
Abstract
Over the past several years, significant progress has been made in the treatment of gynecologic cancers. However, continued improvements to existing therapies, as well as development of novel approaches to treat these diseases, will be necessary to further reduce mortality. With increasing evidence that ovarian cancer in particular is immunogenic, there is a good reason to investigate the potential of immunotherapies. Recent success in a number of immunotherapy clinical trials targeting other tumor types has laid the groundwork necessary to begin using similar therapies against ovarian and other gynecologic cancers. Here we review past experience and future opportunities for immunotherapy in ovarian cancer, with a focus on vaccines and adoptive T-cell therapy, as well as several nonspecific immunomodulators available for immediate clinical testing.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
FormalPara Summary Points-
Active versus passive immunization.
-
Public versus private antigen targeting.
-
Naturally occurring TILs versus engineered T cells.
-
Single therapy versus combinational approach.
Background and Introduction
Although not traditionally considered immunogenic, increasing evidence indicates that ovarian cancers are, in fact, immunogenic tumors and are responsive to immunotherapies. Three distinct categories of data support this claim. First, there is accumulating evidence of spontaneous antitumor immune responses and of their association with longer survival in a proportion of ovarian cancer patients. Second, and conversely, there is evidence of active tumor immune evasion mechanisms and their association with short survival in some ovarian cancer patients. And finally, preclinical as well as clinical data have now demonstrated that immunotherapy can be efficacious against these cancers.
It is now clear that spontaneous antitumor immune responses exist in many ovarian cancer patients. Tumor-reactive T cells and antibodies have been detected in peripheral blood of patients with advanced stage disease at diagnosis [1, 2], while oligoclonal tumor-reactive T cells have been isolated from tumors or ascites [3–11]. The tumor rejection antigens expressed by ovarian cancer have not been thoroughly characterized. Among the most promising candidates are cdr2, mesothelin, and NY-ESO-1 [12–14]. Several additional well-known tumor-associated antigens are recognized by peripheral blood or tumor-associated lymphocytes of many ovarian cancer patients. These include p53; HER2/neu; folate receptor-α; cancer-testis antigens such as the MAGE melanoma antigen family members and sperm surface protein Sp17; mucins or glycoproteins such as Lewis(y), sialylated-Tn, CA-125, and MUC-1; and universal tumor antigens such as survivin and hTERT [15]. Importantly, the detection of an antitumor immune response in the form of intraepithelial (also called intratumoral) tumor-infiltrating lymphocytes (TILs), i.e., T cells infiltrating tumor islets, predicts significantly longer survival in ovarian cancer. We first reported in an Italian cohort that patients whose tumors had intraepithelial T cells experienced longer progression-free and overall survival as compared to patients whose tumors lacked intraepithelial T cells [16]. Survival at 5 years was substantial (38 %) in patients whose tumors had intraepithelial T cells (n = 102) and negligible (4.5 %) in patients lacking them (n = 72), even after complete response to chemotherapy. A signature of antitumor immune response activation was identified in tumors with intraepithelial T cells [16]. The impact of intraepithelial CD3+ or CD8+ T cells was confirmed by multiple independent studies on ethnically and geographically diverse populations [17–21]. Importantly, intraepithelial T cells were more prevalent in tumors with increased proliferation, indicating that improved outcome is not due to indolent tumor cell behavior [17].
Significant progress has been made recently in our understanding of immune evasion mechanisms operating in some patients with ovarian cancer. CD4+ CD25+ FoxP3+ T regulatory (Treg) cells were first demonstrated in ovarian cancer [22, 23], where increased Treg frequency predicts poor patient survival [20, 23]. Immunosuppressive B7-H4 expressing macrophages were recently found to correlate with survival in ovarian cancer [24]. In addition, ovarian cancer cells express programmed death ligand 1 (PD-L1 or B7-H1), a ligand for the immunosuppressive T-cell receptor PD1, which blocks T-cell responses. Expression of PD-L1 by tumor cells predicted paucity of intraepithelial TILs and short overall survival in ovarian cancer [19]. Further, overexpression of the endothelin B receptor (ETBR), which suppresses T-cell-endothelial adhesive interactions and T cell homing to tumor, correlated with absence of TIL and short survival in ovarian cancer [25, 26]. Finally, a recent study segregated high- and low-risk ovarian cancer patients based upon their tumor gene signature and found a strong correlation between decreased expression of immune genes and the development of high-risk tumors. In particular, high-risk tumors often displayed downregulation of genes involved in antigen processing and presentation [27].
The association of antitumor immune responses with prolonged survival and, vice versa, the association of immune escape mechanisms with poor survival suggest that ovarian cancers are intrinsically immunogenic. Indeed, ovarian cancers should no longer be considered immunologically inert tumors. Accordingly, pilot clinical data indicate that ovarian cancer patients can, in fact, respond to the same immunotherapy approaches as patients with other immunogenic tumors [28], including interleukin-2 (IL-2) [29, 30], anti-CTLA-4 antibody [31, 32], and adoptive transfer of ex vivo expanded TIL [33, 34]. Notably, each of these therapies is designed to exploit a preexisting endogenous antitumor immune response. Although, insufficient to reject tumor naturally, these responses can potentially be harnessed therapeutically. Here we will review three categories of immunotherapies which can be used to manipulate natural antitumor immunity or to induce new antitumor immune responses. These include cancer vaccines (active immunization), adoptive T-cell therapy (passive immunization), and nonspecific immunomodulation. Each targets immune cells in different ways. They can be used alone, together, or with conventional approaches for combinatorial tumor therapy.
