
Abstract
An increasing body of experimental evidence suggests that serotonergic neurons play a major role in the production of levodopa-derived dopamine when dopaminergic neurons have degenerated, and that unregulated release of dopamine from serotonergic neurons is responsible for the appearance of levodopa-induced dyskinesia (LID) in animal models of Parkinson’s disease (PD).
Promising preclinical findings show that the activation of 5-HT1 receptors, induced by the administration of 5-HT1A and/or 5-HT1B receptor agonists, suppressed LID in 6-OHDA-lesioned rat, as well as in MPTP-treated nonhuman primate models of PD, suggesting a possible clinical application. This chapter will provide an overview of these preclinical findings concerning the role of serotonergic neurons and serotonergic receptors in the appearance of LID, with a brief review of relevant clinical studies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
The Serotonergic System in Parkinson’s Disease
Serotonin (5-hydroxytryptamine, 5-HT) is an important modulator of the central nervous system (CNS), and its action is mediated by a large variety of receptor subtypes. Serotonin is synthesized from L-tryptophan by a two-step reaction: the tryptophan hydroxylase enzyme generates 5-hydroxytryptophan (5-HTP), which is then converted to serotonin by the l-amino acid decarboxylase enzyme (AADC). The serotonergic system originates from the raphe nuclei and is one of the most widely distributed, innervating virtually all regions of the CNS and participating in mechanisms of cognition, feeding and satiety, mood and emotion, circadian and sleep-wake cycle regulation, pain, and motor functions [1, 2]. An increasing body of experimental evidence demonstrates the involvement of the serotonergic system in modulating the function of basal ganglia circuits and its interaction with the dopaminergic system. Both serotonergic and dopaminergic systems are affected in neurodegenerative disorders such as Parkinson’s disease (PD) and have been implicated in the pathophysiology of depression and schizophrenia [3]. A role of the serotonergic system in the regulation of motor function is suggested by the dense serotonergic input received by areas such as the striatum, substantia nigra pars reticulata, and globus pallidus [4, 5]. Interestingly, postmortem studies of patients with PD have shown loss of serotonergic markers in the caudate, as well as hypothalamus and frontal cortex [6, 7]. However, as also seen in PET imaging studies, the degree of serotonergic terminal loss appears to be less severe than that affecting the dopaminergic system, and there is no correlation with motor disability, or dyskinesia [8]. On the other hand, it has been suggested that the partial loss of serotonergic innervation may contribute to the development of depression in PD patients [9], although consistent evidence is lacking [5].
Serotonin exerts its actions via specific receptors, which are located in most of the brain, especially in the hippocampus, basal ganglia, and striatum. To date, 14 distinct subtypes of the serotonergic receptors have been identified [10, 11]; among all serotonergic receptors, some have been shown to participate in the regulation of motor function and/or induction of dyskinesia in PD, such as the 5-HT1A, 5-HT1B, 5-HT2A, 5-HT2C, and the 5-HT3 receptors.
5-HT1A receptors are located somato-dendritically in the dorsal raphe nuclei, where they regulate cell firing [12]. 5-HT1A receptors have been also identified postsynaptically in other brain regions, such as the cerebral cortex, striatum, and subthalamic nucleus [13], where they serve to control release of other neurotransmitters, such as glutamate [14]. Interestingly, the activation of postsynaptic 5-HT1A receptors located at prefrontal cortex has also been shown to affect serotonin neuron activity through an indirect loop to the raphe nuclei [15–17]. 5-HT1B receptors are also expressed as autoreceptors at serotonergic terminals in target areas, where they contribute to control serotonin release. Moreover, these receptors are also localized in non-serotonergic neurons, such as the striatal medium spiny neurons, and regulate GABA release [18–20].
Among the other 5-HT receptors, the 5-HT2A receptors are involved in a variety of behaviors and diseases including anxiety, hyperactivity disorders, aggression, and social interaction. Moreover, they play a role in drug addiction and in the mechanism of action of anti-psychotic drugs. It has been shown that 5-HT2A receptors are abundant in the neocortex, striatum, and nucleus accumbens, where they appear to have a role in the emergence of motor complications [21, 22].
Another interesting serotonergic receptor that has been shown to play a role in the regulation of motor functions is the 5-HT2C receptor. This receptor subtype shows high and moderate expression in limbic areas and basal ganglia, respectively. The activation or blockade of 5-HT2C receptors results in an opposite effect on dopamine release; indeed, their activation induces a reduction of dopamine release along the dopaminergic pathway, while their blockade increases dopamine release along the meso-cortico-limbic as well as nigrostriatal pathways [23, 24]. Despite the influence of 5-HT2C receptors in modulating dopaminergic system, very little is known on their role in PD and dyskinesia.
