
Abstract
The androgen receptor (AR) is a nuclear hormone receptor that plays a key role in the development of the prostate and progression of prostate cancer (PCa). AR is the initial target for treatment in hormone dependent PCa, where inhibition of AR activity results in a decrease in tumor volume and increased survival. However, PCa often returns as castration-resistant prostate cancer (CRPC) due to several factors including AR overexpression, reactivation of the full-length receptor (AR-FL), and expression of constitutively activate truncated AR-variants (AR-Vs) that lack the ligand binding domain. Upon reactivation of AR-FL and expression of AR-Vs, traditional methods of treatment targeting the ligand binding domain are ineffective. Targeting the evolutionarily conserved N-terminal domain, DNA binding domain (DBD) or using transcription factors that interact with AR will be key in developing new treatments for PCa.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Androgen receptor (AR)
- Prostate cancer (PCa)
- Androgen receptor variants (AR-Vs)
- Castration-resistant prostate cancer (CRPC)
- Phosphorylation
- Reactivation of AR
- Androgen deprivation therapy (ADT)
- Androgen response element (ARE)
- DNA-binding domain (DBD)
Target: Androgen Receptor
The androgen receptor (AR) is a member of the steroid hormone nuclear receptor superfamily; other family members consist of the estrogen (ER), progesterone (PR), mineralocorticoid (MR), and glucocorticoid (GR) receptors. AR plays a vital role in sexual development and in the development of the prostate by regulating cellular proliferation, survival, and apoptosis. The AR gene is located on chromosome Xq11-12 and is transcribed and then translated into a 919 amino acid protein. Full-length AR (AR-FL) is comprised of eight exons that encode four distinct regions of the protein. The N-terminal domain (NTD) contains an activation function motif (AF1) and is highly unstructured. Within the AF1 motif, there are two transcriptional activation units, termed TAU1 and TAU2, that modulate AR transcriptional activity. Adjacent to the NTD is the DNA-binding domain (DBD) that contains two zinc fingers; the first zinc finger binds DNA and the second facilitates dimerization with a second AR monomer. A short hinge region links the DBD to the C-terminal ligand-binding domain (LBD). The LBD encompasses a second transcriptional activation function (AF2) region. Upon ligand binding, AR undergoes a conformational change, exposing functional residues that are critical for dimerization of AR and transcriptional activation. A number of amino acids in AR that are posttranslationally modified are important for regulating transcriptional activity, protein stability, cellular localization, and cellular growth (Bennett et al. 2010; Van der Steen et al. 2013).
Biology of Androgen Receptor
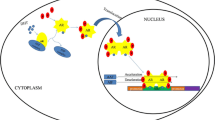
The androgen receptor is shuttled between the cytoplasm and nucleus, depending upon its activation status. Inactive AR resides in the cytoplasm, bound to chaperone and heat shock proteins (HSP90, HSP70, and HSP56) that facilitate folding, impede nuclear localization, and increase stability of the of the nascent inactive receptor. Testosterone, which is one of the ligands for AR, is produced in the Leydig cells of the testis in response to hormonal signals from the hypothalamus and pituitary gland. Testosterone circulates in the serum bound tightly to sex hormone binding globulin and loosely to albumin. After testosterone enters the cell, it is converted to the more potent androgen 5-alpha-dihydrotestosterone (DHT) by 5-alpha-reductase enzymes. The affinity of DHT for AR is 2–3 times greater than that of testosterone. DHT binds to the inactive monomeric AR LDB resulting in a conformational change of AR, release from HSPs, dimerization, and translocation to the nucleus (Vander Griend and Isaacs 2009; Mohler et al 2011).
Nuclear AR acts as a potent transcription factor . The first zinc finger binds to the major groove of DNA to a sequence known as an androgen response element (ARE) that is located within androgen-regulated genes. The full ARE motif is a 15-base pair sequence that consists of two hexameric half-sites, 5′-AGAACA-3′, arranged as a palindromic repeat with a three-base pair spacer sequence, 5′-AGAACA-XXX-ACAAGA-3′, where X is any nucleotide. Additionally, AR can also bind to a half-ARE, where only one of the two hexameric sites is encoded in the DNA. These response elements are located within the enhancer and promoter regions of androgen-regulated genes and are critical for transcriptional activation or silencing of genes (Agoulnik and Weigel 2009; Sahu et al. 2014). More than 300 cofactors can interact with AR depending upon cell type, gene, and stage of the cell cycle (Agoulnik and Weigel 2009; Nuclear Receptor Signaling Atlas 2014). Other transcription machinery such as histone acetyltransferase, histone methyltransferase, and DNA polymerase are recruited to change the conformation of chromatin into an active motif. Activated full-length AR (AR-FL) stimulates cell proliferation via the production and secretion of many growth factors such as epidermal growth factor (EGF), keratinocyte growth factor (KGF), and insulin-like growth factor (IGF), cyclin-dependent kinases (cdks), and cyclins and by the recruitment of replication machinery and checkpoint proteins to ensure proper replication of DNA prior to cell division (Balk and Knudsen 2008; Vander Griend and Isaacs 2009).