Cancer Vaccines
As with many other tumor types, vaccines have been the primary approach to ovarian cancer immunotherapy so far [15, 35–37]. Consistent with experience in other immunogenic tumors [38], vaccines have shown limited efficacy as monotherapy in patients with advanced recurrent disease. Clearly, much work is required to improve their performance. Current efforts to improve vaccines are directed broadly towards (a) optimizing the choice of antigens, (b) improving vaccine delivery systems to maximize the magnitude and quality (phenotype and polarization) of T-cell response, and (c) developing combinatorial approaches with adoptive T cell or immunomodulation therapy to maximize activation and function of vaccine-primed T cells in vivo.
Pros
The results of some studies provide encouragement for further vaccine development. In a retrospective review of patients treated in the adjuvant setting after secondary complete response, Sabbatini and colleagues noted that patients vaccinated with monovalent or heptavalent vaccines against carbohydrate epitopes experienced significantly longer time to progression and higher progression-free survival rates relative to controls from the same institutions treated with alternative consolidation therapies [39]. In addition, vaccination with anti-idiotype ACA-125, an analogue of CA-125, resulted in CA-125-specific antibodies and was associated with prolonged survival [40]. Another study was performed using CEA-MUC-1-TRICOM poxviral-based vaccines in 16 patients including 3 ovarian cancer patients. Immune responses to MUC-1 and/or CEA were seen following vaccination in 9 patients. A patient with clear cell ovarian cancer and symptomatic ascites had a radiographically and biochemically durable (18-month) clinical response [41]. In another study, vaccination against HER2 has resulted in sustained antigen-specific T-cell and humoral immunity as well as epitope spreading in ovarian cancer patients [42].
An alternative to vaccines directed towards specific antigens is whole tumor antigen vaccines created using tumor cells, autologous tumor lysate, or tumor-derived RNA [43–45]. Tumor antigen preparations can be injected into patients directly, or they can be first loaded onto autologous dendritic cells. Advantages of these vaccines include the opportunity to induce immunity to a personalized and broad range of antigens, which could minimize the development of tumor escape variants, the inclusion of yet unidentified tumor rejection antigens, no HLA haplotype restriction, and the simultaneous administration of MHC class I and class II epitopes, which could prove beneficial for immunologic memory. In a pilot study using mature DCs pulsed with whole autologous tumor lysate, three of six subjects demonstrated remission inversion, i.e., their progression-free survival postvaccination was longer than the interval between pre-vaccine recurrence and prior chemotherapy treatment [46]. The use of DC/tumor cell fusion approach is a viable alternative whereby autologous DCs are fused with tumor cells, which allows DCs to express the entire antigen repertoire of the tumor cells to CD4+ and CD8+ T cells. DC/ovarian tumor cell fusions have been generated and demonstrated to be able to induce antitumor CTL activity in vitro [47].
Several groups have used viruses to increase tumor cell immunogenicity for whole tumor cell vaccination. Objective responses have been seen after intracavity delivery of a viral oncolysate vaccine generated with ovarian cancer cell lines infected with influenza A virus [48, 49] or with autologous tumor cells infected with Newcastle disease virus [50]. We also performed preclinical studies using replication-restricted herpes simplex virus (HSV) 1 to infect autologous tumor cells for vaccine preparation. HSV-infected tumor cells used directly or pulsed on dendritic cells elicited potent antitumor immune response in the mouse, which was superior to the use of UV-irradiated tumor cells [51–53]. Thus, whole tumor antigen vaccines can produce objective response if immunogenicity is increased through the use of pathogens.
Cons
A major limitation of cancer vaccines presently stems from the inability to elicit a rapid and overwhelming T-cell response, which is required to reject established tumors. This problem is magnified in ovarian cancer by the paucity of well-characterized rejection antigens to target and by the significant molecular heterogeneity of the disease [54]. Even when a defined target is available, and vaccination successfully induces an immune response, the long-term benefit can be limited by tumor evolution. In a recent study, one patient experienced complete objective response to NY-ESO-1 peptide vaccine, but later recurred with an NY-ESO-1-negative tumor, proving that single-target immunization can result in immune escape tumor variants following initial response [55].
