
Abstract
Ion channel remodeling in heart failure modulates key cellular electrophysiological properties, predisposing to arrhythmias and sudden death. Heart failure induced ion channel dysfunction prolongs the action potential, increases spatio-temporal gradients of repolarization, promotes arrhythmogenic triggers and results in conduction abnormalities. Understanding fundamental ionic mechanisms of normal and abnormal electrogenesis is a key requirement for the development of effective and safe therapies. Elucidating the underlying molecular mechanisms and functional consequences of ion channel remodeling at the cellular and organ levels presents a unique opportunity for the development of novel pharmacological, device, gene, and cell based approaches for the treatment of arrhythmias in heart failure.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Heart failure (HF) claims over 200,000 lives annually in the US alone [1]. Approximately 50 % of these deaths are sudden and unexpected, and presumably the consequence of lethal ventricular arrhythmias [2]. Our limited understanding of fundamental arrhythmia mechanisms at the ionic and molecular levels has hampered the development of effective pharmacological treatments for these patients [3]. In fact, the proarrhythmic tendency of ion channel targeting agents has resulted in the premature termination of the Cardiac Arrhythmia Suppression Trial (CAST) [4]. These proarrhythmic effects are due to the heterogeneity of ion channel expression and function [5, 6], the nonspecific nature of most pharmacological agents that target ion channels, the cross-talk between individual ion channels, and the complex remodeling that occurs dynamically in the context of left ventricular dysfunction. Indeed, elucidation of arrhythmia mechanisms in HF at the basic ionic level is expected to improve existing pharmacological therapies and facilitate the design of novel approaches, including cell- and gene-transfer strategies designed to target ion channels and transporters. In this chapter, we focus on key HF-induced ion channel remodeling at the cellular level and its pro-arrhythmic manifestations at the tissue level, with the intent of identifying molecular targets for anti-arrhythmic therapy in HF.
Ionic Basis of the Ventricular Action Potential
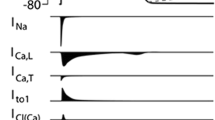
A unique signature of any excitable tissue is its action potential (AP) profile, which reflects a delicate balance in the activity of several depolarizing and repolarizing ion channels, transporters and exchangers. Myocytes are characterized by a long AP, which is initiated by the influx of sodium ions (Na+) when voltage-gated Na+ channels (encoded by Na v 1.5) open. The transient opening of these channels gives rise to a rapid phase of depolarization (phase 0) which is terminated by the inactivation of the fast inward Na+ current (I Na) within a few milliseconds. The AP upstroke is followed by a brief interval of early repolarization (phase 1), caused by the activation of the voltage-gated transient outward potassium (K+) current (I to). Influx of calcium ions via L-type Ca2+ channels carries a small depolarizing current (I Ca-L) which stimulates the release of calcium from the sarcoplasmic reticulum (SR) though ryanodine receptors (RyR2). This regenerative process known as calcium-induced calcium-release results in tropomyosin translocation and myofilament contraction. Cytosolic Ca2+ levels are restored to diastolic levels by the coordinated activities of the Na+–Ca2+ exchanger (NCX) and the SR calcium ATPase (SERCA2a). A delicate balance between inward and outward currents results in the plateau phase of the AP. Finally, AP repolarization is sculpted by the orchestrated activities of the delayed rectifier K+ currents (I Kr and I Ks), the inward rectifier K+ current (I K1) along with a gradual decrease in net depolarizing currents. The membrane potential of fast response myocytes is effectively locked at rest (phase 4) by the background I K1 until the next wave of depolarization activates I Na resulting in a new propagating AP.
Since most ion channels are time- and voltage-dependent, even a subtle change in one ionic transport mechanism can modulate the activity of many other channels and transporters, often profoundly altering the AP duration and profile. As in the long QT syndrome, HF results in a major prolongation of the cellular AP, which translates clinically into QT interval prolongation on the surface electrocardiogram (ECG). Numerous studies using cellular electrophysiology, molecular biology, protein chemistry, high-throughout gene expression profiling and proteomics have considerably advanced our understanding of ion channel remodeling in HF [5, 7]. Complementary studies in multicellular tissue preparations are bridging major gaps in our knowledge of the functional consequences of ion channel remodeling and dysfunction in generating arrhythmias [8–10]. In this Chapter, we summarize some of the key developments in this rapidly evolving field of investigation, with a view towards identifying appropriate ion channel targets for antiarrhythmic therapy in HF.
Potassium Channel Remodeling in Heart Failure
Transient Outward K+ Current
I to inscribes the notch of the AP during early repolarization and sets the membrane potential at which activator calcium is initiated. As such I to indirectly controls excitation–contraction coupling and AP duration by modulating the take-off potential at which subsequent plateau currents are activated [11]. I to varies regionally (right ventricle > left ventricle) and transmurally (epicardium > midmyocardium > endocardium). Functional down-regulation of I to is consistently observed in most models of HF [7]. The effect of HF-induced I to down-regulation on AP duration varies across species. Since I to is the major repolarizing current in rodents (with the exception of guinea pigs), its reduction is directly responsible for AP prolongation in these species [12]. On the other hand, in humans and large animal models reduction of I to influences the early part of the AP (phase 1), reducing Ca2+ entry through I Ca-L and shortening the AP duration [13]. The discrepancy in how I to modulates AP duration highlights the importance of exercising caution when applying knowledge gained from rodent studies of repolarizing currents to humans before carefully investigating large animal models.
Kv4.3 (Shal-related subfamily, member 3) encodes the pore-forming alpha subunit of cardiac I to in large mammals [14]. An additional component of I to (encoded by Kv1.4) with slower inactivation kinetics also exists preferentially in the endocardial layer of rodents [15]. The role of Kv1.4 channels in large animals and humans remains unclear. Despite abundant expression of Kv1.4 in the myocardium, we recently argued against its functional role in the formation of native I to in human and canine ventricular myocytes [16]. Our findings highlight important differences in the molecular composition of I to and other repolarizing currents across species. Such species differences may profoundly impact the response to pharmacological agents targeting ion channel components. I to down-regulation in HF is transcriptionally regulated as Kv4 mRNA levels are reduced in both humans [14] and dogs with HF [6]. Interestingly, gene transfer of Kv4.3 shortened AP duration and limited the hypertrophic response in rat hearts that underwent transaortic constriction [17]. The effects of Kv4.3 over-expression in large animal models whose terminal repolarization is sculpted by delayed rectifier K+ currents, however, remain unknown.
Other accessory subunits participate in the formation of native I to, including Kv-channel interacting proteins (KChIPs) [18] and the CD26-related dipeptidyl aminopeptidase-like protein 6 (DPPX) [19]. Interestingly, both of these accessory subunits likely contribute to the formation of the transmural I to gradient [20, 21]. Recently, another beta subunit (MiRP-1; potassium voltage-gated channel subfamily E member 2) originally known to modulate I Kr has also been shown to physically associate with, and functionally regulate Kv4- and Kv1-encoded channels in mammalian expression systems [22]. Elucidation of the macromolecular complex that forms native I to is expected to offer new opportunities for regulating this current, which is strongly remodeled by HF, and whose dysregulation has been linked to various arrhythmic syndromes [23, 24]. The strong influence of KChIP2 and MiRP-1 on multiple ion channels suggests that macromolecular complexes involving the same accessory subunits could indeed regulate diverse electrophysiological properties [18, 22]. As such, selective targeting of these proteins is expected to produce complex physiological outcomes that are difficult to predict, especially in the setting of HF, in which these subunits are differentially remodeled.