Recently, AR splice variants (AR-Vs) have been identified in prostate cancer cell lines and patient tumor samples. Most AR-Vs contain the NTD and DBD, but have a truncated C-terminus. Due to the absence of the LBD, AR-Vs are regulated by the two TAU domains (TAU1 and TAU2) located within the AF1 region in the N-terminus and are constitutively active. To date, more than 20 AR-Vs have been identified, and their role in prostate cancer is still being investigated (Lonergan and Tindall 2013). It has been speculated that AR-Vs arise from androgen deprivation or antiandrogen treatments, which are the current mainstay treatments for prostate cancer (PCa). There are two hypotheses for their function, the first being that the full-length and variant receptors regulate the same genes, implying that androgen-regulated genes are controlled by both the constitutively active variants and the activated full-length receptor. An alternative hypothesis is that AR-Vs regulate a unique subset of genes. This would suggest that AR-FL and AR-Vs do not regulate the same genes and that AR-Vs may regulate a distinct set of genes in PCa. The coexistence of AR-FL and AR-Vs within the same cells results in the potential for the formation of several different dimers. As with the ligand-activated AR-FL dimer, AR-Vs contain the first and second zinc fingers needed for dimerization and DNA binding. Therefore, AR-Vs can form homodimers with the same AR-V and can potentially form heterodimers between different AR-Vs or AR-Vs and the full-length receptor. However, the extent of the formation of heterodimers is unknown, and their function is still under investigation.
High-Level Overview of Androgen Receptor
Most of what is known about the role of AR in cellular proliferation is based on AR-FL studies. Several protein-protein interactions that occur with AR during cell cycle progression require the binding of androgen to the LBD. This induces conformational change, presumably to expose new binding sites or allow for modifications such as phosphorylation, methylation, SUMOylation, ubiquitination, and acetylation to various AR residues (Balk and Knudsen 2008). Due to the importance of the LBD for protein-protein interaction, it is still unknown how, or if, the AR-Vs that lack this domain are able to regulate the cell cycle in a similar manner when interactions occur within the LBD. Additionally, prostate cells that express truncated AR-Vs, but not AR-FL, are able to proliferate, suggesting that AR-Vs are able to regulate cellular proliferation in the absence of AR-FL. Endogenous interactions with the NTD of AR-FL are found to occur in AR-Vs, which could regulate the cell cycle.
AR regulates several genes that are key for cell cycle progression . Androgen stimulation promotes mTOR-dependent accumulation of cyclins D1 and D3, which are required for progression through the G1/S transition of the cell cycle. The accumulation of cyclin D1 results in phosphorylation of retinoblastoma (RB) that allows for expression of cell cycle genes. Cyclin D1 binds to the N-terminus of AR and disrupts the N-terminal-LBD interaction of AR, thereby limiting AR transactivation and binding to DNA. As a result of cyclin D1 binding to AR, cellular proliferation decreases due to insufficient AR activity. In the absence of androgens, cdk 4/6-cyclin D and cdk 2-cyclin E complexes needed for G1/S transition are mostly inactive. AR-cdk 6 interaction occurs at the chromatin level, suggesting that cdk 6 is part of the AR transcriptional complex that assembles at AREs. AR-cdk 6 interaction is independent of cyclin D. Cell division cycle 25 (Cdc25) is a dual function phosphatase and is a coactivator of AR along with other steroid receptor family members (ER, PR, and GR). Cdc25 mediates the activation of cdk1, which allows for progression through the cell cycle (Balk and Knudsen 2008; Ruscetti and Wu 2013; Kokontis et al. 2014).
Phosphorylation of AR at serine 81 (AR-S81) regulates cellular translocation of AR to the nucleus, protein stability, recruitment to specific AREs, and overall transcriptional activity. Several cdks phosphorylate AR-S81 including cdk 1, cdk 5, and cdk 9. Phosphorylation at AR serine 308 by cdk11-cyclin D3 is delayed upon AR activation and thus acts as an inhibitory site. Alternatively, cyclin E binds to AR at amino acids 419–556 and enhances AR activity. Cyclin H and cdk7 regulate AR by interacting with the N-terminal domain of AR. Cdk7 phosphorylates AR serine 515 allowing for polyubiquitination of AR, resulting in degradation of AR protein (Van der Steen et al. 2013).
