
Abstract
Tumor formation is not a cell autonomous phenomenon, but rather an evolution of disease within and responding to the host environment. In particular, metastatic spread from a primary tumor results from a complex interplay between tumor cells and the host. In order to form successful metastases, tumor cells must escape the primary tumor, enter the host vasculature, travel to and arrest in a distant tissue and survive and grow in that new organ. Cells that progress through these stages must both escape and exploit host systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Tumor formation is not a cell autonomous phenomenon, but rather an evolution of disease within and responding to the host environment. In particular, metastatic spread from a primary tumor results from a complex interplay between tumor cells and the host. In order to form successful metastases, tumor cells must escape the primary tumor, enter the host vasculature, travel to and arrest in a distant tissue and survive and grow in that new organ (Chambers et al. 2002). Cells that progress through these stages must both escape and exploit host systems.
As tumor cells acquire a metastatic phenotype, they do so through interacting with and manipulating host responses (Brooks et al. 2010; Borsig 2008; Lorusso and Ruegg 2008). The tissue microenvironment is significantly altered by the presence of a primary tumor, with changes in stromal cell composition and activation and the presence of infiltrating immune cells . The individual components are specific to tumor type, but the net result is a cycle of mutual stimulation of host and tumor tissue, leading to increased tumor growth and aggressive behavior. For example, in melanoma , direct contact between keratinocytes and melanocytes is essential to maintain normal melanocyte growth characteristics. This contact is maintained by E-cadherin , which is often down-regulated as a first step toward melanoma tumorogenesis (Li et al. 2003). Interestingly, hepatocyte growth factor/scatter factor (HGF/SF) production by fibroblasts is capable of stimulating growth and reducing E-Cadherin expression in normal melanocytes resulting in decreased adhesion to keratinocytes . These normal melanocytes begin to express basic fibroblast growth factor (bFGF) , platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and transforming growth factor-β (TGFβ)—growth factor signals then expanded by neighboring fibroblasts which express insulin-like growth factor-1 (IGF-1), HGF/SF, bFGF and TGFβ in response, further stimulating the melanocytes . Therefore, the initial transformation of melanocytes does not necessarily involve major genetic changes, but is a result of losing contact and regulation from keratinocytes , leading to a cycle of mutual positive feedback between stromal cells and melanocytes (reviewed in (Li et al. 2003)). In breast cancer, a loss of tissue organization and polarity is also seen, with increasing disorganization and decreased cell-cell contact as tumor invasiveness increases (Weaver et al. 1997) (reviewed in (Takeichi 1993)).
After initial tumorigenesis , host systems and pathways are further co-opted by tumor cells. Herein we will focus on how particular tumor types are capable of exploiting host cells, growth factors, pathways and systems during each of the key steps in metastasis.
2 Tumor Cell Invasion and Intravasation
Excessive proliferation of neoplastic cells in a developing cancer leads to hypoxia and necrosis in the tumor microenvironment . Tumor and stromal cells react by secreting growth factors and cytokines such as colony stimulating factor (CSF)-1 and TGF-β , which are chemoattractants for immune cells (Robinson and Coussens 2005). Further host reaction to the developing neoplasm leads to recruitment of mesenchymal stem cells, activated fibroblasts, endothelial precursors, dendritic cells, macrophages, monocytes, lymphocytes, leukocytes and mast cells (Olumi et al. 1999; Le Bitoux and Stamenkovic 2008). Initially, it is likely that this recruitment is a host defense mechanism, but the tumor is able to capitalize on the pro-growth factors and counteract the growth-inhibitory capabilities of the recruited cells (Le Bitoux and Stamenkovic 2008). It would be expected that an abundance of immune cells would be beneficial for the host, yet it often correlates with poor clinical prognosis (Nonomura et al. 2007; Taskinen et al. 2008), a global indicator of a tumor’s ability to subvert the host response .
A major effect of the inflammatory response to tumor development is an increase in tumor invasiveness. Breast cancer cells cultured in macrophage-conditioned media, or co-cultured with macrophages, show a significant increase in invasive behavior in vitro (Wu et al. 2009; Hagemann et al. 2005). This increase was found to be due to nuclear factor kappa B (NF-κB)-mediated stabilization of Snail, a major transcription factor for epithelial—mesenchymal transition (EMT) induction (Nieto 2002). Snail expression by tumor cells conferred metastatic ability to non-metastatic cell lines (MCF7 and T47D) and shRNA knockdown of Snail suppressed both innate and ‘inflammation-enhanced’ invasion and metastasis of MDA-MB-231 and MDA-MB-435 cells (Wu et al. 2009). Additionally, tumor necrosis factor-α (TNFα) produced by macrophages was found to induce expression of macrophage migration inhibiting factor (MIF) in tumor cells, which led to increased matrix metalloproteinase (MMP) production by macrophages, also through stimulation of NF-κB. This increase in MMP activity was found to aid tumor cell invasion (Hagemann et al. 2005). These results indicate that tumor cells can capitalize on the host immune response leading to increased invasiveness and subsequent metastasis.
Tumor-associated macrophages (TAMs) are often the most common immune cell in the tumor microenvironment and play an essential role in tumor metastasis. Using an in vivo model of mammary carcinoma , it was found that TAMs are most likely to be found at the margin of a primary tumor, with decreasing numbers upon imaging deeper into the tumor (Wyckoff et al. 2007). The few TAMs that were found in the tumor core were associated with blood vessels and were essential for tumor cell intravasation (Fig. 2.1a). There was a significant correlation between the number of perivascular TAMs and the number of circulating tumor cells in this rat model of mammary carcinoma. Additionally, time-lapse imaging was only able to detect tumor cell intravasation at the site of TAM association with the vasculature, and this intravasation was found to be dependent on epidermal growth factor (EGF)-CSF-1 signaling (Wyckoff et al. 2007; Wyckoff et al. 2004; Goswami et al. 2005). Further, deletion of CSF-1 in a murine model of mammary carcinogenesis showed limited tumor invasion coupled with decreased angiogenesis, resulting in abrogation of lung metastasis due to deficient macrophage recruitment to the tumor microenvironment (Lin and Pollard 2004). Analysis of murine and clinical samples found that TAMs may guide breast cancer cells toward blood vessels through EGF-CSF-1 signaling, as cancer cells were often found in contact with perivascular macrophages. The density of these interactions in clinical samples correlated with the histological grade of the tumor and positively associated with the risk of distant metastasis formation (Robinson et al. 2009). It has also been noted that macrophages are often present at the site of basement membrane breach and tumor cell dissemination (Pollard 2004).