Inward Rectifying K+ Current
I K1, encoded by the Kir2.x family of genes, maintains the resting membrane potential and contributes to terminal repolarization. Reduced I K1 density in HF promotes AP prolongation and enhanced susceptibility to spontaneous membrane depolarizations [25, 26]. Indeed, the genetic suppression of I K1 converts fast-response myocytes to cells with automatic activity (i.e. biological pacemakers) [27]. Disease-induced downregulation of I K1 could, therefore, enhance automaticity in the failing heart. Interventricular differences in I K1 density (left ventricle > right ventricle) underlie the localization of high-frequency sources of activation during ventricular fibrillation [28]. Moreover, pharmacological suppression of I K1 reduces the dominant frequency of the so-called ‘mother rotor’ underlying arrhythmias in structurally normal hearts [28]. Conversely, overexpression of I K1 leads to the acceleration and maintenance of fibrillatory rotors [29]. While I K1 down-regulation promotes triggers and enhanced automaticity, I K1 over-expression can shorten the AP and the cardiac wavelength, thereby stabilizing reentrant activity. This delicate balance supports the notion that ion channels are regulated to operate within a narrow physiological range, such that excessive modulation (reduction or enhancement) of ion channel activity or expression can have equally deleterious effects. This property adds another challenge to pharmacological and gene transfer approaches targeting ion channels, whose expression is differentially altered across myocardial layers and regions of the failing heart.
The underlying basis for I K1 downregulation in HF remains unknown, as no consistent changes in the steady-state levels of Kir2.1 (inward rectifier potassium channel 2) mRNA [14] or protein [6] have been found. Surprisingly, Kir2.1 mRNA and protein levels are up-regulated in terminally failing human hearts [30]. Clearly, further investigation into the molecular identity and regulation of I K1 by HF is warranted.
Delayed Rectifier K+ Currents
The delayed rectifier K+ currents (I Kr and I Ks) play a prominent role in the control of terminal cardiac repolarization in large animals and humans. In fact, a relatively small reduction of either component leads to significant AP prolongation [31]. The reduction in the outward current, over the plateau range of voltages in various models of HF, predisposes to the development of arrhythmogenic early after-depolarizations (EADs) [8, 32]. Indeed, K+ current downregulation in HF underlies key phenotypic similarities between HF and the long QT syndrome, including AP and QT-interval prolongation, reduced repolarization reserve and arrhythmia propensity [33, 34].
Despite knowledge of the major alpha and beta subunits that form I Kr and I Ks, the molecular mechanisms underlying I Kr and I Ks downregulation in HF remain controversial. We measured the expression of HERG (encoding I Kr) and KvLQT1 (encoding I Ks) in normal and failing canine ventricles. Surprisingly, we found a paradoxical increase in HERG expression with no change in KvLQT1 protein levels [6]. Other groups reported decreased HERG expression in human HF [35]. These discrepant findings highlight the fact that ion channel function is dependent on complex factors that are well beyond the expression levels of the alpha and beta subunits that form these channels. For example, changes in protein assembly, folding, trafficking, membrane insertion, internalization, and degradation can significantly modulate channel behavior.
Evidence is emerging for the existence of macromolecular complexes that include diverse proteins in the proper assembly, trafficking and function of cardiac ion channels, such as I Kr and I Ks. Interestingly, a strong physical and functional interaction between KvLQT1 and HERG was recently demonstrated [36]. Specifically, KvLQT1 was shown to modulate both the distribution and biophysical properties of HERG. Indeed, this elegant demonstration of an interaction between two K+ channel alphasubunits underscores the complexity of selectively targeting individual ion channels without affecting others [36].
Gene transfer of various alpha and beta subunits that encode I Kr and I Ks has been used to modulate AP duration in small animal models [26, 37]. Whether or not these strategies can reverse remodel the failing heart, normalize AP duration, reduce repolarization gradients, and prevent arrhythmias remain to be seen. Indeed, the complex regulation of ion channel function by post-translational modifications may strongly limit the ability of gene transfer of individual alpha and beta subunits to completely and safely restore ion channel function in the failing heart.
ATP-Sensitive K+ Current
Sarcolemmal (Sarc) KATP channels link membrane excitability to metabolism [38]. They are regulated by intracellular nucleotides, membrane phospholipids, protein kinases and phosphatases [38]. The ATP-sensitive K+ current (I K-ATP) is the principal mediator of AP shortening under conditions of increased metabolic demand and/or energy deficit. Due to their abundance in the plasma membrane, the opening of sarcKATP channels causes rapid AP shortening, loss of intracellular K+, and reduction in myocyte excitability. Interestingly, AP shortening in ischemia is exaggerated in cells from hypertrophied compared to normal ventricles [39]. We described a novel mechanism by which oscillations in the mitochondrial membrane potential (Δ(delta)Ψ(psi)m) under conditions of oxidative stress, can produce oscillations in the AP duration via cyclical activation of I K-ATP, setting the stage for conduction block (which was termed ‘metabolic sink’) and arrhythmias [40, 41]. While hypertrophied and failing hearts are associated with oxidative stress and Δ(delta)Ψ(psi)m instability [42, 43], the exact role of sarcKATP channels in HF-mediated arrhythmias remains unclear.
Pacemaker Current
The hyperpolarization-activated pacemaker or ‘funny’ current (I f) is a nonselective cation current originally described in automatic tissues such as the sinoatrial node [44]. Although I f has been observed in human ventricular myocytes [45, 46], it appears to activate at negative voltages outside the physiological range of membrane potential. Nonetheless, increased I f density in hypertrophy suggests its possible role in either promoting disease progression or arrhythmic triggers [47]. The HCN (hyperpolarization-activated mammalian cation channels) family of genes encoding I f has been cloned [48, 49]. In a canine model of HF, HCN expression is significantly decreased in the sinoatrial node and increased in the right atrium, likely contributing to sinus node dysfunction and atrial ectopy [50].
Adenoviral-mediated gene transfer of an engineered HCN construct, which exhibits a shift in its inactivation kinetics to more depolarized potentials, unleashes a biological pacemaking source in guinea pig ventricles and pig atria [51]. Increased I f in the setting of reduced I K1 is likely to predispose the failing heart to enhanced automaticity. A strategy for suppressing ventricular I f could, therefore, be appealing in HF. Indeed, Ivabradine, a potent I f inhibitor that suppresses the spontaneous depolarization rate in sino-atrial nodal cells, improved cardiac structure and function in HF [52]. The Systolic Heart Failure Treatment with the I f Inhibitor Trial (SHIFT) has shown significant efficacy in reversing left ventricular remodeling in a large cohort of patients with HF [53].
Calcium Channel Remodeling in Heart Failure
Excitation–contraction coupling refers to the fundamental principle by which a myocyte’s ionic (excitation) properties tightly coordinate its mechanical function. Defective Ca2+ handling in HF has a profound electrophysiological impact because the intracellular calcium transient and the AP are intricately linked by a variety of Ca2+-mediated channels and transporters.
L-Type and T-Type Ca2+ Channels
Ca2+ entry through I Ca-L triggers SR Ca2+ release. The density of I Ca-L, dictated in part by the stage of HF [54, 55], is increased in mild-to-moderate hypertrophy and decreased in more advanced stages of hypertrophy and failure [56, 57]. Interestingly, myocytes from failing hearts also exhibit attenuated augmentation of I Ca-L in response to beta-adrenergic stimulation [58]. Finally, slowing of I Ca-L inactivation in HF alters Ca2+ handling and prolongs the AP [59]. The molecular mechanisms underlying these changes are unknown, but could involve a dephosphorylation defect that alters open channel probability [60]. Sipido et al. demonstrated that the negative force–frequency relationship in dilated cardiomyopathy is due to impaired recovery of I Ca-L inactivation at fast stimulation frequencies. These findings provided a potential ionic target for improving myocardial contractile reserve [61].
The pore-forming subunit of I Ca-L is encoded by CACNA1C (calcium channel, voltage-dependent, L type, alpha 1C subunit) and is highly regulated by a variety of accessory subunits that affect channel trafficking, current density and kinetics. In vitro and in vivo knockdown of the L-type Ca2+ channel accessory beta subunit, using a short hairpin RNA template sequence, reduced I Ca-L and attenuated the hypertrophic response, without compromising systolic performance [62].