5-HT3 receptors are mostly localized in limbic areas and brainstem nuclei, while they show low expression in the basal ganglia areas. Unlike other serotonergic receptors, 5-HT3 receptors are ion channels. They have been found to be involved in anxiety, schizophrenia, learning, and attention, as well as in craving and pain. Although 5-HT3 receptors have been reported to play a role in controlling striatal dopamine release [25], their involvement in PD and dyskinesia is poorly studied.
Despite the important role of serotonergic receptors in regulating motor functions, selective agonists for these receptors were generally unsuccessful for the treatment of parkinsonian motor symptoms [26]; on the other hand, the serotonergic receptors have been implicated in the development of motor complications induced by drug treatment in parkinsonian patients, such as levodopa-induced dyskinesia (LID).
Involvement of the Serotonergic System in Levodopa-Induced Dyskinesia
The therapeutic efficacy of levodopa during the first few years of treatment is conceivably due to the presence of a sufficient number of spared dopamine neurons that can provide conversion of levodopa and mediate physiological release of dopamine; however, with the progression of the disease, most of dopaminergic neurons are lost, and levodopa-derived dopamine is produced in non-dopaminergic elements, including the serotonergic terminals. In fact, serotonergic neurons are able to convert exogenous levodopa to dopamine and store and release dopamine in an activity-dependent manner in a number of experimental conditions, both in vitro and in vivo [27–29]. Indeed, serotonergic neurons share the same enzymatic machinery as dopaminergic terminals (aromatic amino acid decarboxylase and vesicular monoamine transporter 2 enzymes) and are able to convert levodopa to dopamine and to mediate storage of dopamine into synaptic vesicles.
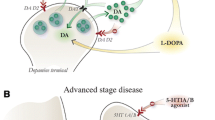
It is conceivable to think that conversion of levodopa takes place into serotonergic neurons also in earlier stages of disease; however, as long as there are sufficient spared dopaminergic terminals, the contribution of serotonergic neurons may be beneficial, as dopaminergic terminals provide a buffering system for the levodopa-derived dopamine (through the dopamine transporter). By contrast, as the disease progresses and spared dopaminergic terminals are lost, the contribution of serotonergic neurons becomes detrimental (see Fig. 11.1).

In the early stage of PD, the ability to release dopamine is partially preserved due to the presence of a sufficient number of spared dopaminergic neurons that have not yet degenerated. At this stage, serotonergic terminals may contribute to levodopa-derived dopamine release, due to an ability to convert exogenous levodopa to dopamine and store and release dopamine in an activity-dependent manner. However, their contribution may be beneficial as synaptic dopamine levels are kept within a physiological range due to the presence of an efficient buffering system provided by the spared dopamine terminals (through the dopamine transporter). With the progression of the disease, when most of dopaminergic terminals are lost, the contribution of serotonergic neurons becomes detrimental due to the lack of autoregulatory feedback mechanisms able to regulate synaptic dopamine levels. The uncontrolled release of dopamine from serotonergic neurons will act in concert with the intermittent nature of the orally administered levodopa to produce swings in synaptic dopamine levels and pulsatile stimulation of striatal dopaminergic receptors, which is responsible for the onset of dyskinesia. 5-HT1 receptor agonists will act to reduce levodopa-derived dopamine release from serotonin neurons, which will dampen swings in synaptic dopamine levels
In fact, unlike dopaminergic neurons, serotonergic neurons cannot regulate the extracellular levels of dopamine due to the lack of an autoregulatory feedback mechanism for dopamine release. As a consequence, levodopa-derived dopamine is released in uncontrolled way following levodopa administration. This will act in concert with the intermittent nature of the orally administered levodopa, to cause swing in synaptic dopamine levels, which is responsible for pulsatile stimulation of striatal dopaminergic receptors and aberrant downstream signaling cascade (see Fig. 11.1). In line with this view, it has been shown that removal of the forebrain serotonergic innervation by the selective toxin 5,7-dihydroxy-tryptamine (5,7-DHT) produces an almost complete suppression of LID [30, 31]. Dramatic reduction of levodopa-derived striatal dopamine levels appears to account for the anti-dyskinetic effect of 5,7-DHT lesions [29].
Support for the important role of serotonergic neurons in dyskinesia also came from a rat PET imaging study, where the administration of the 5-HT1A receptor agonist 8-OH-DPAT (which reduced LID) reversed levodopa-induced decrease of [(11) C]raclopride binding and increase of extracellular dopamine [32].
Interestingly, Rylander and colleagues have demonstrated a correlation between the density of serotonergic terminals in the striatum and the severity of LID across different species. In fact, dyskinetic rats and primates, as well as postmortem tissue of PD patients, have shown a growth-promoting effect on the striatal serotonergic fibers, accompanied by an increase of serotonin transporter (SERT) expression, induced by levodopa treatment. Regenerative sprouting of the serotonergic neurons was also accompanied by increased BDNF (Brain Derived Neurotrophic Factor) levels and increased BDNF-induced levodopa-derived dopamine release in rat striatal slices [33]. This is interesting as intracerebral delivery of BDNF has previously been shown to promote serotonergic terminal sprouting [34].