Androgen Receptor Assessment
Expression of AR in the laboratory is determined largely by western blotting where antibodies are available to detect and delineate between AR-FL and AR-Vs based on molecular weight. In clinical samples, it is possible to examine expression of AR-FL by immunostaining, but there are currently no reliable antibodies that can be used to detect individual AR-Vs. To monitor AR activity in cells in culture, luciferase assays are used with constructs comprised of AR-regulated gene promoters attached to the luciferase gene allowing for detection of AR activity. Additionally, measuring mRNA expression changes of AR-regulated genes as a result of treatment with activators/inhibitors can determine AR activity. In patients, in vivo AR activity is measured by monitoring serum PSA levels, where increased PSA levels may indicate prostatitis, benign prostate hyperplasia (BPH), or prostate cancer. Androgen deprivation therapy (ADT) reduces PSA levels initially, but often PCa recurs in the form of CRPC; recurrence is initially determined by an increase in serum PSA levels (Gupta et al. 2011). There are no current clinical assessments to delineate between expression of AR-FL or AR-Vs in serum samples.
Role of Androgen Receptor in Cancer
Prostate Cancer Target AR
Rank: 10
The discovery that the AR plays a pivotal role in proliferation and apoptosis in the prostate was made over 70 years ago by Charles Huggins and Clarence Hodges. Their work set the stage for the investigation and study of AR in normal and cancerous prostate as a key transcription factor and as a therapeutic target for treatment of PCa. Removal of androgens results in the reduction of prostate tumor volume suggesting that the androgen-AR signaling axis plays a critical role in regulating cellular proliferation and apoptosis (Balk and Knudsen 2008). Limiting the amount of androgens available to activate AR results in decreased tumor burden, but in castration recurrent prostate cancer (CRPC), the AR is reactivated at insignificant levels of androgen or is independent of androgens. In CRPC, reactivated AR allows for proliferation resulting in an increase in tumor size. An increase in kinase activity resulting in increased phosphorylation yields an increase in AR activity and cell proliferation. The recent identification of AR splice variants that lack the LBD and are constitutively active may play a critical role in CRPC (Lonergan and Tindall 2013; Ware et al. 2014). AR-Vs arise as a result of androgen deprivation therapy (ADT) and antiandrogen treatment. Currently, there is great interest in how these variants arise, how they are regulated, if variants regulate the same genes as the full-length receptor, and if all AR-Vs are similar in their function in PCa.
Reactivation of AR is known to occur through several additional mechanisms including (1) AR mutations, (2) AR overexpression, (3) ligand-independent activation, and (4) increased AR protein stability. Mutations to the AR gene are found in approximately 10% of CRPC. Most of these mutations occur in the C-terminus, which results in decreased ligand specificity. These mutations allow for other steroids to bind to the LBD of AR such as adrenal androgens, glucocorticoids, and progesterone and even antiandrogens (The Androgen Receptor Gene Mutation Database World Wide Web Server 2014). Overexpression of AR is observed in approximately 35% of CRPCs after ADT. The increase in AR mRNA expression may occur as a result of either AR gene amplification, which often occurs in cancers, or increased transcriptional expression of the AR gene (Bennett et al. 2010). The third possible mechanism is ligand-independent activation of AR. Noncanonical activation of AR can occur as a result of cytokines and growth factor signaling that activates downstream kinases. For example, interleukin 6 (IL-6) expression increases during progression to CRPC and can transactivate AR. IL-6 signaling through the MAPK pathway can regulate p300, an acetyltransferase, which is a coregulator of AR. The loss of the tumor suppressor PTEN, which is a potent regulator of the PI3K/AKT/mTOR pathway, is often observed in CRPC. Kinases in both of these pathways phosphorylate and regulate AR activity. In CRPC, degradation of AR protein is approximately half of that in androgen-dependent PCa. The increased half-life of AR may result in enhanced AR activity (Ruscetti and Wu 2013; Vander Steen et al. 2013).
Preclinical Summary
The increase of AR-Vs in CRPC and the reactivation of the AR-FL have resulted in the focus of treatment to shift from the CTD of AR toward the NTD of AR, which is conserved between most AR-Vs and AR-FL. One hypothesis is that AR-Vs arise as a result of the current treatment for PCa leading to development of CRPC. Identifying inhibitors of the NTD of AR have the potential to limit the activity of not only the full-length AR but also AR-Vs. Since AR-Vs are present in CRPC, in theory, this would allow for a single type of treatment that would target all forms of AR.