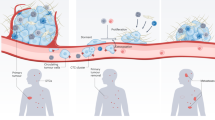
Interaction between metastatic tumor cells and the host environment in early stages of metastasis. a A primary tumor is infiltrated with host-derived macrophages and fibroblasts that aid in tumor cell invasion and intravasation. Upon arrest in a secondary site, tumor cells often stimulate formation of a thrombus b, which provides adhesion contacts and protection from the host immune system. These arrested cells may undergo apoptosis due to release of nitric oxide from the vascular endothelium c or may extravasate, often with assistance from a host macrophage d. Not all metastatic cells extravasate prior to initiating growth in a secondary organ, and intravascular micrometastases are found e, especially in the lung. Extravascular micrometastatic growths f are also common, and often found to be associated with host macrophages. The site of metastatic growth is dependent on many factors, but formation of a pre-metastatic niche g is thought to direct and aid initial growth and survival of metastatic cells
Neutrophils, lymphocytes and TAMs all express and secrete MMPs, which collectively can degrade every extracellular matrix (ECM) protein. The association of these immune cells with the invasive border of a tumor leads to a degradation of the physical barrier that prevents tumor cell dissemination . This degradation releases and activates many growth factors (TGFβ, TNFα, Fas Ligand, heparin bound–epidermal growth factor and others) that are normally sequestered in the ECM (Ii et al. 2006; Hynes 2009). Additionally, the degradation products of many ECM proteins have their own activity. For example, degradation of laminin results in peptides that mimic epidermal growth factor receptor (EGFR) ligands and can result in increased cell migration and invasion in EGFR positive cells (Giannelli et al. 1997; Pirila 2003). It is understood that a tumor is not a uniformly organized mass—each tumor cell will have differential access to nutrients, oxygen and tumor stromal components depending on its individual location (Kedrin 2008). Direct imaging of murine mammary tumor growth using a mammary window was able to visualize individual cells longitudinally and evaluate differences in their behavior depending on their initial location. It was found that those cells in close proximity to blood vessels showed increased migration and invasion and were more likely to spread from the primary tumor to the lung than those cells that did not have immediate access to the vasculature (Kedrin 2008).
Immune cells are a key component of tumor stroma , but the most abundant stromal cell is the carcinoma associated fibroblast (CAF) (Orimo and Weinberg 2006) (Fig. 2.1a), which is also associated with an increase in tumor cell invasion . These fibroblasts have been recruited as normal fibroblasts and are activated to become myofibroblasts, or have been recruited as bone marrow derived cells (BMDCs) and differentiate into fibroblasts at the tumor site (Direkze et al. 2004). Using a 3D in vitro model of the epidermal/dermal microenvironment, it was found that invasion of squamous cell carcinoma (SCC) cells always followed a leading CAF (Gaggioli et al. 2007). This leading fibroblast was able to create a track in the matrigel matrix through both protease- and force-mediated remodeling that the SCC cells would follow. The track was found to be necessary and sufficient for SCC cell invasion as removal of the fibroblasts after track formation still allowed SCC cells to invade. These SCC cells have not undergone an epithelial—mesenchymal transition (EMT) and are non-invasive. It had been questioned how tumors that maintained an epithelial phenotype were able to intravasate; this work illustrates that those tumor cells that are not invasive are able to co-opt host cells in order to metastasize (Gaggioli et al. 2007).
During melanoma development, melanocytes lose expression of E-cadherin thereby losing regulatory contact with keratinocytes , and gain expression of N-cadherin and melanoma cell adhesion molecule (MCAM) which mediate adhesion between melanoma cells and fibroblasts, vascular endothelial cells and other melanoma cells (Hsu et al. 2002; Li et al. 2001). Signaling between melanoma cells, which produce PDGF, bFGF and TGFβ, and fibroblasts which produce IGF-1, HGF/SF , bFGF and TGFβ, results in increased melanoma tumor growth and invasiveness (Li et al. 2003; Hsu et al. 2002; Lee and Herlyn 2007). It has also been shown that TGFβ expression decreases E-cadherin expression, up-regulates β1 and β3 integrin expression and increases MMP-9 activity leading to increased migration (Janji et al. 1999) and enhanced adhesion of melanoma cells to the endothelium (Teti 1997). Re-expresion of E-cadherin in melanoma cells led to reduced invasion in vitro and tumorigenicitiy in vivo (Hsu et al. 2000). Additionally, during the transition of melanomas from the radial growth phase (RGP, flat, non-invasive tumor) to vertical growth phase (VGP, invasive growth), significant matrix remodeling is required; the majority of the enzymes and MMPs utilized are contributed by host fibroblasts and TAMs (Liotta and Kohn 2001).
Components of the host coagulation system are also involved in regulating tumor cell invasiveness. Tissue Factor (TF) is consistently upregulated in many human malignancies and is found to contribute to many facets of tumor aggressiveness (Rak et al. 2009). TF is expressed by tumor cells, often at high levels, but also by many host cells such as endothelial cells, TAMs and CAFs. The main function of TF is to activate thrombin which potentiates clot formation, but thrombin is also essential for activating protease activated receptor (PAR)-1 and -2. Activation of PAR-1 expressed by tumor cells leads to increased tumor invasion and metastasis through induction of proteases and cell adhesion molecules (Melnikova and Bar-Eli 2009).