Further evidence for the involvement of serotonergic neurons in the appearance of LID came from recent experimental studies, where increased serotonergic tone by administration of either selective serotonin reuptake blockers (SSRIs) or the serotonergic precursor 5-hydroxytryptophan significantly reduced LID expression in hemiparkinsonian rats, without compromising the levodopa therapeutic efficacy [35–37].
Swings in synaptic dopamine levels, rather than high dopamine levels, are suggested to be a key factor for the development of LID. Indeed, postsynaptic dopaminergic receptors are highly plastic and can adapt to either low or high synaptic dopamine levels by altering the efficiency of intracellular signal transduction; by contrast, rapid changes in synaptic neurotransmitter levels, with high dopaminergic receptor occupancy (following each levodopa dose), followed by dramatic reduction (few hours after each levodopa dose), would impair this ability. In agreement with this view, it has been shown that dyskinetic patients present higher synaptic dopamine levels 1 h after levodopa administration compared to stable responders, while this difference disappeared few hours later [38]. Furthermore, dyskinesia is significantly dampened in advanced dyskinetic patients receiving continuous intra-duodenal infusion of levodopa, which is effective in reducing swing in synaptic dopamine levels [39].
All together, these experimental findings provide strong evidences supporting the important role of serotonergic neurons in the induction and expression of LID, at least in animal models of PD.
5-HT1A and 5-HT1B Receptor Agonists in the Treatment of Dyskinesia
The activation of serotonergic autoreceptors is a physiological mechanism meant to avoid excessive synaptic neurotransmitter release. Thus, while in normal conditions this mechanism is able to keep synaptic serotonin levels within a physiological range, under levodopa treatment in parkinsonian states, the activation of serotonergic autoreceptors is also able to inhibit serotonergic neuron-derived dopamine release. In fact, accumulating evidence support the efficacy of 5-HT1 receptor agonists for the treatment of LID, with the 5-HT1A receptors having received most of the attention, as they are able to control serotonin neuron firing and release (See Fig. 11.1).
Several 5-HT1A receptor agonists have displayed acute and chronic anti-dyskinetic effects in both animal models and in clinical studies [30, 31, 40–44]. Unfortunately, some of these studies have also shown that the administration of 5-HT1 agonists is associated with the induction of side effects, including worsening of the anti-parkinsonian efficacy of levodopa (See Table 11.1). For instance, the partial 5-HT1A receptor agonist sarizotan demonstrated potent efficacy in reducing dyskinesias in primate and rodent models of PD and in idiopathic PD patients in early open-label studies [41]; however, the anti-dyskinetic efficacy of sarizotan could not be shown to be significant compared to placebo in a following study, where the drug increased also off-time duration [45]. Moreover, in rats, high doses of sarizotan resulted in the induction of “serotonin syndrome” components, i.e., body posture associated with motor depression. Furthermore, the selective 5-HT1A receptor agonist 8-OH-DPAT inhibited LID in dyskinetic MPTP primates but only with increased motor disability [46, 47]. The partial, non-selective 5-HT1A receptor agonist buspirone was found to reduce LID in patients [48], but two other studies have found that this effect was again associated with a reduction of the anti-parkinsonian efficacy of levodopa [49, 50].
It has recently been demonstrated that 5-HT1B receptor agonists can also elicit anti-dyskinetic effects in animal models of PD [30, 51, 52]. However, to date, no clinical trials have been performed with these agonists.
It should be stressed that the anti-dyskinetic effect of 5-HT1 receptor agonists, as seen in the studies reported above, is likely not exclusively due to the activation of 5-HT1 autoreceptors. In fact, the activation of postsynaptic 5-HT1A and 5-HT1B receptors has also been shown to produce anti-dyskinetic effect by reducing striatal release of glutamate and GABA, respectively [30, 53–55]. Accordingly, relatively high doses of 5-HT1 receptor agonists have been shown to reduce dyskinesia induced by direct dopamine receptor agonists, such as apomorphine, which is not dependent on serotonergic neuron release [30, 52, 56].
Recent results have also shown that simultaneous activation of 5-HT1A and 5-HT1B receptors, using low doses of 8-OH-DPAT and CP-94253, respectively, produced a synergistic effect on suppression of LID in 6-OHDA-lesioned rats and in MPTP-treated macaques, with near to full inhibition at doses of the two drugs that were ineffective when given individually [30, 56]. In contrast, the same combined doses did not affect dyskinesia induced by apomorphine, suggesting that the observed effect is mainly due to the activation of presynaptic receptors [30]. Accordingly, combination of 8-OH-DPAT and CP-94253 was found to reduce extracellular dopamine levels following levodopa administration [57].
Recently, the mixed 5-HT1A/5-HT1B receptor agonist eltoprazine has been characterized for its anti-dyskinetic properties, both in rat and monkey models of PD. Eltoprazine, developed for the treatment of aggression, exhibited a safe toxicological profile and lack of serious side effects [58, 59], and is currently investigated in a clinical trial for the treatment of attention-deficit hyperactivity disorder (ADHD) (ClinicalTrials.gov Identifier: NCT01266174). In the rat 6-OHDA as well as macaque MPTP lesion models of PD, eltoprazine displayed high effectiveness in blocking LID at doses that were ineffective to reduce dyskinesia induced by apomorphine, supporting a presynaptic effect of this drug; however, the anti-dyskinetic effect of eltoprazine was accompanied by a partial reduction of the levodopa therapeutic efficacy [44, 60]. Similarly, anpirtoline, a mixed 5-HT1A/5-HT1B receptor agonist, with higher affinity for the 5-HT1B receptor, was very effective in reducing dyskinesia in rats and monkeys but at the expense of PD score at significantly effective doses [43].
Thus, as seen with other selective 5-HT1 receptor agonists, the maintenance of the levodopa therapeutic effect may represent a concern in this approach, not only for eltoprazine but, possibly, for any drug with similar profile. In spite of this, eltoprazine is under investigation in a phase 2 double-blind clinical trial employing a limited number of patients, with encouraging preliminary results (see http://www.psychogenics.com/press2012.html).
Based on the preclinical observation that combination of 5-HT1A and 5-HT1B receptor agonists showed anti-dyskinetic effects, without worsening levodopa therapeutic properties [56], it is also possible that the ideal compound has to possess the right affinity for the two serotonergic autoreceptors to be clinically useful. Nevertheless, the narrow therapeutic window is a concern and may require a careful titration of the selected compound in each patient.
A plausible explanation for the worsening of the levodopa therapeutic effect observed after the administration of 5-HT1 receptor agonists is the advanced stage of dopaminergic neuron degeneration characterizing the animal models employed in these investigations, as well as the patients recruited in the sarizotan trial. In fact, under this condition, the serotonergic neurons may mediate not only the pro-dyskinetic effect of levodopa but also its residual therapeutic efficacy. If so, the anti-dyskinetic effect of 5-HT1 receptor agonists should unavoidably lead to parallel reduction of the therapeutic effect of levodopa. Therefore, 5-HT1 receptor agonists may find better clinical efficacy in a situation of less severe dopaminergic neuron degeneration, where spared dopamine neurons can mediate the effect of levodopa, and silencing of serotonergic neurons should be less detrimental. PET imaging studies could be useful to identify patients retaining some residual dopaminergic innervation that may be more likely to benefit from 5-HT1 receptor agonists. On the other hand, for the same reason, these patients are also less likely to suffer from severe dyskinesia.
It should also be mentioned that sarizotan, like other 5-HT1 receptor agonists, such as buspirone, has antagonistic activity for the dopaminergic D2 receptors that may have contributed to the negative outcome of the clinical trial [61, 62]. Moreover, these compounds are able to target only one autoreceptor subtype, and sufficient anti-dyskinetic efficacy may be obtained at relatively high doses of the drugs, which are likely to also affect postsynaptic 5-HT1 receptors.
Therefore, new large clinical investigations employing more selective 5-HT1A/5-HT1B receptor agonists are warranted to clarify the role of serotonin neurons in the appearance of LID in PD patients and to verify whether pharmacological silencing of serotonin neurons is a feasible therapeutic approach for the treatment of LID.
5-HT2A Receptors Antagonists in the Treatment of Dyskinesia
Serotonergic 5-HT2A receptors are localized at postsynaptic level, and in general they exert an excitatory effect. The role of 5-HT2A receptors in the expression of dyskinesia has been recently confirmed in a study by Riahi et al. [63] where they showed an increase at striatal level of these receptors in levodopa dyskinetic compared to non-dyskinetic monkeys, suggesting a possible use of drugs targeting these receptors for the treatment of dyskinesia. Preclinical and clinical studies have demonstrated the effect of drugs acting on 5-HT2A receptors in controlling levodopa-induced motor complications [64, 65]; interestingly, one clinical study reported the ability of aripiprazole, a 5-HT2A receptor antagonist and partial 5-HT1A and dopamine D2 receptor agonist, to reduce LID without worsening motor performance in PD patients [66]; however, it is difficult to establish the contribution of each receptor subtype, given that the effect may also be due to the activation of 5-HT1A receptors. In MPTP-treated primates, the selective 5-HT2A inverse agonist pimavanserin was recently demonstrated to reduce LID by 36 % without worsening motor scores [67]. However, it has also been reported that the selective 5-HT2A antagonist ritanserin alleviated LID but at the expense of levodopa-induced therapeutic action [68, 69]. By contrast, the selective 5-HT2A antagonist volinanserin was not effective in reducing LID in 6-OHDA-lesioned rats [70].
Further work is required to establish whether 5-HT2A antagonists could be useful in dyskinetic patients.