One potential treatment directed toward the NTD of AR is small molecule inhibitor EPI-001. EPI-001 interacts with the AF1 region, which is the regulatory region for AR-Vs, and inhibits protein-protein interactions. The loss of these interactions results in decreased recruitment of both AR-FL and ARv567es to AREs, thereby limiting expression of AR target genes. Treatment of cells that express either AR-FL and/or AR-Vs with EPI-002, an analog of EPI-001, results in a decrease in growth of CRPC xenografts expressing either AR-FL or ARv567es (Myung et al. 2013). These studies have demonstrated the potential to target AR-Vs and limit cell cycle progression; however, additional studies using other AR-Vs are needed.
A second region that is highly conserved between AR-FL and AR-Vs is the DNA-binding domain. Inhibiting AR interaction with chromatin by targeting its DBD is the focus of further research. One small molecule inhibitor, pyrvinium pamoate, that is a noncompetitive inhibitor directed toward the AR DBD has therapeutic potential (Dalal et al. 2014). Xenografts treated with pyrvinium pamoate result in inhibition of AR-V constitutive activity and cellular growth. Other molecules that are derivatives of morpholine have been shown to bind to a pocket in the DBD and disrupt AR-chromatin interactions (Lu et al. 2015). Through inhibition of these interactions, there is therapeutic potential to treat all forms of AR that bind DNA.
Other potential means of inhibiting AR activity include targeting heat shock proteins, which bind AR in the cytoplasm and affect protein stability. Through inhibition of HSP90, there is improper folding and destabilization of AR protein. However, two variants (AR-V7 and ARv567es) have exhibited resistance to HSP90 inhibitors due to the lack of interaction of AR-Vs with HSP90 (Azad et al 2015).
Clinical Summary
Currently, limiting the activity of AR is used for treatment of advanced PCa. Impeding AR activity is obtained through (1) removal of the prostate, (2) chemical/surgical castration, (3) treatment with antiandrogens, and (4) use of an AR antagonist. Initially, androgen deprivation therapy (ADT) is used to reduce androgen levels in the body and ultimately leads to suppression of AR activity. The decrease in androgen synthesis does not cure prostate cancer, but is used if PCa has metastasized and cannot be contained by surgery or radiation. ADT is also used upon recurrence of PCa to the refractory state but in these cases only results in a remission of 2–3 years from the time of initiation of ADT.
To decrease testosterone, men with PCa may undergo surgical castration (orchiectomy) to remove the testes where most of the androgens are produced. A second option that is used is treatment with luteinizing hormone-releasing hormone (LHRH) analogs, also known as chemical or medical castration, to lower androgen levels, similar to orchiectomy. Due to the loss of testicular androgens, some prostate cells synthesize their own androgens to activate AR. CYP17 is a key enzyme for the biosynthesis of androgens and is increased in CRPC when compared to primary prostate tumors of untreated men. Abiraterone is often given as a treatment to inhibit CYP17 activity in cells making their own androgens (Mohler et al 2011; Stein et al. 2014).
Inhibition of AR is also achieved using antiandrogens, which bind to AR and prevent androgens from binding and activating AR. Antiandrogens include flutamide, bicalutamide, and nilutamide as a daily treatment. Antiandrogens are usually given in conjunction with ADT treatment to inactivate AR. Use of these treatments results in decreased cell proliferation. Enzalutamide (MDV3100) is a newer antiandrogen that binds to AR, thereby inhibiting its interaction with endogenous androgens. The use of enzalutamide as a therapeutic for CRPC resulted in a decrease in PSA levels in 50% of men and was found to result in an increased life span. Current clinical studies are being conducted to determine the effectiveness of enzalutamide versus bicalutamide in men with CRPC who have progressed while receiving LHRH therapy or orchiectomy (Suzman and Antonarakis 2014).
Current treatments for advanced PCa all target the LBD of AR, either through treatment with antiandrogens or ADT. Once PCa progresses to CRPC, the AR-FL can be activated at castrate levels of androgens by a number of mechanisms, and in many cases there is a rise in the expression of AR-Vs. AR-Vs lack any regulation by androgens due to the truncation of the C-terminal domain and are unresponsive toward these methods of treatment. New treatments that focus on the NTD and DBD of AR are being developed but have yet to be approved for clinical use.
Anticipated High-Impact Results
-
Determine mechanistically how AR-Vs arise in prostate cancer and if they contribute to the development of CRPC.
-
Delineate pathways allowing for constitutive activity of AR-Vs.
-
Develop methods for detecting the presence of AR-Vs in PCa patient samples.