3 Survival and Arrest in the Vasculature
The host coagulation system is known to play a significant role in tumor cell arrest and survival in the vasculature. Tumor cells activate or produce many components of the coagulation cascade such as thrombin, PAR-1, TF, fibrinogen, von Willebrand factor, and platelet-activating factor (PAF), leading to a ‘platelet mimicry’ phenotype (Timar et al. 2005). The hypoxic environment increases TF expression by endothelial cells , TAMs and CAFs leading to thrombin production within the primary tumor. This ‘pre-treatment’ with thrombin increases tumor cell adhesion to platelets and the vascular endothelium following tumor cell intravasation (Nierodzik and Karpatkin 2006).
Through expression of TF, tumor cells are able to exploit the host coagulation system to increase metastatic efficiency. In an elegant series of papers, Palumbo et al. (Palumbo et al. 2000; Palumbo et al. 2002; Palumbo et al. 2005; Palumbo et al. 2007; Palumbo et al. 2008) evaluated the interplay between metastatic cells and the individual components of coagulation. They found that loss of host fibrinogen significantly decreased lung metastasis formation, yet had no impact on the number of cells that originally arrested in the lung following experimental metastasis cell injection. However, fibrinogen was essential for sustained adherence of tumor cells in the lung vasculature (Palumbo et al. 2000). This role for fibrinogen also held true in a spontaneous model of metastasis, with reduced lung metastasis despite equivalent primary tumor formation in fibrinogen-null and wild type animals (Palumbo et al. 2002). Evaluation of metastasis in animals with activation-resistant platelets (platelets present in normal number, but not able to be activated by thrombin, adenosine diphosphate (ADP), or other coagulation stimulants) showed a significant decrease in experimental and spontaneous metastasis, again due to reduced survival or retention in the lung vasculature (Palumbo et al. 2005). Depletion of circulating natural killer (NK) immune cells prior to metastatic cell introduction resulted in equivalent metastasis number in platelet mutant, fibrinogen knock-out and wild type animals, indicating that platelet- and fibrinogen-mediated thrombus formation protects tumor cells from NK cell surveillance in the lung vasculature (Palumbo et al. 2005). The role of NK-mediated cell killing was strengthened through work on Factor XIII, which stabilizes fibrin and other ECM matricies through catalysis of crosslinkages. FXIII was found to be essential in preventing NK cell immunosurveillance of tumor cells (Palumbo et al. 2008).
To specifically evaluate the role of TF and TF signaling in metastasis, tumor cells were derived from TF knock-out animals and cell lines with and without TF, or TF lacking the cytoplasmic tail responsible for cell signaling. It was found that while TF expression was not essential for primary tumor formation, it was critical for lung metastasis but dependent on functional coagulation in the host. Interestingly, blockage of TF signaling had no effect on metastasis generation (Palumbo and Degen 2007).
The formation of a thrombus at the surface of an arrested tumor cell has also been linked to increased metastasis through maintenance of cell adherence in the pulmonary vasculature (Fig. 2.1b)(Borsig 2008; Kim et al. 1998; Im et al. 2004). Metastatic cells protected in a fibrin clot were able to change from a rounded morphology and spread along the inside of a vessel. Those cells that showed stable adherence to the lung vasculature were able to form significantly more lung metastases than those prevented from spreading through treatment with anticoagulant agents (Im et al. 2004). Treatment of animals with the clot-stabilizing drug aprotinin was found to increase metastasis of B16F10 melanoma cells through prolonging the interaction between tumor cells arrested in the pulmonary vasculature and cell surface thrombi (Kirstein et al. 2009). In accordance with this, prevention of thrombus formation with heparin (Kirstein et al. 2009) or hiruden (Esumi et al. 1991) is linked with reduced pulmonary metastasis due to decreased cell retention in the lung.
Stable adherence of tumor cells to the vasculature upon arrest appears to be a major determinant of metastatic efficiency. Comparison of metastatic and non-metastatic cells injected into the circulation showed no difference in the original number of cells that arrested in the lung, however only those cell lines that had a metastatic phenotype were able to persist and form micrometastases in the lung (Kim et al. 2004). Tumor cell arrest is also influenced by host expression of P-selectin. Platelets isolated from P-selectin knock-out mice were unable to bind to tumor cells in vitro, and experimental metastasis assays found that there was a decrease in the initial seeding of the lung tissue in P-selectin-null animals (Kim et al. 1998). Additionally, P-selectin was found to facilitate tumor cell tethering and rolling along the pulmonary vasculature, but further binding by αIIbβ3 was required to stabilize tumor cell adhesion (McCarty et al. 2000). Integrin α3β1 is also involved in tumor cell adhesion to the vascular endothelium through sections of exposed basement membrane. Adhesion and migration of tumor cells was also stimulated by binding of TF on tumor cells to tissue factor pathway inhibitor −1 on tumor associated vessels which was a surprising consequence of receptor-inhibitor binding (Fischer et al. 1999).
Tumor cell-associated thrombus formation may also increase metastatic cell survival in the vasculature, as activation of PAR-1 by thrombin leads to transmission of survival signals and prevention of apoptosis (Shi et al. 2004). Additionally, many growth and survival factors are released from platelets upon activation and are therefore present within thrombi. Tumor cells are able to bind to the provisional matrix provided by a fibrin clot thereby increasing metastasis (Fig. 2.1b) (Palumbo et al. 2002; Reijerkerk et al. 2000; Dvorak et al. 1995). Further, plasmin-mediated clot dissolution may aid tumor cells with the next step in metastasis—extravasation from the host vasculature.