Another class of sarcolemmal calcium channels is the low-voltage-activated transient Ca2+ channel (I Ca-T), encoded by the Cav3.x family of genes [63]. Since these channels activate at hyperpolarized potentials, they might contribute to enhanced automaticity [64], especially in the failing heart, in which I Ca-T density is considerably enhanced [65]. Interestingly, I Ca-T was observed in myocytes from endocardial, but not epicardial, layers of the normal canine ventricle [66]. As such, I Ca-T could also contribute to the electrical and contractile heterogeneity seen across the transmural wall. Whether or not this intrinsic heterogeneity is increased or decreased in HF remains unknown.
Since abnormal calcium cycling is a central feature of mechano-electrical dysfunction of the failing heart, pharmacological and genetic modulation of individual Ca2+ channel subunits could be useful strategies for restoring defective excitation-contraction coupling and electrophysiological properties. However, these strategies have the strong potential to exacerbate intracellular calcium overload and associated dysfunction (increased calcium entry) or result in atrioventricular conduction delays and block (decreased calcium entry).
Sarcoplasmic Reticulum (SR) Ca2+ Pump
The amplitude and rate of decay of the intracellular calcium transient are blunted in cells and tissues from failing hearts [67]. These changes are caused by defective sequestration of Ca2+ by the SR due, in large part, to reduced SERCA2a expression and function [68]. Since compromised SR Ca2+ re-uptake causes abnormal contractile and electrical function in HF, increasing the expression and activity of SERCA2a could be clinically beneficial. Pharmacological stimulation of the pump enhances mechanical function [69]. Furthermore, numerous studies using adenoviral-mediated gene transfer approaches to increase myocardial SERCA2a expression, have demonstrated the efficacy of restoring impaired intracellular Ca2+ handling and normalizing contractile dysfunction [70]. The safety of this approach has been confirmed by experiments that demonstrated improved contractility at no metabolic cost (i.e. cost of oxygen) in normal [71] and failing hearts [72]. Targeted gene-transfer techniques to increase the expression levels of SERCA2a using adeno-associated vectors have also been developed [72], and a First-in-man multicenter trial (CUPID, Calcium Upregulation by Percutaneous Administration of Gene Therapy In Cardiac Disease) has been completed [73, 74]. Whether or not enhancing SERCA2a expression and/or function in the failing heart will protect against or exacerbate arrhythmias remains to be fully determined, although no adverse electrical outcomes were reported in the Phase 1 and Phase 2a CUPID trials [73, 74]. Finally, SERCA2a over-expression using gene transfer has been recently shown to suppress arrhythmogenic repolarization alternans in structurally normal hearts [75].
Phospholamban
Phospholamban (PLB), a key regulator of SERCA2a, blunts the rate of SR Ca2+ re-uptake [76]. The inhibitory influence of PLB on SERCA2a is reduced when the protein is phosphorylated [77]. Therefore, restoration of intracellular calcium cycling in HF could be achieved by targeting PLB. To that end, PLB silencing using an anti-sense strategy successfully enhanced SERCA2a activity and improved contractile function [78]. In contrast, PLB gene ablation in a transgenic mouse model failed to improve global cardiac function, possibly because of compromised metabolic properties in that animal model [79]. Pharmacological manipulation aimed at dissociating PLB from SERCA2a has also been successfully tested in the laboratory [80]. Potential differences in the electrophysiological consequences of enhancing SR calcium uptake by targeting SERCA2a versus PLB requires direct investigation. Indeed, it is expected that these complementary approaches may be associated with unique benefits and potential pitfalls depending on the disease etiology and the sympatho-adrenergic state, as these calcium regulatory proteins are modulated by local kinase and phosphatase signaling mechanisms.
Na+/Ca+ Exchanger
In its ‘forward’ mode of operation, NCX extrudes intracellular Ca2+ via an electrogenic exchange for extracellular Na+ (one Ca2+ for three Na+ ions), thereby generating a net inward current [81]. Indeed, forward-mode NCX function compensates for defective SR Ca2+ uptake at the expense of depleting the releasable pool of Ca2+ with repetitive stimulation. In contrast, reverse-mode exchange (Na+ out and Ca2+ in) could provide inotropic support to the failing ventricle while shortening AP duration [82].
NCX expression and function are increased in HF [83, 84], contributing to AP prolongation and repolarization instability. Importantly, partial inhibition of NCX enhances SR Ca2+ load by shifting the balance of Ca2+ flux away from trans-sarcolemmal efflux [85]. As such, NCX blockade could represent an effective therapeutic strategy for improving contractility in HF [85]. Unfortunately, the efficacy of abrogating arrhythmias by targeting NCX will have to await the development of more selective pharmacological agents with improved lipophilicity and bioavailability properties.
Ryanodine Receptors
RyR2 channels are downregulated in HF, both at the mRNA [86] and protein [87] levels. Hyperphosphorylation of RyR2 causes FKBP12.6 (FK506-binding protein 1B) dissociation resulting in diastolic Ca2+ leak and the generation of Ca2+ waves underlying triggered activity [88]. Whether protein kinase A [88], CaMKII [89], or both mediate RyR2 hyperphosporylation and diastolic calcium leak in HF has been the subject of intense debate. Identification of the specific molecular sites of RyR2 phosphorylation might lead to pharmacological strategies designed to suppress diastolic Ca2+ leak and associated arrhythmias. Marks and colleagues identified a class of small molecules that enhance the binding affinity of FKBP12.6 for RyR2 [90]. While these compounds are effective in suppressing catecholaminergic polymorphic ventricular tachycardia in mouse models [91, 92], their utility in preventing HF-related arrhythmias remains to be tested in clinically relevant large animal models of acquired structural heart diseases.
Calmodulin and Calcium/Calmodulin-Dependent Protein Kinase II
Calmodulin kinase II (CaMKII), the main target of Calmodulin binding, is a multifunctional protein capable of phosphorylating several Ca2+-handling proteins, including RyR2, PLB, and SERCA2a. In human HF, CaMKII activity is markedly increased [93, 94], possibly as a compensatory mechanism for altered Ca2+ homeostasis. Since phosphatase activity is also enhanced in human HF, the net effect with respect to the phosphorylation state of any given target protein is locally controlled within the subcellular milieu [95]. Over-expression of CaMKII has been shown to increase SR Ca2+ leak through RYR2, without altering myocyte contractility [96]. Anderson and colleagues demonstrated that CaMKII inhibition could represent an effective strategy for improving myocardial function and preventing atrial and ventricular arrhythmias in various animal models [97].
Sodium Channel Remodeling in Heart Failure
In light of the important interplay between Na+ and Ca2+ in the cardiac cell, changes in intracellular Na+ levels are expected to alter both mechanical and electrophysiological properties [98]. Moreover, as Na+ channels are critical to normal impulse propagation in the heart, perturbations in I Na could lead to conduction slowing, block, and arrhythmias. In what follows, we summarize some of the major changes in Na+ transport mechanisms that occur in HF.
Na+ Channels
Normal impulse formation and conduction depend on the fast inward I Na. An increase in the late component of this current can markedly prolong AP duration and promote polymorphic ventricular tachycardia [99]. Changes in I Na density and kinetics could, therefore, promote arrhythmias, either by disrupting conduction or prolonging repolarization. In a canine model of myocardial infarction, major downregulation of I Na, acceleration of its inactivation kinetics and slowing of its recovery from inactivation were observed in myocytes isolated from the infarct border zone [100]. In another canine model of repeated microembolization-induced HF, a considerable increase in the late component of I Na was demonstrated [101]. As such, changes in I Na are likely to depend on the specific disease etiology, and could have profound implications for arrhythmogenesis given the relative abundance and importance of this current to myocyte excitability.
Over-expression of Na V 1.5 (also known as SCN5A), which forms the alpha subunit of I Na exerted an antifibrillatory effect by enhancing excitability in monolayers of neonatal rat ventricular myocytes [102]. In addition to Nav1.5, I Na activity is modulated by a variety of auxiliary beta subunits, kinases, phosphatases and cytoskeletal proteins. HF-induced disruption of the macromolecular complex that regulates I Na results in marked changes in current density and kinetics, which alter conduction (by affecting the amplitude of I Na) and repolarization (by affecting the late component of I Na). Finally, pharmacological inhibition of the late I Na with Ranolazine has shown significant promise in improving mechano-electrical function and suppressing atrial and ventricular arrhythmias [103–106]. Indeed, a prospective, proof-of-concept study (RAnolazine for the Treatment of Diastolic Heart Failure, RALI-DHF) was recently initiated to determine the efficiacy of late I Na blockade in improving diastolic function in HF patients with preserved ejection fraction [107].