-
Determine if there is a difference between AR-FL and AR-V gene targets that allow for the proliferation of cells to be uncontrolled due to the constitutive activity of AR-Vs.
-
Development of novel therapeutic agents that are directed toward the N-terminal domain of AR allowing for targeting of both the full-length and variant forms of AR.
References
Agoulnik IU, Weigel NL. Coregulators and the regulation of androgen receptor action in prostate cancer. In: Tindall D, Mohler J, editors. Androgen action in prostate cancer. New York: Springer; 2009. p. 315–40.
Azad AA, Zoubeidi A, Gleave ME, Chi KN. Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nat Rev Urol. 2015;12(1):26–36.
Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6:e001.
Bennett NC, Gardiner RA, Hopper JD, Johnson DW, Gobe GC. Molecular cell biology of androgen receptor signaling. Int J Biochem Cell Biol. 2010;42(6):813–27.
Dalal K, Roshan-Moniri M, Sharma A, Li H, Ban F, Hassona MD, et al. Selectively targeting DNA-binding domain of the androgen receptor as a prospective therapy for prostate cancer. J Biol Chem. 2014;289(38):26417–29.
Gupta A, Lilja H, Schroeder FH. Detection of prostate cancer: PSA. In: Tindall DJ, Scardino PT, editors. Recent advances in prostate cancer basic science discoveries and clinical advances. Singapore: World Scientific; 2011. p. 283–334.
Kokontis JM, Lin H, et al. Androgen suppresses the proliferation of androgen receptor-positive castration-resistant prostate cancer cells via inhibition of the cdk2, cyclin A, and Skp2. Plos One. 2014;9(10):e109170.
Lonergan PE, Tindall DJ. Truncated androgen receptor splice variants in prostate cancer. In: Tindall DJ, editor. Prostate cancer biochemistry, molecular biology and genetics. New York: Springer; 2013. p. 351–82.
Lu J, Van der Steen T, Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor? Nat Rev Urol. 2015;12(3):137–44.
Mohler JL, Titus MA, Godoy AS, Oka D, Nelson PS. Intracrine synthesis of androgens by prostate cancer in response to androgen deprivation therapy. In: Tindall DJ, Scardino PT, editors. Recent advances in prostate cancer basic science discoveries and clinical advances. Singapore: World Scientific; 2011. p. 193–218.
Myung J, Banuelos CA, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123(7):2948–60.
Nuclear Receptor Signaling Atlas. Baylor College of Medicine, Houston. 2014. http://www.nursa.org. Accessed 10 Mar 2015.
Ruscetti MA, Wu H. PTEN in prostate cancer. In: Tindall DJ, editor. Prostate cancer biochemistry, molecular biology and genetics. New York: Springer; 2013. p. 87–138.
Sahu B, Pihaljamaa P, Dubois V, Kerkhofs S, Claessen F, Janne OA. Androgen receptor uses relaxed response element stringency for selective chromatin binding and transcriptional regulation in vivo. Nucleic Acids Res. 2014;42(7):4230–40.
Stein MN, Patel N, Bershadskiy A, et al. Androgen synthesis inhibitors in the treatment of castration-resistant prostate cancer. Asian J Androl. 2014;16(3):387–400.
Suzman DL, Antonarakis ES. Castration-resistant prostate cancer: latest evidence and therapeutic implications. Ther Adv Med Oncol. 2014;6(4):167–79.
The Androgen Receptor Gene Mutations Database World Wide Web Server. McGill University, Montreal. 2014. http://www.androgenb.mcgil.ca. Accessed 24 Feb 2015.
Van der Steen T, Tindall DJ, Huang H. Posttranslational modification of the androgen receptor in prostate cancer. Int J Mol Sci. 2013;14(7):14833–59.
Vander Griend DJ, Isaacs JT. Androgen receptor as a licensing factor for DNA replication. In: Tindall D, Mohler J, editors. Androgen action in prostate cancer. New York: Springer; 2009. p. 619–30.
Ware KE, Garcia-Blanco MA, Armstrong AJ, Dehm SM. Biological and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr Relat Cancer. 2014;21(4):T87–103.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Science+Business Media New York
About this entry
Cite this entry
Van der Steen, T., Schmidt, L.J., Tindall, D.J. (2017). AR, Overview. In: Marshall, J. (eds) Cancer Therapeutic Targets. Springer, New York, NY. https://doi.org/10.1007/978-1-4419-0717-2_111
Download citation
DOI: https://doi.org/10.1007/978-1-4419-0717-2_111
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4419-0716-5
Online ISBN: 978-1-4419-0717-2
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences