrated that Akt dependent phosphorylation and cytoplasmic mislocalization functionally inactivates p27 in human breast cancer. They examined the cellular localization of T157 phosphorylated p21 in 54 human primary breast cancers. Furthermore, they classified the primary tumors into three groups based on the ratio of p-Akt:Akt. Group 1 tumors, of which there were 15, had a pAkt:Akt ratio < 0.1; group 2 tumors, of which 10 were identified, demonstrated an p-Akt:Akt between 0.1–0.8; group 3 tumors, of which there were 15, demonstrated a p-Akt:Akt > 0.8. P27 was absent in 14 tumors. The authors subsequently noted that there was a statistically significant rise of T157 phosphorylated p27 from group 1 to group 3 tumors with 6 % expression in group 1,40 % expression in group 2 and 65 % expression in group 3. Furthermore, T157 phosphorylated Akt was almost exclusively localized to the cytoplasm. Thus, functional inactivation of p27 may occur via Akt mediated cytoplasmic sequestration in human breast cancer (Viglietto et al. 2002).
6 Summary
Progression through the cell cycle is a defining characteristic of a cancer cell that wishes to metastasize. Despite metastatic inefficiency, growth factor signals are numerous and redundant. These signals can arise from the cancer cell itself as well as the inflammatory tumor microenvironment and from the host organ tissue. Cross talk between various growth factor pathways like EGFR/IGF-1R transactivation may bypass the need for autocrine or paracrine growth factor stimulation. Mutations in the signaling pathway may lead to constitutive activation. Inflammatory cells of the microenvironment provide a nurturing source of growth signals. These myriad pathways lead to a similar outcome of proliferation. This obvious complexity highlights the importance of improved understanding of these processes. Only tailored therapies that address organ tropism during metastasis and the numerous portals via which cancer cells gain access to the cell cycle and proliferate out of control will yield a healthy therapeutic response.
References
Acosta JC et al (2008) Myc inhibits p27-induced erythroid differentiation of leukemia cells by repressing erythroid master genes without reversing p27-mediated cell cycle arrest. Mol Cell Biol 28(24):7286–7295
Adams TE, McKern NM, Ward CW (2004) Signalling by the type 1 insulin-like growth factor receptor: interplay with the epidermal growth factor receptor. Growth Factors 22(2):89–95
Albanell J et al (2001) Activated extracellular signal-regulated kinases: association with epidermal growth factor receptor/transforming growth factor alpha expression in head and neck squamous carcinoma and inhibition by anti-epidermal growth factor receptor treatments. Cancer Res 61(17):6500–6510
Alt JR et al (2000) Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev 14(24):3102–3114
Bernards R, Weinberg RA (2002) A progression puzzle. Nature 418(6900):823
Besson A, Dowdy SF, Roberts JM (2008) CDK inhibitors: cell cycle regulators and beyond. Dev Cell 14(2):159–169
Bhattacharjee A et al (2001) Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci U S A 98(24):13790–13795
Bierie B, Moses HL (2006) Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer 6(7):506–520
Bierie B, Moses HL (2009) Gain or loss of TGFbeta signaling in mammary carcinoma cells can promote metastasis. Cell Cycle 8(20):3319–3327
Brodt P et al (2001) Cooperative regulation of the invasive and metastatic phenotypes by different domains of the type I insulin-like growth factor receptor beta subunit. J Biol Chem 276(36):33608–33615
Bunt SK et al (2006) Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176(1):284–290
Burgering BM, Kops GJ (2002) Cell cycle and death control: long live Forkheads. Trends Biochem Sci 27(7):352–360
Calvisi DF et al (2010) The degradation of cell cycle regulators by SKP2/CKS1 ubiquitin ligase is genetically controlled in rodent liver cancer and contributes to determine the susceptibility to the disease. Int J Cancer 126(5):1275–1281