Na+/K+ ATPase
Intracellular Na+ is increased in ventricular hypertrophy and HF by approximately two to three fold [108]. This causes a secondary rise in Ca2+ influx via reverse-mode NCX [108], which in turn increases SR Ca2+ load and enhances contractility. This seemingly beneficial, positive inotropic effect comes at a price — enhanced susceptibility to spontaneous SR Ca2+ release through RyR2, activation of the transient inward current and the development of delayed afterdepolarizations, especially in the setting of beta-adrenergic stimulation [81]. Hence, tight regulation of intracellular Na+ is required for normal electrogenesis.
The Na+/K+ ATPase exchanges extracellular K+ for intracellular Na+, with a stoichiometry of 2:3, thereby generating a net outward repolarizing current. Reduced Na+/K+ ATPase activity in some models of HF contributes to intracellular Na+ and Ca2+ overload, AP prolongation and arrhythmias. Molecular mechanisms underlying altered Na+/K+ ATPase activity may include changes in the expression of its three alpha isoforms. In addition, major changes in the expression and phosphorylation of Phospholemman, a key regulatory subunit of the Na+/K+ ATPase pump have been identified in a rabbit model and in human HF samples. Indeed, recent advances in our understanding of the regulation and physiological properties of Phospholemman under normal and stress conditions are likely to yield new opportunities for controlling the Na+/K+ ATPase pump, intarcellular Na+ homeostasis and electromechanical function in HF.
Na+–H+ Exchanger
The Na+–H+ exchanger (NHE) regulates intracellular pH via proton extrusion, driven by the transmembrane Na+ gradient. NHE inhibition is cardioprotective against myocardial ischemia–reperfusion injury [109]. Prevention of intracellular Na+ accumulation and excessive Ca2+ influx via reverse-mode NCX, have been proposed as the mechanism of cardioprotection by NHE inhibition. Since HF is associated with elevated intracellular Na+ levels, NHE could represent a promising target for antiarrhythmic therapy. The GUARD During Ischemia Against Necrosis (GUARDIAN) trial was designed to determine the efficacy of Cariporide, a selective NHE inhibitor, in reducing mortality. Although GUARDIAN failed to demonstrate an overall clinical benefit in patients at risk of myocardial necrosis [110], it is clear that NHE inhibition preserves mitochondrial function and therefore the metabolic status of the myocyte. As such, NHE inhibition is likely to play a critical role in preventing cellular apoptosis and related dysfunction under conditions of oxidative stress, such ischemia-reperfusion injury. The role of NHE inhibition in modulating the electrophysiological substrate and preventing arrhythmias in HF requires direct exploration. Given the central role of mitochondria in regulating excitability and arrhythmias, it is conceivable that NHE inhibition may play a prominent role in restoring normal electrogenesis to the failing heart.
In what follows, we illustrate how ion channel remodeling at the cellular level alters the electrophysiological substrate at the organ level. Particularly, we focus on mechanisms by which transmural and intra-ventricular repolarization gradients predispose the failing heart to lethal arrhythmias.
Electrophysiological Remodeling at the Tissue Level
Abnormal AP prolongation at the cellular level readily translates to the level of the intact organ resulting in a long and variable QT interval on the surface ECG [2, 111, 112]. Key changes in the early and late phases of AP repolarization have been documented in numerous studies using the patch clamp technique in isolated cardiomyocytes from various small and large animal models of HF. At the opposite end of the spectrum, studies in humans and animal models showed delayed global repolarization and enhanced temporal repolarization instability using clinical non-invasive metrics, such as the QT-interval variability index and T wave alternans on the surface ECG. Rosenbaum, Berger, and others have developed sophisticated algorithms that detect various ECG metrics of global cardiac repolarization to identify patients at high risk of sudden cardiac death (SCD) [113–122]. Of key importance are recent findings of a multi-center clinical trial (Alternans Before Cardioverter Defibrillator, ABCD), in which non-invasive T wave alternans testing was shown to significantly enhance the predictability of impending SCD in patients with HF when combined with standard electrophysiological testing [116].
These cellular and clinical studies highlight the importance of repolarization changes occurring at the tissue level for arrhythmia genesis in HF. Until relatively recently, efforts to investigate the mechanistic link between repolarization changes and reentrant arrhythmias were hampered by technical difficulties in assessing spatio-temporal repolarization gradients across the heart. With the advent of optical imaging techniques using voltage sensitive dyes, a high-resolution measurement of cardiac repolarization at a cellular level within the intact syncytium has become possible [8, 9, 87, 123, 124]. Importantly, a quantitative relationship between altered spatio-temporal repolarization gradients and the incidence of arrhythmias in various animal models of HF have recently emerged [8, 125]. In what follows, we focus on the role of spatial heterogeneities of repolarization on the incidence of reentrant arrhythmias in clinically relevant large animal models of HF that are prone to arrhythmias. Specifically, we discuss changes in transmural and intra-ventricular repolarization gradients as mechanisms for ventricular arrhythmias in non-ischemic dilated cardiomyopathy and dyssynchronous heart failure, respectively.
Transmural Repolarization Heterogeneity in Dilated Cardiomyopathy
Antzelevitch and colleagues [126, 127] advanced the notion that heterogeneities of cellular repolarization in different cell types (epicardial, mid-myocardial, and endocardial) represent a unifying mechanism underlying a host of arrhythmias in congenital and/or acquired cardiac diseases, such as the long QT, short QT [128], Brugada [129], Andersen-Tawil [130], and Timothy syndromes [131, 132]. Of particular importance was the role of mid-myocardial (M) cells in the establishment of transmural repolarization heterogeneity under conditions of prolonged QT interval in various ex vivo models of the long QT syndrome [127, 133, 134]. Because QT interval prolongation represents an electrophysiological hallmark of the failing heart, we hypothesized that both disease states (long QT syndrome and HF) may share important phenotypic properties at the multi-cellular tissue level that predispose them to arrhythmias via similar mechanisms [8]. This hypothesis was directly investigated by determining the functional expression of repolarization gradients across myocardial layers and their potential role in the mechanism(s) of arrhythmias in a model of non-ischemic dilated cardiomyopathy produced by chronic rapid pacing in the dog [8]. As expected, HF was associated with a marked AP prolongation across all myocardial layers, consistent with findings in isolated myocytes and whole animals. Interestingly, AP prolongation was heterogeneous across the left ventricular (LV) wall, affecting mid-myocardial and endocardial muscle layers more selectively; thereby, increasing the effective transmural repolarization gradient by ∼2-fold [8]. In support of transmural dispersion of repolarization as a unifying mechanism for arrhythmias associated with various disease etiologies, Yan et al. elegantly demonstrated that LV hypertrophy in a rabbit model of renovascular hypertension was also associated with significant enhancement of transmural dispersion of repolarization because of selective prolongation of subendocardial APs [135]. Transmural repolarization heterogeneity in HF produced an arrhythmic substrate, as premature stimuli introduced at a critical window during AP repolarization resulted in intramural decremental conduction and block. Importantly, conduction block which was localized at the interface of the mid-myocardial layer was followed by the initiation of ventricular tachycardia.
Mechanisms underlying increased transmural repolarization heterogeneity in HF remain unresolved. These changes, however, likely involve multiple factors, including heterogeneous remodeling of cell-to-cell coupling, ionic currents/exchangers, and calcium handling proteins. Li et al. [32] investigated the ionic basis of transmural AP remodeling in HF by measuring the density of key repolarizing K+ currents, including I to, I K1, I Kr and I Ks. In general, K+ current changes were uniform in epicardial, mid-myocardial, and endocardial myocytes of failing hearts, indicating that transmural repolarization heterogeneity observed at the tissue level could not be explained by cell-type specific remodeling of repolarizing K+ currents. In a subsequent study, we measured the expression levels of key alpha and beta subunits encoding these K+ currents in the three principal myocardial layers of normal and failing hearts [136]. In support of the findings of Li et al. [32] we also did not find a K+ channel molecular basis (neither at the mRNA or protein levels) for the enhanced transmural repolarization heterogeneity observed in the failing heart.