4 Extravasation and Growth Initiation in Secondary Tissue
Compared with the other steps in metastasis, relatively little is known about tumor cell extravasation at a secondary site. Using cell accounting techniques Luzzi, et al. (Luzzi et al. 1998) found that the majority of B16F1 murine melanoma cells had extravasated from the liver vasculature within 3 days of cell injection (Luzzi et al. 1998). Importantly, very few of these cells went on to form micrometastases (2 %) and even fewer were able to form macrometastases (0.02 %). Two weeks following tumor cell injection, over one-third of injected cells remained in the liver as solitary, extravasated cells and 95 % of those identifiable cells were not apoptotic or proliferating (as determined by histological staining for TUNEL and Ki67), indicating that in the liver, extravasation may not be an essential part of metastatic inefficiency. Additionally, using the chick chorioallantoic membrane (CAM) found that nearly all B16F1 cells were able to survive and extravasate following arrest. Tissue inhibitor of metalloproteinases-1 (TIMP-1) overexpressing B16F1 cells were poorly metastatic, and yet they were still able to successfully extravasate in the chick CAM model (Koop et al. 1995). Using ras-transformed and control fibroblasts, it was also found that extravasation was independent of metastatic ability (Koop et al. 1996). Nearly all ras-transformed fibroblasts and control fibroblasts (89 and 96 %, respectively) had extravasated from the chick CAM within 24 h of initial injection. Additionally, migration of both cell types within the mesenchymal layer was equivalent, despite having differential invasion capabilities in vitro (Koop et al. 1996).
Direct visualization of tumor cell extravasation was performed recently in a murine model of brain metastasis (Kienast et al. 2010). Using a cranial window, single cancer cells were visualized throughout arrest and extravasation . MDA-MD-435 cells were found to arrest in microvessel branch points and extravasate as single cells. These cells began to proliferate only after successful extravasation and only when extravasated cells maintained contact with an abluminal endothelial cell of a brain capillary (Kienast et al. 2010).
Study of metastasis to the lung vasculature shows a distinct difference from that seen in the liver and chick CAM, however. Using the 4T1 murine mammary carcinoma cell line it was found that these cells arrest in the lung as individuals attached to the vascular endothelium. The cells were able to form small colonies within three weeks, some entirely maintained within the vasculature. Following growth initiation, the colonies were able to extravasate as micro or macrometastases (Wong et al. 2002). Further to this, fewer than 2 % of HT1080 cells had extravasated from the lung vasculature within 24 h of tumor cell injection, and were found to form colonies within the lung vasculature within three days. These colonies showed tumor cells that projected outwards from the central focus as ‘strings’ following within the capillaries (Fig. 2.1e) (Al-Mehdi et al. 2000). Analysis of experimental metastasis of B16F10 melanoma cells in the mouse lung found that the majority of injected cells had extravasated, with no identifiable clusters or single cells within the pulmonary vasculature within 4 days of injection (Cameron et al. 2000). Using an orthotopic prostate cancer model, however, the majority of metastatic tumor cells and tumor cell clusters were found within the vasculature of both the liver and the lung (Zhang et al. 2010). Taken together, these data indicate that the role of extravasation in successful metastasis formation may be specific to the model, cell type and secondary organ of study.
Recent work has found a subset of macrophages (CD11b + Gr−) recruited to tumor cells arrested in the lung aids in tumor cell extravasation (Qian 2009). The timing of tumor cell extravasation was directly linked to macrophage recruitment, as depletion of macrophages at various times following tumor cell injection resulted in either reduced metastasis number and size, if macrophages were depleted prior to tumor cell injection, or equivalent number of metastases with reduced size, if macrophages were depleted after successful seeding of the lung with PyMT induced or MDA-MB-231 tumor cells. Ex vivo imaging of intact lung tissue at various times following tumor cell injection found that macrophages associated with tumor cells in the vasculature and increased extravasation (Fig. 2.1d). Five minutes after cell injection all tumor cells were retained in the lung vasculature. Within 24 h, there was a significant increase in macrophage association with arrested tumor cells, and ~75 % of tumor cells were outside of a vessel, and within 3 days no cells were found completely within a vessel. Some of the extravasated cells had begun to proliferate as several colonies were visualized, and these colonies showed extensive macrophage association (Fig. 2.1f). Macrophage depletion reduced tumor cell extravasation from 75–25 % within 24 h and within 48 h many fewer cells had survived in the lung. These studies found that macrophage recruitment to the lung promoted tumor cell extravasation and survival (Qian et al. 2009).
It is known that arrest of tumor cells is associated with the formation of a fibrin clot at the arrested cell site. These clots do not persist indefinitely—clot dissolution is mediated by the powerful protease plasmin (Reijerkerk et al. 2000). This clot breakdown may aid tumor cell extravasation through activation of MMPs and other proteinases. Tumor cells that express high amounts of urokinase type plasminogen activator (uPA) tend to be more aggressive and metastatic (reviewed in (Kramer et al. 1994)). Clinically, high levels of uPA, uPA receptor (uPAR), plasminogen activator inhibitor (PAI)-1 and PAI-2 is linked to poor prognosis and increased metastasis development (Duffy et al. 2008; Harbeck et al. 2004).
The site of metastatic cell arrest and growth has been debated for some time—from Stephen Paget’s theory of ‘seed and soil’ whereby the tumor cell (seed) must arrest in a permissible secondary tissue (soil) in order to develop into a tumor (Chambers et al. 2002; Ribatti et al. 2006). This century-old theory still has merit as metastatic cells grow in different tissues depending on the tumor type they originated from. A type of hospitable ‘soil’ has been identified as a pre-metastatic niche. These regions of secondary tissue show recruitment of clusters of BMDCs prior to the arrival of tumor cells. It is thought that primary tumor and tumor stromal secretion of chemokines direct the migration of these cells, as in vivo injection of media conditioned by melanoma cells led to similar recruitment and pattern of metastasis as the presence of a melanoma primary tumor (Kaplan et al. 2005). The primary tumor stimulates pre-metastatic niche formation through secretion of VEGF and placental growth factor (PlGF), which recruit VEGF receptor 1 (VEGFR1)-positive cells. PlGF in particular increases the proliferation of fibroblast-like cells and stimulates their production of fibronectin (Ruzinova 2003).