SERT as a Possible Target for Dyskinesia
Recent evidence has demonstrated the implication of the SERT in the pathophysiology of LID; Rylander et al. [33] observed a significant upregulation of SERT expression in the striatum of dyskinetic animals, both in rats and nonhuman primates, as well as in dyskinetic patients, which provided support to the idea that the serotonergic system is involved in the appearance of LID also in PD patients. Accordingly, few preclinical studies have shown a significant reduction of dyskinesia after blockade of the serotonin transporter by SSRIs. Thus, acute [35] and chronic [37, 71] administration of the serotonin reuptake inhibitors citalopram and paroxetine in levodopa-dyskinetic rats resulted in a significant reduction in AIMs severity, without affecting the anti-parkinsonian action of levodopa [35]. The anti-dyskinetic effect of SSRIs is likely to be due to a combination of different mechanisms: (i) activation of presynaptic 5-HT1 receptors by serotonin, which may reduce dopamine release, as seen for selective 5-HT1 agonists; (ii) blockade of dopamine reuptake by serotonergic neurons, which may reduce swings in synaptic dopamine levels; and (iii) activation of postsynaptic serotonin receptors. In fact, the activation of postsynaptic 5-HT1 receptors by selective agonists has been shown to provide anti-dyskinetic effect in parkinsonian rats. In support of a possible postsynaptic effect of SSRIs, a significant 47 % reduction of apomorphine-induced dyskinesias was observed in patients treated with fluoxetine, without modification of parkinsonian motor disability [72]. In contrast, short-term paroxetine treatment did not affect dyskinesia induced by intravenous levodopa [73]. Clinical investigations are needed to further explore the use of SSRIs as anti-dyskinetic agents.
Conclusions
An overwhelming body of experimental evidence suggests that the serotonin system is implicated in the appearance of LID in animal models of PD. Moreover, accumulating clinical results appear to support a key role of this system also in PD patients. Dampening the release from the serotonin neurons has been shown to be a promising approach in preclinical models. Concerns have been raised about the clinical feasibility of this approach, as not only dyskinesia but also the therapeutic effect of levodopa may depend on dopamine release from the serotonin neurons in advanced stages of disease. However, few compounds have been tested in patients so far, and most of them also presented antagonist activity for the dopamine receptors, which may have played a role in the observed worsening of the levodopa therapeutic efficacy. Thus, new clinical trials employing more selective serotonin 5-HT1 receptor agonists are warranted. The effect of SSRIs should also be further investigated.
References
Benarroch EE. Serotonergic modulation of basal ganglia circuits: complexity and therapeutic opportunities. Neurology. 2009;73:880–6.
Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355–66.
Di Giovanni G, Di Matteo V, Esposito E. Serotonin-dopamine interaction: experimental evidence and therapeutic relevance. Preface. Prog Brain Res. 2008;172:ix.
Lavoie B, Parent A. Immunohistochemical study of the serotonergic innervation of the basal ganglia in the squirrel monkey. J Comp Neurol. 1990;299:1–16.
Fox SH, Chuang R, Brotchie JM. Serotonin and Parkinson’s disease: on movement, mood, and madness. Mov Disord. 2009;24:1255–66.
Scatton B, Javoy-Agid F, Rouquier L, Dubois B, Agid Y. Reduction of cortical dopamine, noradrenaline, serotonin and their metabolites in Parkinson’s disease. Brain Res. 1983;275:321–8.
Kish SJ, Tong J, Hornykiewicz O, Rajput A, Chang LJ, Guttman M, Furukawa Y. Preferential loss of serotonin markers in caudate versus putamen in Parkinson’s disease. Brain. 2008;131:120–31.
Boileau I, Warsh JJ, Guttman M, Saint-Cyr JA, McCluskey T, Rusjan P, Houle S, Wilson AA, Meyer JH, Kish SJ. Elevated serotonin transporter binding in depressed patients with Parkinson’s disease: a preliminary PET study with [11C]DASB. Mov Disord. 2008;23:1776–80.
Jellinger KA. Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol. 1991;14:153–97.
Peroutka SJ. 5-HT receptors: past, present and future. Trends Neurosci. 1995;18:68–9.
Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083–152.
Riad M, Garcia S, Watkins KC, Jodoin N, Doucet E, Langlois X, el Mestikawy S, Hamon M, Descarries L. Somatodendritic localization of 5-HT1A and preterminal axonal localization of 5-HT1B serotonin receptors in adult rat brain. J Comp Neurol. 2000;417:181–94.
Pompeiano M, Palacios JM, Mengod G. Distribution and cellular localization of mRNA coding for 5-HT1A receptor in the rat brain: correlation with receptor binding. J Neurosci. 1992;12:440–53.
Antonelli T, Fuxe K, Tomasini MC, Bartoszyk GD, Seyfried CA, Tanganelli S, Ferraro L. Effects of sarizotan on the corticostriatal glutamate pathways. Synapse. 2005;58:193–9.