Poelzing and Rosenbaum [137] attributed the location of the maximum transmural repolarization gradient to increased electrical resistivity at that location. Furthermore, they converted transmural repolarization gradients measured across normal preparations into gradients that mimicked those in HF simply by perfusing normal preparations with the gap junction inhibitor, Carbenoxolone [137]. These findings highlight the potential importance of gap junction uncoupling in the mechanism of increased transmural dispersion of repolarization in HF. We and others have investigated the molecular basis for gap junction uncoupling in HF and have found major changes in the expression, distribution, and phosphorylation state of the main ventricular gap junction protein, Cx43 that develop with varying time-courses during disease progression [9, 125, 138, 139]. Specifically, end-stage HF was associated with over-all Cx43 downregulation, dephosphorylation, and lateralization. In addition, we recently reported the loss of interaction between Cx43 and the cytoskeletal protein, ZO-1 as a potentially critical event underlying severe conduction slowing and therefore gap junction uncoupling at late stages of remodeling in a model of pressure overload hypertrophy [125]. Interestingly, hyperphosphorylation of Cx43 also occurred at earlier stages of remodeling that were associated with a milder form of conduction slowing [125]. As such disrupted phosphorylation (either increased or decreased) at critical residues within the carboxyl domain of Cx43 may lead to loss of gap junction function via distinct mechanisms. The individual contribution of these complex molecular changes to the establishment of transmural repolarization heterogeneity across the failing heart requires direct investigation.
Intra-ventricular Repolarization Heterogeneity in Dyssynchronous Heart Failure
Left ventricular (LV) dyssynchrony caused by delayed activation of the lateral free wall results in heterogeneous mechanical stress across the ventricle [140]. We hypothesized that regional differences in local mechanical function across the dyssynchronously contracting failing ventricle produce important changes in electrophysiological, metabolic, and molecular properties, which ultimately worsen outcome in these patients. We further hypothesized that re-establishing LV synchrony using bi-ventricular pacing in cardiac resynchronization therapy (CRT) could potentially reverse-remodel the electrophysiological substrate. Indeed, the late activating lateral LV endocardium of dyssynchronously failing hearts is distinguished by selective remodeling of calcium handling, gap junction, and stress related molecules [141]. These regionally heterogeneous molecular changes were linked to key electrophysiological abnormalities. Specifically, Aiba et al. [142] found that myocytes isolated from the lateral LV of dyssynchonrously failing hearts were characterized by excessive AP prolongation and enhanced propensity for early after depolarization mediated triggered beats. The ion channel basis of these changes was also investigated. Specifically, dyssynchronous mechanical contraction of the failing heart reduced K+ current densities (I to, I K, and I K1) in both anterior and lateral regions. In contrast, I Ca-L was differentially remodeled across the dyssynchronously failing heart, potentially underpinning differences in repolarization properties and susceptibility to early afterdepolarizations. In particular, a significantly greater reduction in I Ca-L amplitude and decay rate in myocytes isolated from the high-stress lateral compared to the low-stress anterior LV wall was described. Interestingly, CRT partially restored abnormal repolarization of lateral LV myocytes, reducing the regional disparity in electrophysiological function across the failing ventricle.
In a subsequent study, the effect of mechanical dyssynchrony on changes in regional gene expression was also investigated [143]. Electromechanical dyssynchrony resulted in complex cardiac transcriptome remodeling, causing major gene expression changes in the anterior wall. Once again, CRT corrected the region specific alterations in gene expression, highlighting the importance of mechanical synchrony/dyssynchrony on global and regional gene expression remodeling, which is likely to impact diverse cellular processes. As such, the electrophysiological and molecular changes induced by CRT may indeed suppress ventricular arrhythmias and potentially promote mechanical and metabolic function.
Finally, the influence of mechanical dyssynchrony per se on key electrophysiological properties was investigated independently of LV dysfunction [144]. Normal hearts were subjected to chronic left bundle branch block, inducing mechanical dyssynchrony with preserved LV function. We found that mechanical dyssynchrony in non-failing canine hearts was sufficient to cause regional changes in conduction and repolarization properties [144]. In addition, the distribution of the main ventricular gap junction protein, Cx43 was significantly more lateralized in the late-activating lateral LV, which was associated with slower conduction and faster repolarization [144]. Taken together, these findings suggest an important mechano-sensitive component to chronic remodeling associated with dyssynchronous LV contraction which causes regional changes in protein expression and electrophysiological properties that enhance intra-ventricular repolarization gradients and potentially predispose to reentrant arrhythmias. Understanding mechanisms by which local mechanical stresses and strains regulate electrophysiological properties offers the potential for designing novel device-based strategies for the control of arrhythmias in heart failure.
Conclusions
Complex remodeling of a host of ion channels and Ca2+-cycling proteins modulates key cellular electrophysiological properties, predisposing to arrhythmias and sudden death. HF-induced ion channel dysfunction prolongs the AP, increases spatiotemporal gradients of repolarization, promotes arrhythmogenic triggers and results in conduction abnormalities. Understanding fundamental ionic mechanisms of normal and abnormal electrogenesis is a key requirement for the development of modern therapies. Elucidating the role of individual current components and their underlying molecular identities presents a unique opportunity for the development of novel pharmacological, device, gene, and cell based approaches for the treatment of arrhythmias in HF.
References
Schocken DD, Arrieta MI, Leaverton PE, Ross EA. Prevalence and mortality rate of congestive heart failure in the United States. J Am Coll Cardiol. 1992;20(2):301–6.
Tomaselli GF, Beuckelmann DJ, Calkins HG, Berger RD, Kessler PD, Lawrence JH, Kass D, Feldman AM, Marban E. Sudden cardiac death in heart failure. The role of abnormal repolarization. Circulation. 1994;90(5):2534–9.
Estes 3rd NA, Weinstock J, Wang PJ, Homoud MK, Link MS. Use of antiarrhythmics and implantable cardioverter-defibrillators in congestive heart failure. Am J Cardiol. 2003;91(6A):45D–52.
Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The cardiac arrhythmia suppression trial. N Engl J Med. 1991;324(12):781–8.
Schram G, Pourrier M, Melnyk P, Nattel S. Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res. 2002;90(9):939–50.
Akar FG, Xiong W, Juang GJ, Tian Y, DiSilvestre D, Tomaselli GF. Molecular mechanisms underlying potassium current down-regulation in heart failuire. Circulation. 2003;108(17):82.
Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42(2):270–83.
Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res. 2003;93(7):638–45.
Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res. 2004;95(7):717–25.
Pajouh M, Wilson LD, Poelzing S, Johnson NJ, Rosenbaum DS. IKs blockade reduces dispersion of repolarization in heart failure. Heart Rhythm. 2005;2(7):731–8.
Carmeliet E. K+ channels and control of ventricular repolarization in the heart. Fundam Clin Pharmacol. 1993;7(1):19–28.
Rozanski GJ, Xu Z, Zhang K, Patel KP. Altered K+ current of ventricular myocytes in rats with chronic myocardial infarction. Am J Physiol. 1998;274(1 Pt 2):H259–65.
Greenstein JL, Wu R, Po S, Tomaselli GF, Winslow RL. Role of the calcium-independent transient outward current I(to1) in shaping action potential morphology and duration. Circ Res. 2000;87(11):1026–33.
Kaab S, Dixon J, Duc J, Ashen D, Nabauer M, Beuckelmann DJ, Steinbeck G, McKinnon D, Tomaselli GF. Molecular basis of transient outward potassium current downregulation in human heart failure: a decrease in Kv4.3 mRNA correlates with a reduction in current density. Circulation. 1998;98(14):1383–93.