BMDCs expressing VEGFR1 and α 4β1 arrest in regions of increased fibronectin synthesis by fibroblasts and fibroblast-like cells. These arrested BMDCs secrete MMP-9 which may degrade the basement membrane to allow extravasation of more BMDCs and/or metastatic cells. They are also found to express Id3, which is involved in proliferation and mobilization of hematopoietic progenitor cells (HPCs) from the bone marrow and maintains an activated state within the BMDC clusters. These clusters alter the local microenvironment and activate integrins and chemokines such as stromal derived factor –1 (SDF-1). This activation leads to further recruitment of BMDCs and increased attachment, survival, and growth of tumor cells (Fig. 2.1g) (Kaplan et al. 2005).
Pre-metastatic niche formation can also be directed by platelet aggregation (Massberg et al. 2006). At a site of endothelium disruption, platelet activation was essential for recruitment of BMDCs, which adhere to P-selectin and α IIbβ3 on the platelet surface, rather than to exposed ECM. Additionally, SDF-1 released from platelets leads to ongoing retention of BMDC and tumor cell arrest .
5 Angiogenesis and Sustained Growth
Sustained primary tumor and metastatic growth beyond ~1mm3 requires the recruitment of a blood supply (Folkman 1995). Vascularization of tumors promotes growth by providing oxygen and nutrients and increases metastasis by providing an entry point into the circulation. Normal tissues undergo angiogenesis during development, wound healing and tissue regeneration, through a tightly regulated system leading to structured, hierarchical branching of vessels (Carmeliet 2005). Tumor vascularization is characterized by highly tortuous dysfunctional vessels due to improper regulation of angiogenesis (McDonald and Choyke 2003).
High levels of VEGF-A in the tumor microenvironment expressed by tumor cells, macrophages (Barbera-Guillem et al. 2002; Harmey et al. 1998; Evans 1992), neutrophils (McCourt et al. 1999), platelets (McCabe et al. 2006), fibroblasts (Hlatky et al. 1994) and endothelial cells (Nilsson et al. 2004) tips the balance of pro- and anti-angiogenic factors and leads to activation of angiogenesis. VEGF-A is elevated in response to hypoxia and inflammation , which are common in during tumor formation. Solid tumors tend to have a hypoxic core due to poorly functioning vasculature leading to constant stimulation of pro-angiogenic factors such as VEGF-A (Byrne et al. 2005).
The initial reaction to high levels of VEGF-A is destabilization of existing blood vessels. The number of endothelial cell interactions with stabilizing mural cells decreases, leading to leaky, dialated vessels (Carmeliet und Jain 2000; Bach et al. 2007). This destabilization is mediated by angiopoietin-2 (Ang-2) (Tait and Jones 2004; Maisonpierre et al. 1997), partly through up-regulation of MMP-1 and -9 (Etoh et al. 2001) and is found to increase tumor cell entry into the vasculature (Chung et al. 2006; Nakayama et al. 2005).
Following vessel destabilization, endothelial tip cells begin sprouting through tightly regulated signaling between Delta-like 4 (Dll4) and Notch (Suchting et al. 2007) which may be de-regulated in the tumor microenvironment resulting in incomplete vascular remodeling (Noguera-Troise 2006). Tip cells lead the migration of endothelial cells following chemotactic signals, especially VEGF-A. HPCs and endothelial progenitor cells (EPCs) are also recruited by VEGF-A signaling to further increase vessel growth (Hattori et al. 2001). In order for proper vascular function, vessel growth is followed by vasculature stabilization by pericytes due to PDGF and Ang-1 signaling (Abramsson et al. 2003; Milner et al. 2009). Ang-1 and PDGF can be overexpressed in the tumor microenvironment (Tait and Jones 2004; Nakamura 2008) yet tumor vessels are poorly stabilized, showing leakiness and poor pericyte coverage. This indicates that the balance of pro-angiogenic, destabilizing factors such as VEGF-A are present in higher functional concentration than Ang-1 and PDGF (Hall and Ran 2010). Recent work has also linked MMP-14 and TGFβ to vascular stability, as MMP inhibition was found to increase vascular leakiness due to reduced activation of TGFβ present in the ECM. TGFβ was found to signal through ALK5 to control leakage of small (10 kDa dextran) and large (70 kDa dextran) molecules in tumor and normal tissue though control of vasodilation and venular openings (Sounni et al. 2010). These data indicates that TGFβ in the ECM can modulate host response depending on its bioavailability, and its release by MMP activity in the tumor stroma may increase tumor cell extravasation through increased vascular leakiness.
Deregulated angiogenesis in a tumor is due to an imbalance between pro- and anti-angiogenic factors in the tumor microenvironment. The over-expression of pro-angiogenic factors VEGF-A, Ang-2, bFGF and TGFβ leads to constant stimulation of angiogenesis and a reduction in stabilized vessels. This leads to poor tissue perfusion, high vasculature permeability and chronic inflammation and an increase in metastasis due to ease of metastatic cell entry into the vasculature (Hall and Ran 2010).
The process of angiogenesis in the metastatic setting is thought to proceed through a similar path as seen in the primary tumor. Initial growth of a micrometastasis is halted without the recruitment of a blood supply, leading to a functionally dormant metastasis with balanced levels of proliferation and apoptosis (Naumov et al. 2008). Tumor cells and macrophages present at the metastatic site stimulate expression of VEGF-A leading to the same cascade of angiogenic events as seen in the primary tumor setting.
Blood clot formation at the metastatic site provides further angiogenic and growth signals as platelet activation results in the release of many growth and pro-angiogenic factors such as VEGF, PDGF, Ang-1, TGFβ, IGF-1, EGF, and platelet-derived epidermal growth factor (PD-EGF). Additionally, thrombin activity is linked to increased angiogenesis through up-regulation of cathepsin-D which increases endothelial cell growth, migration and tube formation in vitro (Hu et al. 2008). Thrombin may also play an important role in angiogenesis through induction of VEGF-A in tumor cells (Huang et al. 2002) and platelets (Mohle et al. 1997), as well as Ang-1 and -2 from platelets (Li et al. 2001) and endothelial cells (Huang et al. 2002) respectively.