Colotta F et al (2009) Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30(7):1073–1081
Cools-Lartigue JJ et al (2008) Immunomagnetic isolation and in vitro expansion of human uveal melanoma cell lines. Mol Vis 14:50–55
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420(6917):860–867
Criscitiello C, Sotiriou C, Ignatiadis M (2010) Circulating tumor cells and emerging blood biomarkers in breast cancer. Curr Opin Oncol 22(6):552–558
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature Rev Cancer 3:453–458
Fleitas T et al (2010) Circulating endothelial and endothelial progenitor cells in non-small-cell lung cancer. Clin Transl Oncol 12(8):521–525
Gladden AB et al (2006) Expression of constitutively nuclear cyclin D1 in murine lymphocytes induces B-cell lymphoma. Oncogene 25(7):998–1007
Grandis JR et al (1998) Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth In vitro. J Clin Invest 102(7):1385–1392
Groner B, Lucks P, Borghouts C (2008) The function of Stat3 in tumor cells and their microenvironment. Semin Cell Dev Biol 19(4):341–350
Heron M et al (2009) Deaths: final data for 2006. Natl Vital Stat Rep 57(14):1–134
Hong Y et al (2010) A ‘metastasis-prone’ signature for early-stage mismatch-repair proficient sporadic colorectal cancer patients and its implications for possible therapeutics. Clin Exp Metastasis 27(2):83–90
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5(5):341–354
Kalyankrishna S, Grandis JR (2006) Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol 24(17):2666–2672
Karin M (2006) Nuclear factor-kappaB in cancer development and progression. Nature 441(7092):431–436
Kelly-Spratt KS et al (2009) Inhibition of PI-3K restores nuclear p27Kip1 expression in a mouse model of Kras-driven lung cancer. Oncogene 28(41):3652–3662
Kim CJ et al (2009) Cyclin D1b variant promotes cell invasiveness independent of binding to CDK4 in human bladder cancer cells. Mol Carcinog 48(10):953–964
Kornblau SM et al (2010) Highly phosphorylated FOXO3A is an adverse prognostic factor in acute myeloid leukemia. Clin Cancer Res 16(6):1865–1874
Koutsilieris M, Polychronakos C (1992) Proteinolytic activity against IGF-binding proteins involved in the paracrine interactions between prostate adenocarcinoma cells and osteoblasts. Anticancer Res 12(3):905–910
Krieger S et al (2006) Relevance of cyclin D1b expression and CCND1 polymorphism in the pathogenesis of multiple myeloma and mantle cell lymphoma. BMC Cancer 6:238
Liang J et al (2002) PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 8(10):1153–1160
Liu S, Yamauchi H (2009) p27-Associated G1 arrest induced by hinokitiol in human malignant melanoma cells is mediated via down-regulation of pRb, Skp2 ubiquitin ligase, and impairment of Cdk2 function. Cancer Lett 286(2):240–249
Lurje G et al (2010) Circulating tumor cells in gastrointestinal malignancies: current techniques and clinical implications. J Oncol 2010:392652
McDonald B et al (2009) Systemic inflammation increases cancer cell adhesion to hepatic sinusoids by neutrophil mediated mechanisms. Int J Cancer 125(6):1298–1305
Medema RH et al (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404(6779):782–787
Minn AJ et al (2005) Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest 115(1):44–55
Motti ML et al (2005) Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol 166(3):737–749
Naka K et al (2010) TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 463(7281):676–680
Opitz OG et al (2002) A mouse model of human oral-esophageal cancer. J Clin Invest 110(6):761–769
Paik JH et al (2007) FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128(2):309–323
Pantel K, Brakenhoff RH (2004) Dissecting the metastatic cascade. Nat Rev Cancer 4(6):448–456
Qi CF et al (2006) Expression of the cyclin-dependent kinase inhibitor p27 and its deregulation in mouse B cell lymphomas. Leuk Res 30(2):153–163
Ramaswamy S et al (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33(1):49–54