Guo W, Xu H, London B, Nerbonne JM. Molecular basis of transient outward K+ current diversity in mouse ventricular myocytes. J Physiol. 1999;521(Pt 3):587–99.
Akar FG, Wu RC, Deschenes I, Armoundas AA, Piacentino 3rd V, Houser SR, Tomaselli GF. Phenotypic differences in transient outward K+ current of human and canine ventricular myocytes: insights into molecular composition of ventricular Ito. Am J Physiol Heart Circ Physiol. 2004;286(2):H602–9.
Lebeche D, Kaprielian R, del Monte F, Tomaselli G, Gwathmey JK, Schwartz A, Hajjar RJ. In vivo cardiac gene transfer of Kv4.3 abrogates the hypertrophic response in rats after aortic stenosis. Circulation. 2004;110(22):3435–43.
Pourrier M, Schram G, Nattel S. Properties, expression and potential roles of cardiac K+ channel accessory subunits: MinK, MiRPs, KChIP, and KChAP. J Membr Biol. 2003;194(3):141–52.
Jerng HH, Pfaffinger PJ, Covarrubias M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci. 2004;27(4):343–69.
An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403(6769):553–6.
Rosati B, Pan Z, Lypen S, Wang HS, Cohen I, Dixon JE, McKinnon D. Regulation of KChIP2 potassium channel beta subunit gene expression underlies the gradient of transient outward current in canine and human ventricle. J Physiol. 2001;533(Pt 1):119–25.
McCrossan ZA, Abbott GW. The MinK-related peptides. Neuropharmacology. 2004;47(6):787–821.
Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660–6.
Antzelevitch C, Yan GX. J wave syndromes. Heart Rhythm. 2010;7(4):549–58.
Wang Z, Yue L, White M, Pelletier G, Nattel S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation. 1998;98(22):2422–8.
Nuss HB, Kaab S, Kass DA, Tomaselli GF, Marban E. Cellular basis of ventricular arrhythmias and abnormal automaticity in heart failure. Am J Physiol. 1999;277(1 Pt 2):H80–91.
Miake J, Marban E, Nuss HB. Biological pacemaker created by gene transfer. Nature. 2002;419(6903):132–3.
Warren M, Guha PK, Berenfeld O, Zaitsev A, Anumonwo JM, Dhamoon AS, Bagwe S, Taffet SM, Jalife J. Blockade of the inward rectifying potassium current terminates ventricular fibrillation in the guinea pig heart. J Cardiovasc Electrophysiol. 2003;14(6):621–31.
Noujaim SF, Pandit SV, Berenfeld O, Vikstrom K, Cerrone M, Mironov S, Zugermayr M, Lopatin AN, Jalife J. Up-regulation of the inward rectifier K+ current (I K1) in the mouse heart accelerates and stabilizes rotors. J Physiol. 2007;578(Pt 1):315–26.
Soltysinska E, Olesen SP, Christ T, Wettwer E, Varro A, Grunnet M, Jespersen T. Transmural expression of ion channels and transporters in human nondiseased and end-stage failing hearts. Pflugers Arch. 2009;459(1):11–23.
Nattel S. Acquired delayed rectifier channelopathies: how heart disease and antiarrhythmic drugs mimic potentially-lethal congenital cardiac disorders. Cardiovasc Res. 2000;48(2):188–90.
Li GR, Lau CP, Ducharme A, Tardif JC, Nattel S. Transmural action potential and ionic current remodeling in ventricles of failing canine hearts. Am J Physiol Heart Circ Physiol. 2002;283(3):H1031–41.
Tsuji Y, Zicha S, Qi XY, Kodama I, Nattel S. Potassium channel subunit remodeling in rabbits exposed to long-term bradycardia or tachycardia: discrete arrhythmogenic consequences related to differential delayed-rectifier changes. Circulation. 2006;113(3):345–55.
Han W, Chartier D, Li D, Nattel S. Ionic remodeling of cardiac Purkinje cells by congestive heart failure. Circulation. 2001;104(17):2095–100.
Choy A-M, Kuperschmidt S, Lang CC, Pierson RN, Roden DM. Regional expression of HERG and KvLQT1 in heart failure. Circulation. 1996;94:164.
Ehrlich JR, Pourrier M, Weerapura M, Ethier N, Marmabachi AM, Hebert TE, Nattel S. KvLQT1 modulates the distribution and biophysical properties of HERG. A novel alpha-subunit interaction between delayed rectifier currents. J Biol Chem. 2004;279(2):1233–41.
Mazhari R, Nuss HB, Armoundas AA, Winslow RL, Marban E. Ectopic expression of KCNE3 accelerates cardiac repolarization and abbreviates the QT interval. J Clin Invest. 2002;109(8):1083–90.
Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–6.
Kimura S, Bassett AL, Furukawa T, Furukawa N, Myerburg RJ. Differences in the effect of metabolic inhibition on action potentials and calcium currents in endocardial and epicardial cells. Circulation. 1991;84(2):768–77.
Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115(12):3527–35.
Aon MA, Cortassa S, Akar FG, O’Rourke B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochim Biophys Acta. 2006;1762(2):232–40.
Jin H, Nass RD, Joudrey PJ, Lyon AR, Chemaly ER, Rapti K, Akar FG. Altered spatiotemporal dynamics of the mitochondrial membrane potential in the hypertrophied heart. Biophys J. 2010;98(10):2063–71.
Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115(3):500–8.
Baruscotti M, Difrancesco D. Pacemaker channels. Ann N Y Acad Sci. 2004;1015:111–21.
Cerbai E, Pino R, Porciatti F, Sani G, Toscano M, Maccherini M, Giunti G, Mugelli A. Characterization of the hyperpolarization-activated current, I(f), in ventricular myocytes from human failing heart. Circulation. 1997;95(3):568–71.
Hoppe UC, Jansen E, Sudkamp M, Beuckelmann DJ. Hyperpolarization-activated inward current in ventricular myocytes from normal and failing human hearts. Circulation. 1998;97(1):55–65.
Cerbai E, Barbieri M, Mugelli A. Occurrence and properties of the hyperpolarization-activated current If in ventricular myocytes from normotensive and hypertensive rats during aging. Circulation. 1996;94(7):1674–81.
Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393(6685):587–91.
Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93(5):717–29.
Zicha S, Fernandez-Velasco M, Lonardo G, L’Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res. 2005;66(3):472–81.
Tse HF, Xue T, Lau CP, Siu CW, Wang K, Zhang QY, Tomaselli GF, Akar FG, Li RA. Bioartificial sinus node constructed via in vivo gene transfer of an engineered pacemaker HCN Channel reduces the dependence on electronic pacemaker in a sick-sinus syndrome model. Circulation. 2006;114(10):1000–11.
Mulder P, Thuillez C. Heart rate slowing for myocardial dysfunction/heart failure. Adv Cardiol. 2006;43:97–105.
Terracciano CM, Yacoub MH. Heart failure: a SHIFT from ion channels to clinical practice. Nat Rev. 2010;7(12):669–70.
Brooksby P, Levi AJ, Jones JV. The electrophysiological characteristics of hypertrophied ventricular myocytes from the spontaneously hypertensive rat. J Hypertens. 1993;11(6):611–22.
Cerbai E, Barbieri M, Li Q, Mugelli A. Ionic basis of action potential prolongation of hypertrophied cardiac myocytes isolated from hypertensive rats of different ages. Cardiovasc Res. 1994;28(8):1180–7.
Richard S, Leclercq F, Lemaire S, Piot C, Nargeot J. Ca2+ currents in compensated hypertrophy and heart failure. Cardiovasc Res. 1998;37(2):300–11.
Hill JA. Electrical remodeling in cardiac hypertrophy. Trends Cardiovasc Med. 2003;13(8):316–22.
Ouadid H, Albat B, Nargeot J. Calcium currents in diseased human cardiac cells. J Cardiovasc Pharmacol. 1995;25(2):282–91.
Ryder KO, Bryant SM, Hart G. Membrane current changes in left ventricular myocytes isolated from guinea pigs after abdominal aortic coarctation. Cardiovasc Res. 1993;27(7):1278–87.
Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98(10):969–76.
Sipido KR, Stankovicova T, Vanhaecke J, Flameng W, Verdonck F. A critical role for L-type Ca2+ current in the regulation of Ca2+ release from the sarcoplasmic reticulum in human ventricular myocytes from dilated cardiomyopathy. Ann N Y Acad Sci. 1998;853:353–6.
Cingolani E, Ramirez Correa GA, Kizana E, Murata M, Cheol Cho H, Marban E. Gene therapy to inhibit the calcium channel {beta} subunit. Physiological consequences and pathophysiological effects in models of cardiac hypertrophy. Circ Res. 2007;101:166–75.
Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–55.
Clozel JP, Ertel EA, Ertel SI. Voltage-gated T-type Ca2+ channels and heart failure. Proc Assoc Am Physicians. 1999;111(5):429–37.
Nuss HB, Houser SR. T-type Ca2+ current is expressed in hypertrophied adult feline left ventricular myocytes. Circ Res. 1993;73(4):777–82.
Wang HS, Cohen IS. Calcium channel heterogeneity in canine left ventricular myocytes. J Physiol. 2003;547(Pt 3):825–33.
Winslow RL, Rice J, Jafri S, Marban E, O’Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res. 1999;84(5):571–86.
O’Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res. 1999;84(5):562–70.
Ohizumi Y, Sasaki S, Shibusawa K, Ishikawa K, Ikemoto F. Stimulation of sarcoplasmic reticulum Ca(2+)-ATPase by gingerol analogues. Biol Pharm Bull. 1996;19(10):1377–9.
del Monte F, Hajjar RJ, Harding SE. Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ Res. 2001;88(11):E66–7.
Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, Takewa Y, Jeong D, Park WJ, Kawase Y, Hajjar RJ. Targeted gene transfer increases contractility and decreases oxygen cost of contractility in normal rat hearts. Am J Physiol Heart Circ Physiol. 2007;292(5):H2356–63.
Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, Tsuji T, Takewa Y, del Monte F, Peluso R, Zsebo K, Jeong D, Park WJ, Kawase Y, Hajjar RJ. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007;42(4):852–61.
Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, Borow K, Dittrich H, Zsebo KM, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009;15(3):171–81.
Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+ −ATPase in patients with advanced heart failure. Circulation. 2010;124(3):304–13.
Cutler MJ, Wan X, Laurita KR, Hajjar RJ, Rosenbaum DS. Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ Arrhythm Electrophysiol. 2009;2(6):686–94.
Levine BA, Patchell VB, Sharma P, Gao Y, Bigelow DJ, Yao Q, Goh S, Colyer J, Drago GA, Perry SV. Sites on the cytoplasmic region of phospholamban involved in interaction with the calcium-activated ATPase of the sarcoplasmic reticulum. Eur J Biochem. 1999;264(3):905–13.
Hasenfuss G, Meyer M, Schillinger W, Preuss M, Pieske B, Just H. Calcium handling proteins in the failing human heart. Basic Res Cardiol. 1997;92 Suppl 1:87–93.
del Monte F, Harding SE, Dec GW, Gwathmey JK, Hajjar RJ. Targeting phospholamban by gene transfer in human heart failure. Circulation. 2002;105(8):904–7.
Janczewski AM, Zahid M, Lemster BH, Frye CS, Gibson G, Higuchi Y, Kranias EG, Feldman AM, McTiernan CF. Phospholamban gene ablation improves calcium transients but not cardiac function in a heart failure model. Cardiovasc Res. 2004;62(3):468–80.
McKenna E, Smith JS, Coll KE, Mazack EK, Mayer EJ, Antanavage J, Wiedmann RT, Johnson Jr RG. Dissociation of phospholamban regulation of cardiac sarcoplasmic reticulum Ca2+ ATPase by quercetin. J Biol Chem. 1996;271(40):24517–25.
Bers DM, Pogwizd SM, Schlotthauer K. Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res Cardiol. 2002;97 Suppl 1:I36–42.
Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. Contribution of reverse-mode sodium-calcium exchange to contractions in failing human left ventricular myocytes. Cardiovasc Res. 1998;37(2):424–31.
Houser SR, Piacentino 3rd V, Mattiello J, Weisser J, Gaughan JP. Functional properties of failing human ventricular myocytes. Trends Cardiovasc Med. 2000;10(3):101–7.
Sipido KR, Volders PG, Vos MA, Verdonck F. Altered Na/Ca exchange activity in cardiac hypertrophy and heart failure: a new target for therapy? Cardiovasc Res. 2002;53(4):782–805.
Hobai IA, Maack C, O’Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95(3):292–9.
Meyer M, Schillinger W, Pieske B, Holubarsch C, Heilmann C, Posival H, Kuwajima G, Mikoshiba K, Just H, Hasenfuss G, et al. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation. 1995;92(4):778–84.
Heerdt PM, Holmes JW, Cai B, Barbone A, Madigan JD, Reiken S, Lee DL, Oz MC, Marks AR, Burkhoff D. Chronic unloading by left ventricular assist device reverses contractile dysfunction and alters gene expression in end-stage heart failure. Circulation. 2000;102(22):2713–19.
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–76.
Curran J, Hinton MJ, Rios E, Bers DM, Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100(3):391–8.
Marks AR. Novel therapy for heart failure and exercise-induced ventricular tachycardia based on ‘fixing’ the leak in ryanodine receptors. Novartis Found Symp. 2006;274:132–47; discussion 147–55, 272–6.
Lehnart SE, Wehrens XH, Laitinen PJ, Reiken SR, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109(25):3208–14.
Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science (New York). 2004;304(5668):292–6.
Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42(1):254–61.
Maier LS, Bers DM. Calcium, calmodulin, and calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34(8):919–39.
Lokuta AJ, Rogers TB, Lederer WJ, Valdivia HH. Modulation of cardiac ryanodine receptors of swine and rabbit by a phosphorylation-dephosphorylation mechanism. J Physiol. 1995;487(Pt 3):609–22.
Kohlhaas M, Zhang T, Seidler T, Zibrova D, Dybkova N, Steen A, Wagner S, Chen L, Brown JH, Bers DM, Maier LS. Increased sarcoplasmic reticulum calcium leak but unaltered contractility by acute CaMKII overexpression in isolated rabbit cardiac myocytes. Circ Res. 2006;98(2):235–44.
Anderson ME, Higgins LS, Schulman H. Disease mechanisms and emerging therapies: protein kinases and their inhibitors in myocardial disease. Nat Clin Pract Cardiovasc Med. 2006;3(8):437–45.
Pogwizd SM, Sipido KR, Verdonck F, Bers DM. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc Res. 2003;57(4):887–96.
Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade des pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96(6):2038–47.
Pu J, Boyden PA. Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res. 1997;81(1):110–19.
Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55(3):494–505.
Auerbach D, Berenfeld O, Jalife J. Antifibrillatory action of increased excitability in neonatal rat ventricular monolayers overexpressing hSCN5A. Circulation. 2006;114(18):268–9.
Antzelevitch C, Burashnikov A, Sicouri S, Belardinelli L. Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm. 2010;8(8):1281–90.
Hoyer K, Song Y, Wang D, Phan D, Balschi J, Ingwall JS, Belardinelli L, Shryock JC. Reducing the late sodium current improves cardiac function during sodium pump inhibition by ouabain. J Pharmacol Exp Ther. 2011;337(2):513–23.
Antoons G, Oros A, Beekman JD, Engelen MA, Houtman MJ, Belardinelli L, Stengl M, Vos MA. Late na(+) current inhibition by ranolazine reduces torsades de pointes in the chronic atrioventricular block dog model. J Am Coll Cardiol. 2010;55(8):801–9.
Wu Y, Song Y, Belardinelli L, Shryock JC. The late Na+ current (INa) inhibitor ranolazine attenuates effects of palmitoyl-L-carnitine to increase late INa and cause ventricular diastolic dysfunction. J Pharmacol Exp Ther. 2009;330(2):550–7.