6 Host-Mediated Inhibition of Metastasis
Successful metastasis formation results when tumor cells are able to exploit and avoid natural host defenses. Yet metastasis is an exceptionally inefficient process, (Chambers et al. 2002) indicating that the host is capable of preventing progression of the majority of metastatic cells. The mechanisms behind this prevention are largely unknown, yet several interesting examples of host triumph over tumor have been established.
Following tumor cell arrest in the liver vasculature, nitric oxide (NO) is released and induces apoptosis in B16F1 cells (Wang et al. 2000). B16F1 cell arrest in the pulmonary vasculature was also found to lead to an eNOS-dependent release of NO. NO may represent a natural host defense mechanism as it triggers apoptosis in melanoma cells and reduced the growth of metastatic tumors (Fig. 2.1c) (Qiu et al. 2003). Accordingly, comparison of metastatic (isolated from VGP tumor) and non-metastatic (isolated from RGP tumor) melanoma cells following arrest in the murine lung showed that non-metastatic cells were unable to survive in the pulmonary vasculature. Within 8 h of tumor cell injection, non-metastatic cells had apoptosed and were cleared from the lung, whereas metastatic cells persisted and were able to form metastatic colonies within 7 days (Kim et al. 2004).
Given the extensive interaction between tumors and the host, there is the potential to alter the microenvironment to create an anti-tumor rather than pro-tumor interface. It has been proposed that the large number of TAMs present in tumor stroma could be ‘re-educated’ to target tumor cells (Hagemann et al. 2008). Using NF-κB signaling, tumor cells are able to keep TAMs in an immunosuppressive state. By introducing a dominant negative inhibitor of nuclear factor kappa B kinase β (IKKβ) into bone marrow derived macrophages, TAMs became tumoricidal through release of NO and through promotion of NK cell-mediated killing (Hagemann et al. 2008). The extensive interaction between TAMs and metastatic cells throughout invasion and extravasation as discussed earlier illustrates the great potential for manipulation of TAM activity to reduce tumor progression.
Normal tissue structure and function is maintained through proper ECM adhesion and tissue polarity. In breast and melanoma tumor development, dysregulation of cell adhesion represents an initiating step in tumor formation (Li et al. 2003; Takeichi 1993). Therefore the effect of re-establishing proper tissue architecture and adhesion in tumor tissues has been investigated (Bissell et al. 2002). It was found that restoration of proper integrin signaling within a 3D culture setting led to phenotypic reversion of breast cancer cells. Without alterations to tumor cell genotype, tumor cells were induced to form normal breast structures. Metastatic breast cancer cells could also be reverted to a non-malignant phenotype in 3D culture following treatment with anti-integrin antibodies (Wang et al. 2002). The global switch in cellular behavior as a direct result of modulation of environmental interaction indicates the powerful role that the tumor stroma and microenvironment has on tumor development and progression and illustrates that many treatment options are available beyond direct targeting of tumor tissue.
7 Summary
The study of tumor biology and metastasis has long been investigated from the perspective of the individual tumor cell. However, the importance of tumor cell interaction with host cells and systems has also been recognized. Tumor cells are unable to form metastases without interaction with many microenvironments—from the primary tumor stroma , through the host vasculature and host coagulation systems, to an entirely new environment in a secondary organ. The metastatic cell’s ability to survive and proliferate in each of these new environments depends on the ability to influence and often exploit the host. Fundamental to this is the interaction of tumor cells with the host coagulation pathway and avoidance of the host immune system. These two major systems exist to maintain tissue homeostasis and health though elimination of non-normal cells, yet tumor cells are able to circumvent these host responses and turn them from anti-tumor to pro-metastatic. Full understanding of the interplay between tumor progression and host responses is essential for understanding metastatic disease and successful patient treatment.
References
Abramsson A, Lindblom P, Betsholtz C (2003) Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest 112:1142–1151
Al-Mehdi AB et al (2000) Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med 6:100–102
Bach F, Uddin FJ, Burke D (2007) Angiopoietins in malignancy. Eur J Surg Oncol 33:7–15
Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW (2002) Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res 62:7042–7049
Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW (2002) The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 70:537–546
Borsig L (2008) The role of platelet activation in tumor metastasis. Expert Rev Anticancer Ther 8:1247–1255
Brooks SA, Lomax-Browne HJ, Carter TM, Kinch CE, Hall DM (2010) Molecular interactions in cancer cell metastasis. Acta Histochem 112:3–25
Byrne AM, Bouchier-Hayes DJ, Harmey JH (2005) Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF). J Cell Mol Med 9:777–794
Cameron MD et al (2000) Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res 60:2541–2546
Carmeliet P (2005) VEGF as a key mediator of angiogenesis in cancer. Oncology 69(Suppl 3):4–10
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407:249–257
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2:563–572
Chung YC, Hou YC, Chang CN, Hseu TH (2006) Expression and prognostic significance of angiopoietin in colorectal carcinoma. J Surg Oncol 94:631–638
Direkze NC et al (2004) Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res 64:8492–8495
Duffy MJ, McGowan PM, Gallagher WM (2008) Cancer invasion and metastasis: changing views. J Pathol 214:283–293
Dvorak HF, Brown LF, Detmar M, Dvorak AM (1995) Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 146:1029–1039
Esumi N, Fan D, Fidler IJ (1991) Inhibition of murine melanoma experimental metastasis by recombinant desulfatohirudin, a highly specific thrombin inhibitor. Cancer Res 51:4549–4556