Rodriguez-Puebla ML, Robles AI, Conti CJ (1999) Ras activity and cyclin D1 expression: an essential mechanism of mouse skin tumor development. Mol Carcinog 24(1):1–6
Samani AA et al (2007) The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev 28(1):20–47
Schmitz R et al (2005) Insights into the multistep transformation process of lymphomas: IgH-associated translocations and tumor suppressor gene mutations in clonally related composite Hodgkin’s and non-Hodgkin’s lymphomas. Leukemia 19(8):1452–1458
Shi Y, Massague J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113(6):685–700
Shinozaki H et al (1996) Cyclin D1 amplification as a new predictive classification for squamous cell carcinoma of the esophagus, adding gene information. Clin Cancer Res 2(7):1155–1161
Sica A et al (2008) Macrophage polarization in tumour progression. Semin Cancer Biol 18(5):349–355
Slingerland J, Pagano M (2000) Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 183(1):10–17
Song Y, Washington MK, Crawford HC (2010) Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res 70(5):2115–2125
Spicer JD, Brodt P, Ferri LE (2010) Role of inflammation in the early stages of liver metastasis. In: Brodt P (Ed) Liver metastasis – Biology and Clinical Management. Spinger, Berlin
Sumegi J et al (2010) Recurrent t(2;2) and t(2;8) translocations in rhabdomyosarcoma without the canonical PAX-FOXO1 fuse PAX3 to members of the nuclear receptor transcriptional coactivator family. Genes Chromosomes Cancer 49(3):224–236
Thomas SM et al (2003) Epidermal growth factor receptor-stimulated activation of phospholipase Cgamma-1 promotes invasion of head and neck squamous cell carcinoma. Cancer Res 63(17):5629–5635
Tothova Z et al (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128(2):325–339
van de Vijver MJ et al (2002) A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347(25):1999–2009
Viglietto G (2002) Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med 8(10):1136–1144
Vivanco I, Sawyers CL (2002) The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer 2(7):489–501
Weigelt B et al (2003) Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci U S A 100(26):15901–15905
Weigelt B, van’t Veer LJ (2004) Hard-wired genotype in metastatic breast cancer. Cell Cycle 3(6):756–757
Witzel II, Koh LF, Perkins ND (2010) Regulation of cyclin D1 gene expression. Biochem Soc Trans 38(Pt 1):217–222
Woelfle U et al (2003) Molecular signature associated with bone marrow micrometastasis in human breast cancer. Cancer Res 63(18):5679–5684
Wu Y, Zhou BP (2009) Inflammation: a driving force speeds cancer metastasis. Cell Cycle 8(20):3267–3273
Wu Y et al (2002) Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res 62(4):1030–1035
Wu GQ et al (2009) Cell cycle-related kinase supports ovarian carcinoma cell proliferation via regulation of cyclin D1 and is a predictor of outcome in patients with ovarian carcinoma. Int J Cancer 125(11):2631–2642
Yamamoto S et al (2009) Aberrant expression of p27(Kip1)-interacting cell-cycle regulatory proteins in ovarian clear cell carcinomas and their precursors with special consideration of two distinct multistage clear cell carcinogenetic pathways. Virchows Arch 455(5):413–422
Zhou Q, He Q, Liang LJ (2003) Expression of p27, cyclin E and cyclin A in hepatocellular carcinoma and its clinical significance. World J Gastroenterol 9(11):2450–2454
Zou Y et al (2008) Forkhead box transcription factor FOXO3a suppresses estrogen-dependent breast cancer cell proliferation and tumorigenesis. Breast Cancer Res 10(1):R21
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Cools-Lartigue, J., Spicer, J. (2013). Cell Cycle Control and Growth Factor Systems in Metastasis. In: Burnier, J., Burnier, Jr., M. (eds) Experimental and Clinical Metastasis. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-3685-0_7
Download citation
DOI: https://doi.org/10.1007/978-1-4614-3685-0_7
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-3684-3
Online ISBN: 978-1-4614-3685-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)