Ceci A, Baschirotto A, Borsini F. The inhibitory effect of 8-OH-DPAT on the firing activity of dorsal raphe serotonergic neurons in rats is attenuated by lesion of the frontal cortex. Neuropharmacology. 1994;33:709–13.
Casanovas JM, Hervas I, Artigas F. Postsynaptic 5-HT1A receptors control 5-HT release in the rat medial prefrontal cortex. Neuroreport. 1999;10:1441–5.
Hajos M, Hajos-Korcsok E, Sharp T. Role of the medial prefrontal cortex in 5-HT1A receptor-induced inhibition of 5-HT neuronal activity in the rat. Br J Pharmacol. 1999;126:1741–50.
Mathur BN, Capik NA, Alvarez VA, Lovinger DM. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–11.
Mathur BN, Lovinger DM. Serotonergic action on dorsal striatal function. Parkinsonism Relat Disord. 2012;18 Suppl 1:S129–31.
Rylander D. The serotonin system: a potential target for anti-dyskinetic treatments and biomarker discovery. Parkinsonism Relat Disord. 2012;18 Suppl 1:S126–8.
Pazos A, Probst A, Palacios JM. Serotonin receptors in the human brain–III. Autoradiographic mapping of serotonin-1 receptors. Neuroscience. 1987;21:97–122.
Bishop C, Tessmer JL, Ullrich T, Rice KC, Walker PD. Serotonin 5-HT2A receptors underlie increased motor behaviors induced in dopamine-depleted rats by intrastriatal 5-HT2A/2C agonism. J Pharmacol Exp Ther. 2004;310:687–94.
Alex KD, Yavanian GJ, McFarlane HG, Pluto CP, Pehek EA. Modulation of dopamine release by striatal 5-HT2C receptors. Synapse. 2005;55:242–51.
Di Matteo V, De Blasi A, Di Giulio C, Esposito E. Role of 5-HT(2C) receptors in the control of central dopamine function. Trends Pharmacol Sci. 2001;22:229–32.
Benloucif S, Keegan MJ, Galloway MP. Serotonin-facilitated dopamine release in vivo: pharmacological characterization. J Pharmacol Exp Ther. 1993;265:373–7.
Jenner P, Sheehy M, Marsden CD. Noradrenaline and 5-hydroxytryptamine modulation of brain dopamine function: implications for the treatment of Parkinson’s disease. Br J Clin Pharmacol. 1983;15 Suppl 2:277S–89.
Arai R, Karasawa N, Geffard M, Nagatsu T, Nagatsu I. Immunohistochemical evidence that central serotonin neurons produce dopamine from exogenous levodopa in the rat, with reference to the involvement of aromatic L-amino acid decarboxylase. Brain Res. 1994;667:295–9.
Arai R, Karasawa N, Geffard M, Nagatsu I. levodopa is converted to dopamine in serotonergic fibers of the striatum of the rat: a double-labeling immunofluorescence study. Neurosci Lett. 1995;195:195–8.
Tanaka H, Kannari K, Maeda T, Tomiyama M, Suda T, Matsunaga M. Role of serotonergic neurons in levodopa-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport. 1999;10:631–4.
Carta M, Carlsson T, Kirik D, Bjorklund A. Dopamine released from 5-HT terminals is the cause of levodopa-induced dyskinesia in parkinsonian rats. Brain. 2007;130:1819–33.
Eskow KL, Dupre KB, Barnum CJ, Dickinson SO, Park JY, Bishop C. The role of the dorsal raphe nucleus in the development, expression, and treatment of levodopa-induced dyskinesia in hemiparkinsonian rats. Synapse. 2009;63:610–20.
Nahimi A, Holtzermann M, Landau AM, Simonsen M, Jakobsen S, Alstrup AK, Vang K, Moller A, Wegener G, Gjedde A, Doudet DJ. Serotonergic modulation of receptor occupancy in rats treated with levodopa after unilateral 6-OHDA lesioning. J Neurochem. 2012;120:806–17.
Rylander D, Parent M, O’Sullivan SS, Dovero S, Lees AJ, Bezard E, Descarries L, Cenci MA. Maladaptive plasticity of serotonin axon terminals in levodopa-induced dyskinesia. Ann Neurol. 2010;68:619–28.
Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci. 2000;20:771–82.
Bishop C, George JA, Buchta W, Goldenberg AA, Mohamed M, Dickinson SO, Eissa S, Eskow Jaunarajs KL. Serotonin transporter inhibition attenuates levodopa-induced dyskinesia without compromising levodopa efficacy in hemi-parkinsonian rats. Eur J Neurosci. 2012;36:2839–48.
Tronci E, Lisci C, Stancampiano R, Fidalgo C, Collu M, Devoto P, Carta M. 5-Hydroxy-tryptophan for the treatment of levodopa-induced dyskinesia in the rat Parkinson’s disease model. Neurobiol Dis. 2013;60:108–14.
Conti MM, Ostock CY, Lindenbach D, Goldenberg AA, Kampton E, Dell’isola R, Katzman AC, Bishop C. Effects of prolonged selective serotonin reuptake inhibition on the development and expression of levodopa-induced dyskinesia in hemi-parkinsonian rats. Neuropharmacology. 2014;77:1–8.
de la Fuente-Fernandez R, Sossi V, Huang Z, Furtado S, Lu JQ, Calne DB, Ruth TJ, Stoessl AJ. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain. 2004;127:2747–54.
Antonini A, Odin P, Opiano L, Tomantschger V, Pacchetti C, Pickut B, Gasser UE, Calandrella D, Mancini F, Zibetti M, Minafra B, Bertaina I, De Deyn P, Cras C, Wolf E, Spielberger S, Poewe W. Effect and safety of duodenal levodopa infusion in advanced Parkinson’s disease: a retrospective multicenter outcome assessment in patient routine care. J Neural Transm. 2013;120:1553–8.
Bibbiani F, Oh JD, Chase TN. Serotonin 5-HT1A agonist improves motor complications in rodent and primate parkinsonian models. Neurology. 2001;57:1829–34.
Bara-Jimenez W, Bibbiani F, Morris MJ, Dimitrova T, Sherzai A, Mouradian MM, Chase TN. Effects of serotonin 5-HT1A agonist in advanced Parkinson’s disease. Mov Disord. 2005;20:932–6.
Eskow KL, Gupta V, Alam S, Park JY, Bishop C. The partial 5-HT(1A) agonist buspirone reduces the expression and development of levodopa-induced dyskinesia in rats and improves levodopa efficacy. Pharmacol Biochem Behav. 2007;87:306–14.
Bezard E, Munoz A, Tronci E, Pioli EY, Li Q, Porras G, Bjorklund A, Carta M. Anti-dyskinetic effect of anpirtoline in animal models of levodopa-induced dyskinesia. Neurosci Res. 2013;77:242–6.
Bezard E, Tronci E, Pioli EY, Li Q, Porras G, Bjorklund A, Carta M. Study of the antidyskinetic effect of eltoprazine in animal models of levodopa-induced dyskinesia. Mov Disord. 2013;28:1088–96.
Goetz CG, Damier P, Hicking C, Laska E, Muller T, Olanow CW, Rascol O, Russ H. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: a double-blind placebo-controlled trial. Mov Disord. 2007;22:179–86.
Carey RJ, Depalma G, Damianopoulos E, Muller CP, Huston JP. The 5-HT1A receptor and behavioral stimulation in the rat: effects of 8-OHDPAT on spontaneous and cocaine-induced behavior. Psychopharmacology (Berl). 2004;177:46–54.
Iravani MM, Tayarani-Binazir K, Chu WB, Jackson MJ, Jenner P. In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated primates, the selective 5-hydroxytryptamine 1a agonist (R)-(+)-8-OHDPAT inhibits levodopa-induced dyskinesia but only with\ increased motor disability. J Pharmacol Exp Ther. 2006;319:1225–34.
Bonifati V, Fabrizio E, Cipriani R, Vanacore N, Meco G. Buspirone in levodopa-induced dyskinesias. Clin Neuropharmacol. 1994;17:73–82.
Hammerstad JP, Carter J, Nutt JG, Casten GC, Shrotriya RC, Alms DR, Temple D. Buspirone in Parkinson’s disease. Clin Neuropharmacol. 1986;9:556–60.
Kleedorfer B, Lees AJ, Stern GM. Buspirone in the treatment of levodopa induced dyskinesias. J Neurol Neurosurg Psychiatry. 1991;54:376–7.
Zhang X, Andren PE, Greengard P, Svenningsson P. Evidence for a role of the 5-HT1B receptor and its adaptor protein, p11, in levodopa treatment of an animal model of Parkinsonism. Proc Natl Acad Sci U S A. 2008;105:2163–8.
Jaunarajs KL, Dupre KB, Steiniger A, Klioueva A, Moore A, Kelly C, Bishop C. Serotonin 1B receptor stimulation reduces D1 receptor agonist-induced dyskinesia. Neuroreport. 2009;20:1265–9.
Bishop C, Krolewski DM, Eskow KL, Barnum CJ, Dupre KB, Deak T, Walker PD. Contribution of the striatum to the effects of 5-HT1A receptor stimulation in levodopa-treated hemiparkinsonian rats. J Neurosci Res. 2009;87:1645–58.
Dupre KB, Eskow KL, Barnum CJ, Bishop C. Striatal 5-HT1A receptor stimulation reduces D1 receptor-induced dyskinesia and improves movement in the hemiparkinsonian rat. Neuropharmacology. 2008;55:1321–8.
Dupre KB, Ostock CY, Eskow Jaunarajs KL, Button T, Savage LM, Wolf W, Bishop C. Local modulation of striatal glutamate efflux by serotonin 1A receptor stimulation in dyskinetic, hemiparkinsonian rats. Exp Neurol. 2011;229:288–99.
Munoz A, Li Q, Gardoni F, Marcello E, Qin C, Carlsson T, Kirik D, Di Luca M, Bjorklund A, Bezard E, Carta M. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of levodopa-induced dyskinesia. Brain. 2008;131:3380–94.
Lindgren HS, Andersson DR, Lagerkvist S, Nissbrandt H, Cenci MA. levodopa-induced dopamine efflux in the striatum and the substantia nigra in a rat model of Parkinson’s disease: temporal and quantitative relationship to the expression of dyskinesia. J Neurochem. 2010;112:1465–76.
Miczek KA, Mos J, Olivier B. Brain 5-HT and inhibition of aggressive behavior in animals: 5-HIAA and receptor subtypes. Psychopharmacol Bull. 1989;25:399–403.
Mos J, Olivier B, Poth M, van Aken H. The effects of intraventricular administration of eltoprazine, 1-(3-trifluoromethylphenyl)piperazine hydrochloride and 8-hydroxy-2-(di-n-propylamino)tetralin on resident intruder aggression in the rat. Eur J Pharmacol. 1992;212:295–8.
Tronci E and Carta M. 5-HT1 receptor agonists for the treatment of levodopa-induced dyskinesia: from animal models to clinical investigation. Basal Ganglia. 2013;3:9–13.
Olanow CW, Damier P, Goetz CG, Mueller T, Nutt J, Rascol O, Serbanescu A, Deckers F, Russ H. Multicenter, open-label, trial of sarizotan in Parkinson disease patients with levodopa-induced dyskinesias (the SPLENDID Study). Clin Neuropharmacol. 2004;27:58–62.
Rijnders HJ, Slangen JL. The discriminative stimulus properties of buspirone involve dopamine-2 receptor antagonist activity. Psychopharmacology (Berl). 1993;111:55–61.
Riahi G, Morissette M, Parent M, Di Paolo T. Brain 5-HT(2A) receptors in MPTP monkeys and levodopa-induced dyskinesias. Eur J Neurosci. 2011;33:1823–31.
Oh JD, Bibbiani F, Chase TN. Quetiapine attenuates levodopa-induced motor complications in rodent and primate parkinsonian models. Exp Neurol. 2002;177:557–64.
Roberts C. ACP-103, a 5-HT2A receptor inverse agonist. Curr Opin Investig Drugs. 2006;7:653–60.
Meco G, Stirpe P, Edito F, Purcaro C, Valente M, Bernardi S, Vanacore N. Aripiprazole in Levodopa-induced dyskinesias: a one-year open-label pilot study. J Neural Transm. 2009;116:881–4.
Vanover KE, Betz AJ, Weber SM, Bibbiani F, Kielaite A, Weiner DM, Davis RE, Chase TN, Salamone JD. A 5-HT2A receptor inverse agonist, ACP-103, reduces tremor in a rat model and levodopa-induced dyskinesias in a monkey model. Pharmacol Biochem Behav. 2008;90:540–4.
Maertens de Noordhout A, Delwaide PJ. Open pilot trial of ritanserin in parkinsonism. Clin Neuropharmacol. 1986;9:480–4.
Meco G, Frascarelli M, Pratesi L, Linfante I, Rocchi L, Formisano R. Headache in Parkinson’s disease. Headache. 1988;28:26–9.
Taylor JL, Bishop C, Ullrich T, Rice KC, Walker PD. Serotonin 2A receptor antagonist treatment reduces dopamine D1 receptor-mediated rotational behavior but not levodopa-induced abnormal involuntary movements in the unilateral dopamine-depleted rat. Neuropharmacology. 2006;50:761–8.
Kuan WL, Zhao JW, Barker RA. The role of anxiety in the development of levodopa-induced dyskinesias in an animal model of Parkinson’s disease, and the effect of chronic treatment with the selective serotonin reuptake inhibitor citalopram. Psychopharmacology (Berl). 2008;197:279–93.
Durif F, Vidailhet M, Bonnet AM, Blin J, Agid Y. Levodopa-induced dyskinesias are improved by fluoxetine. Neurology. 1995;45:1855–8.
Chung KA, Carlson NE, Nutt JG. Short-term paroxetine treatment does not alter the motor response to levodopa in PD. Neurology. 2005;64:1797–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Tronci, E., Fidalgo, C., Carta, M. (2014). The Serotonergic System in Levodopa-Induced Dyskinesia. In: Fox, S., Brotchie, J. (eds) Levodopa-Induced Dyskinesia in Parkinson's Disease. Springer, London. https://doi.org/10.1007/978-1-4471-6503-3_11
Download citation
DOI: https://doi.org/10.1007/978-1-4471-6503-3_11
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-6502-6
Online ISBN: 978-1-4471-6503-3
eBook Packages: MedicineMedicine (R0)