Etoh T et al (2001) Angiopoietin-2 is related to tumor angiogenesis in gastric carcinoma: possible in vivo regulation via induction of proteases. Cancer Res 61:2145–2153
Evans R et al (1992) Tumor cell IL-6 gene expression is regulated by IL-1 alpha/beta and TNF alpha: proposed feedback mechanisms induced by the interaction of tumor cells and macrophages. J Leukoc Biol 52:463–468
Fischer EG et al (1999) Tumor cell adhesion and migration supported by interaction of a receptor-protease complex with its inhibitor. J Clin Invest 104:1213–1221
Folkman J (1995) Angiogenesis inhibitors generated by tumors. Mol Med 1:120–122
Gaggioli C et al (2007) Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9:1392–1400
Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V (1997) Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science 277:225–228
Goswami S et al (2005) Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res 65:5278–5283
Hagemann T et al (2005) Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J Immunol 175:1197–1205
Hagemann T et al. (2008) “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med 205:1261–1268
Hall K, Ran S (2010) Regulation of tumor angiogenesis by the local environment. Front Biosci 15:195–212
Harbeck N et al (2004) Urokinase-type plasminogen activator (uPA) and its inhibitor PAI-I: novel tumor-derived factors with a high prognostic and predictive impact in breast cancer. Thromb Haemost 91:450–456
Harmey JH, Dimitriadis E, Kay E, Redmond HP, Bouchier-Hayes D (1998) Regulation of macrophage production of vascular endothelial growth factor (VEGF) by hypoxia and transforming growth factor beta-1. Ann Surg Oncol 5:271–278
Hattori K et al (2001) Vascular endothelial growth factor and angiopoietin-1 stimulate postnatal hematopoiesis by recruitment of vasculogenic and hematopoietic stem cells. J Exp Med 193:1005–1014
Hlatky L, Tsionou C, Hahnfeldt P, Coleman CN (1994) Mammary fibroblasts may influence breast tumor angiogenesis via hypoxia-induced vascular endothelial growth factor up-regulation and protein expression. Cancer Res 54:6083–6086
Hsu MY et al (2000) E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am J Pathol 156:1515–1525
Hsu MY, Meier F, Herlyn M (2002) Melanoma development and progression: a conspiracy between tumor and host. Differentiation 70:522–536
Hu L, Roth JM, Brooks P, Luty J, Karpatkin S (2008) Thrombin up-regulates cathepsin D which enhances angiogenesis, growth, and metastasis. Cancer Res 68:4666–4673
Huang YQ, Li JJ, Hu L, Lee M, Karpatkin S (2002) Thrombin induces increased expression and secretion of angiopoietin-2 from human umbilical vein endothelial cells. Blood 99:1646–1650
Hynes RO (2009) The extracellular matrix: not just pretty fibrils. Science 326:1216–1219
Ii M, Yamamoto H, Adachi Y, Maruyama Y, Shinomura Y (2006) Role of matrix metalloproteinase-7 (matrilysin) in human cancer invasion, apoptosis, growth, and angiogenesis. Exp Biol Med (Maywood) 231:20–27
Im JH et al (2004) Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res 64:8613–8619
Janji B, Melchior C, Gouon V, Vallar L, Kieffer N (1999) Autocrine TGF-beta-regulated expression of adhesion receptors and integrin-linked kinase in HT-144 melanoma cells correlates with their metastatic phenotype. Int J Cancer 83:255–262
Kaplan RN et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438:820–827
Kedrin D et al (2008) Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods 5:1019–1021
Kienast Y et al (2010) Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 16:116–122
Kim JW et al (2004) Rapid apoptosis in the pulmonary vasculature distinguishes non-metastatic from metastatic melanoma cells. Cancer Lett 213:203–212
Kim YJ, Borsig L, Varki NM, Varki A (1998) P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci U S A 95:9325–9330
Kirstein JM et al (2009) Effect of anti-fibrinolytic therapy on experimental melanoma metastasis. Clin Exp Metastasis 26:121–131
Koop S et al (1995) Fate of melanoma cells entering the microcirculation: over 80 % survive and extravasate. Cancer Res 55:2520–2523
Koop S et al (1996) Independence of metastatic ability and extravasation: metastatic ras-transformed and control fibroblasts extravasate equally well. Proc Natl Acad Sci U S A 93:11080–11084
Kramer MD, Reinartz J, Brunner G, Schirrmacher V (1994) Plasmin in pericellular proteolysis and cellular invasion. Invasion Metastasis 14:210–222
Le Bitoux MA, Stamenkovic I (2008) Tumor-host interactions: the role of inflammation. Histochem Cell Biol 130:1079–1090
Lee JT, Herlyn M (2007) Microenvironmental influences in melanoma progression. J Cell Biochem 101:862–872
Li G. et al. (2003) Function and regulation of melanoma-stromal fibroblast interactions: when seeds meet soil. Oncogene 22:3162–3171
Li G, Satyamoorthy K, Herlyn M (2001a) N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res 61:3819–3825
Li JJ, Huang YQ, Basch R, Karpatkin S (2001b) Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost 85:204–206
Lin EY, Pollard JW (2004) Role of infiltrated leucocytes in tumour growth and spread. Br J Cancer 90:2053–2058
Liotta LA, Kohn EC (2001) The microenvironment of the tumour-host interface. Nature 411:375–379
Lorusso G, Ruegg C (2008) The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol 130:1091–1103
Luzzi KJ et al (1998) Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153:865–873
Maisonpierre PC et al (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 277:55–60
Massberg S et al (2006) Platelets secrete stromal cell-derived factor 1alpha and recruit bone marrow-derived progenitor cells to arterial thrombi in vivo. J Exp Med 203:1221–1233
McCabe NP, Selman SH, Jankun J (2006) Vascular endothelial growth factor production in human prostate cancer cells is stimulated by overexpression of platelet 12-lipoxygenase. Prostate 66:779–787
McCarty OJ, Mousa SA, Bray PF, Konstantopoulos K (2000) Immobilized platelets support human colon carcinoma cell tethering, rolling, and firm adhesion under dynamic flow conditions. Blood 96:1789–1797
McCourt M, Wang JH, Sookhai S, Redmond HP (1999) Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch Surg 134:1325–1331; (discussion 1331–2)
McDonald DM, Choyke PL (2003) Imaging of angiogenesis: from microscope to clinic. Nat Med 9:713–725
Melnikova VO, Bar-Eli M (2009) Inflammation and melanoma metastasis. Pigment Cell Melanoma Res 22:257–267