Jacobshagen C, Belardinelli L, Hasenfuss G, Maier LS. Ranolazine for the treatment of heart failure with preserved ejection fraction: background, aims, and design of the RALI-DHF study. Clin Cardiol. 2011;34(7):426–32.
Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. [Na+]i and the driving force of the Na+/Ca2+ −exchanger in heart failure. Cardiovasc Res. 2003;57(4):986–95.
Karmazyn M, Sostaric JV, Gan XT. The myocardial Na+/H+ exchanger: a potential therapeutic target for the prevention of myocardial ischaemic and reperfusion injury and attenuation of postinfarction heart failure. Drugs. 2001;61(3):375–89.
Erhardt LR. GUARD during ischemia against necrosis (GUARDIAN) trial in acute coronary syndromes. Am J Cardiol. 1999;83(10A):23G–5.
Pak PH, Nuss HB, Tunin RS, Kaab S, Tomaselli GF, Marban E, Kass DA. Repolarization abnormalities, arrhythmia and sudden death in canine tachycardia-induced cardiomyopathy. J Am Coll Cardiol. 1997;30(2):576–84.
Atiga WL, Calkins H, Lawrence JH, Tomaselli GF, Smith JM, Berger RD. Beat-to-beat repolarization lability identifies patients at risk for sudden cardiac death. J Cardiovasc Electrophysiol. 1998;9(9):899–908.
Amit G, Costantini O, Rosenbaum DS. Can we alternate between T-wave alternans testing methods? Heart Rhythm. 2009;6(3):338–40.
Amit G, Rosenbaum DS, Super DM, Costantini O. Microvolt T-wave alternans and electrophysiological testing predict distinct arrhythmia substrates: implications for identifying patients at risk for sudden cardiac death. Heart Rhythm. 2010;7(6):763–8.
Costantini O, Drabek C, Rosenbaum DS. Can sudden cardiac death be predicted from the T wave of the ECG? A critical examination of T wave alternans and QT interval dispersion. Pacing Clin Electrophysiol. 2000;23(9):1407–16.
Costantini O, Hohnloser SH, Kirk MM, Lerman BB, Baker 2nd JH, Sethuraman B, Dettmer MM, Rosenbaum DS. The ABCD (alternans before cardioverter defibrillator) trial: strategies using T-wave alternans to improve efficiency of sudden cardiac death prevention. J Am Coll Cardiol. 2009;53(6):471–9.
Cutler MJ, Rosenbaum DS. Risk stratification for sudden cardiac death: is there a clinical role for T wave alternans? Heart Rhythm. 2009;6(8 Suppl):S56–61.
Cutler MJ, Rosenbaum DS. Explaining the clinical manifestations of T wave alternans in patients at risk for sudden cardiac death. Heart Rhythm. 2009;6(3 Suppl):S22–8.
Laurita KR, Rosenbaum DS. Cellular mechanisms of arrhythmogenic cardiac alternans. Prog Biophys Mol Biol. 2008;97(2–3):332–47.
Rosenbaum DS. T-wave alternans in the sudden cardiac death in heart failure trial population: signal or noise? Circulation. 2008;118(20):2015–18.
Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med. 1994;330(4):235–41.
Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, Laurita KR, Rosenbaum DS. Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm. 2009;6(2):251–9.
Akar FG, Laurita KR, Rosenbaum DS. Cellular basis for dispersion of repolarization underlying reentrant arrhythmias. J Electrocardiol. 2000;33(Suppl):23–31.
Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105(10):1247–53.
Jin H, Chemaly ER, Lee A, Kho C, Hadri L, Hajjar RJ, Akar FG. Mechanoelectrical remodeling and arrhythmias during progression of hypertrophy. FASEB J. 2010;24(2):451–63.
Antzelevitch C. Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol. 2007;293(4):H2024–38.
Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov A, Di Diego J, Saffitz J, Thomas GP. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10(8):1124–52.
Antzelevitch C. Cardiac repolarization. The long and short of it. Europace. 2005;7 Suppl 2:3–9.
Antzelevitch C, Brugada P, Brugada J, Brugada R. Brugada syndrome: from cell to bedside. Curr Probl Cardiol. 2005;30(1):9–54.
Tsuboi M, Antzelevitch C. Cellular basis for electrocardiographic and arrhythmic manifestations of Andersen-Tawil syndrome (LQT7). Heart Rhythm. 2006;3(3):328–35.
Sicouri S, Glass A, Ferreiro M, Antzelevitch C. Transseptal dispersion of repolarization and its role in the development of torsade de pointes arrhythmias. J Cardiovasc Electrophysiol. 2010;21(4):441–7.
Sicouri S, Timothy KW, Zygmunt AC, Glass A, Goodrow RJ, Belardinelli L, Antzelevitch C. Cellular basis for the electrocardiographic and arrhythmic manifestations of Timothy syndrome: effects of ranolazine. Heart Rhythm. 2007;4(5):638–47.
Antzelevitch C, Yan GX, Shimizu W. Transmural dispersion of repolarization and arrhythmogenicity: the Brugada syndrome versus the long QT syndrome. J Electrocardiol. 1999;32(Suppl):158–65.
Yan GX, Shimizu W, Antzelevitch C. Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation. 1998;98(18):1921–7.
Yan GX, Rials SJ, Wu Y, Liu T, Xu X, Marinchak RA, Kowey PR. Ventricular hypertrophy amplifies transmural repolarization dispersion and induces early afterdepolarization. Am J Physiol Heart Circ Physiol. 2001;281(5):H1968–75.
Akar FG, Wu RC, Juang GJ, Tian Y, Burysek M, Disilvestre D, Xiong W, Armoundas AA, Tomaselli GF. Molecular mechanisms underlying K+ current downregulation in canine tachycardia-induced heart failure. Am J Physiol Heart Circ Physiol. 2005;288(6):H2887–96.
Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287(4):H1762–70.
Akar FG, Nass RD, Hahn S, Cingolani E, Shah M, Hesketh GG, DiSilvestre D, Tunin RS, Kass DA, Tomaselli GF. Dynamic changes in conduction velocity and gap junction properties during development of pacing-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293(2):H1223–30.
Qu J, Volpicelli FM, Garcia LI, Sandeep N, Zhang J, Marquez-Rosado L, Lampe PD, Fishman GI. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009;104(3):365–71.
Spragg DD, Kass DA. Pathobiology of left ventricular dyssynchrony and resynchronization. Prog Cardiovasc Dis. 2006;49(1):26–41.
Spragg DD, Leclercq C, Loghmani M, Faris OP, Tunin RS, DiSilvestre D, McVeigh ER, Tomaselli GF, Kass DA. Regional alterations in protein expression in the dyssynchronous failing heart. Circulation. 2003;108(8):929–32.
Aiba T, Hesketh GG, Barth AS, Liu T, Daya S, Chakir K, Dimaano VL, Abraham TP, O’Rourke B, Akar FG, Kass DA, Tomaselli GF. Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation. 2009;119(9):1220–30.
Barth AS, Aiba T, Halperin V, DiSilvestre D, Chakir K, Colantuoni C, Tunin RS, Dimaano VL, Yu W, Abraham TP, Kass DA, Tomaselli GF. Cardiac resynchronization therapy corrects dyssynchrony-induced regional gene expression changes on a genomic level. Circ Cardiovasc Genet. 2009;2(4):371–8.
Spragg DD, Akar FG, Helm RH, Tunin RS, Tomaselli GF, Kass DA. Abnormal conduction and repolarization in late-activated myocardium of dyssynchronously contracting hearts. Cardiovasc Res. 2005;67(1):77–86.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag London
About this chapter
Cite this chapter
Akar, F.G., Tomaselli, G.F. (2013). Electrophysiological Remodeling in Heart Failure. In: Gussak, I., Antzelevitch, C. (eds) Electrical Diseases of the Heart. Springer, London. https://doi.org/10.1007/978-1-4471-4881-4_22
Download citation
DOI: https://doi.org/10.1007/978-1-4471-4881-4_22
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-4880-7
Online ISBN: 978-1-4471-4881-4
eBook Packages: MedicineMedicine (R0)