Milner CS, Hansen TM, Singh H, Brindle NP (2009) Roles of the receptor tyrosine kinases Tie1 and Tie2 in mediating the effects of angiopoietin-1 on endothelial permeability and apoptosis. Microvasc Res 77:187–191
Mohle R, Green D, Moore MA, Nachman RL, Rafii S (1997) Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc Natl Acad Sci U S A 94:663–668
Nakamura Y et al (2008) PDGF-BB is a novel prognostic factor in colorectal cancer. Ann Surg Oncol 15:2129–2136
Nakayama T et al (2005) Expression and significance of Tie-1 and Tie-2 receptors, and angiopoietins-1, 2 and 4 in colorectal adenocarcinoma: Immunohistochemical analysis and correlation with clinicopathological factors. World J Gastroenterol 11:964–969
Naumov GN, Folkman J, Straume O, Akslen LA (2008) Tumor-vascular interactions and tumor dormancy. Apmis 116:569–585
Nierodzik ML, Karpatkin S (2006) Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 10:355–362
Nieto MA (2002) The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 3:155–166
Nilsson I, Shibuya M, Wennstrom S (2004) Differential activation of vascular genes by hypoxia in primary endothelial cells. Exp Cell Res 299:476–485
Noguera-Troise I et al (2006) Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444 1032–1037
Nonomura N et al (2007) Decreased number of mast cells infiltrating into needle biopsy specimens leads to a better prognosis of prostate cancer. Br J Cancer 97:952–956
Olumi AF et al (1999) Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 59:5002–5011
Orimo A, Weinberg RA (2006) Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 5:1597–1601
Palumbo JS et al (2000) Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood 96:3302–3309
Palumbo JS et al (2002) Spontaneous hematogenous and lymphatic metastasis, but not primary tumor growth or angiogenesis, is diminished in fibrinogen-deficient mice. Cancer Res 62:6966–6972
Palumbo JS et al (2005) Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 105:178–185
Palumbo JS et al (2007) Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood 110:133–141
Palumbo JS et al (2008) Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J Thromb Haemost 6:812–819
Palumbo JS, Degen JL (2007) Mechanisms linking tumor cell-associated procoagulant function to tumor metastasis. Thromb Res 120(Suppl 2):S22–8
Pirila E et al (2003) Matrix metalloproteinases process the laminin-5 gamma 2-chain and regulate epithelial cell migration. Biochem Biophys Res Commun 303:1012–1017
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78
Qian B et al (2009) A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One 4:e6562
Qiu H et al. (2003) Arrest of B16 melanoma cells in the mouse pulmonary microcirculation induces endothelial nitric oxide synthase-dependent nitric oxide release that is cytotoxic to the tumor cells. Am J Pathol 162:403–412
Rak J, Milsom C, Magnus N, Yu J (2009) Tissue factor in tumour progression. Best Pract Res Clin Haematol 22:71–83
Reijerkerk A, Voest EE, Gebbink MF (2000) No grip, no growth: the conceptual basis of excessive proteolysis in the treatment of cancer. Eur J Cancer 36:1695–1705
Ribatti D, Mangialardi G, Vacca A (2006) Stephen Paget and the ‘seed and soil’ theory of metastatic dissemination. Clin Exp Med 6:145–149
Robinson BD et al (2009) Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res 15:2433–2441
Robinson SC, Coussens LM (2005) Soluble mediators of inflammation during tumor development. Adv Cancer Res 93:159–187
Ruzinova MB et al (2003) Effect of angiogenesis inhibition by Id loss and the contribution of bone-marrow-derived endothelial cells in spontaneous murine tumors. Cancer Cell 4:277–289
Shi X, Gangadharan B, Brass LF, Ruf W, Mueller BM (2004) Protease-activated receptors (PAR1 and PAR2) contribute to tumor cell motility and metastasis. Mol Cancer Res 2:395–402
Sounni NE et al (2010) Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis Model Mech 3:317–332
Suchting S et al (2007) The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci U S A 104:3225–3230
Tait CR, Jones PF (2004) Angiopoietins in tumours: the angiogenic switch. J Pathol 204:1–10
Takeichi M (1993) Cadherins in cancer: implications for invasion and metastasis. Curr Opin Cell Biol 5:806–811
Taskinen M, Karjalainen-Lindsberg ML, Leppa S (2008) Prognostic influence of tumor-infiltrating mast cells in patients with follicular lymphoma treated with rituximab and CHOP. Blood 111:4664–4667
Teti A et al (1997) Transforming growth factor-beta enhances adhesion of melanoma cells to the endothelium in vitro. Int J Cancer 72:1013–1020
Timar J et al (2005) Platelet-mimicry of cancer cells: epiphenomenon with clinical significance. Oncology 69:185–201
Wang F et al (2002) Phenotypic reversion or death of cancer cells by altering signaling pathways in three-dimensional contexts. J Natl Cancer Inst 94:1494–1503
Wang HH et al (2000) B16 melanoma cell arrest in the mouse liver induces nitric oxide release and sinusoidal cytotoxicity: a natural hepatic defense against metastasis. Cancer Res 60:5862–5869
Weaver VM et al. (1997) Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol 137:231–245
Wong CW et al (2002) Intravascular location of breast cancer cells after spontaneous metastasis to the lung. Am J Pathol 161:749–753
Wu Y et al (2009) Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell 15:416–428
Wyckoff J et al (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64:7022–7029
Wyckoff JB et al (2007) Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res 67:2649–2656
Zhang Q. et al (2010) The role of the intravascular microenvironment in spontaneous metastasis development. Int J Cancer 126:2534–2541
Acknowledgements
We would like to thank Kyle MacLean for his assistance with figure design. JMK is the recipient of a Translational Breast Cancer Studentship from the London Regional Cancer Program. AFC is a Cancer Research Chair in Oncology and receives salary support from the Canada Research Chairs Program.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Kirstein, J., Chambers, A. (2013). Interactions of Normal Tissues and Systems with Metastatic Cells: Impact on Location, Survival and Growth. In: Burnier, J., Burnier, Jr., M. (eds) Experimental and Clinical Metastasis. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-3685-0_2
Download citation
DOI: https://doi.org/10.1007/978-1-4614-3685-0_2
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-3684-3
Online ISBN: 978-1-4614-3685-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)