A recent meta-analysis of 173 published, peer-reviewed immunotherapy trials revealed the low success rate of cancer vaccines to date. The trials involved patients with a variety of tumor types, including melanoma, renal cell and hepatocellular carcinomas, lung, prostate, breast, colorectal, cervical, pancreatic, and ovarian cancers. Patients received either molecular-defined antigens (synthetic peptides or proteins and viral or plasmid vectors encoding peptides or proteins; 1,711 patients) or whole tumor antigen (autologous or allogeneic tumor cells, dendritic cells pulsed with tumor extracts or mRNA; 1,733 patients). Overall, the authors calculated that 8.1 % of patients vaccinated with whole tumor antigen had objective clinical responses while 3.6 % of patients vaccinated with molecularly defined tumor antigens had objective clinical responses (p < 0.0001, chi-square test) [56].
Although whole tumor vaccines offer distinct advantages, some drawbacks warrant consideration. First, surgical procurement of large numbers of autologous tumor cells may not be possible in many patients. Alternatives to this limitation exist, including use of allogeneic cell lines or the use of tumor mRNA. RNA electroporation of DCs is a convenient approach to generate a potent tumor vaccine [52]. An additional concern with whole tumor vaccination relates to the inclusion of a large number of “self” antigens, which could potentially drive tolerogenic responses, i.e., expand Treg rather than cytotoxic lymphocyte responses. Recent work has demonstrated that DCs can be polarized ex vivo with the use of interferons, Toll-like receptor agonists, or p38 mitogen-activated protein kinase (MAPK) inhibitors to drive cytotoxic lymphocytes and Th17 effector cells at the expense of Treg [57]. On the other hand, if immunization is successful, there may be increased concern for breaking tolerance to “self” antigens, leading to immunopathology. To date, pilot studies with whole tumor vaccines have reported no autoimmunity in patients with ovarian cancer.
There is a controversy in the choice of target antigen with cancer vaccines and adoptively transferred T cells, as well. In the past few decades, shared (also known as “public”) tumor-associated antigens have been the favored target of various immunotherapy strategies. This approach has been based largely on studies with melanoma [58]. This leads to the concept of “dispensable tissues,” meaning that in order to achieve tumor eradication, it was necessary to expect tissue-specific toxicity damaging normal tissues [59]. As the expression of these antigens was shared between most individuals, this would make the manufacturing of a universal vaccine a possibility. However, recent advances in the clinical application of immunotherapy suggest that immunotherapy with “personalized” antigens (that arise from mutations) with preexisting immunity, which are designed to stimulate antigen-specific memory T cells, could also be expected to induce rapid and strong secondary immune responses (reviewed in [60, 61]). The current view is that both approaches, targeting public or targeting private antigens, can be beneficial either in cancer vaccines or adoptive T-cell therapy, but to increase the clinical benefits, special attention should be paid to the immunological status of each patient by characterizing the preexisting immune responses to the targeted antigens before immunotherapy.
Adoptive T-Cell Therapy
Effective cancer immunotherapy is dependent on the presence of large numbers of antitumor lymphocytes with appropriate homing and effector functions that enable them to seek out and destroy cancer cells in vivo. The adoptive transfer of ex vivo expanded tumor-reactive T cells holds the potential of achieving this condition in a short period of time. Clinical trials testing spontaneous or induced polyclonal or oligoclonal T cells conducted in the past two decades have provided crucial lessons that can guide further optimization. The use of ex vivo expanded TILs has yielded promising clinical results. Based on animal studies showing that host lymphodepletion prior to T-cell transfer enhances persistence of T cells and antitumor responses, a scheme of incremental lymphodepletion through high dose non-myeloablating chemotherapy and added whole-body radiation was tested. Infused cells were both long lived and highly penetrating, showing regression of voluminous metastatic tumors, with up to 16 % complete response and 72 % overall objective response rates in recent reports with maximal lymphodepletion and radiation. T-cell persistence correlated with long lasting responses [38, 62]. Although these are phase I studies involving a highly selected cohort of patients with metastatic melanoma with preexisting antitumor immunity, whose tumors yield tumor-reactive TILs, the results clearly demonstrate the power of adoptive immunotherapy and dispel the assumption that immunotherapy can only control small tumors [28]. Furthermore, although the role of CD8+ T cells has been well established in adoptive immunotherapy [38, 62], CD4+ cells can also produce objective responses [63].
Currently, attempts to improve the efficacy of adoptive TIL therapy are focused on two areas: (a) optimizing methods to select tumor-reactive TIL and expand them under optimal costimulation conditions and (b) optimizing host and/or tumor conditioning. Findings from melanoma trials argue that use of memory rather than effector cells may be more efficacious for adoptive transfer [64]. In these key studies, although infused cells dominantly displayed a highly differentiated effector cell phenotype (CD27– CD28– CD45RA– CD62L– CCR7–), TILs persisting 2 months after infusion in patients who exhibited tumor regression were characterized by a less differentiated phenotype (CD27+ CD28+ CD45RA+ but CD62L– CCR7–) and longer telomeres [65–69]. Mouse models confirm these findings [70]. Because TILs comprise a large number of tumor-reactive effector cells, identification of culture conditions that preferentially expand memory phenotypes is a priority. Recent technological advances with the development of artificial antigen-presenting cells (aAPCs) expressing a variable repertoire of costimulatory molecules and cytokines have generated new opportunities to provide the desired costimulatory molecules and cytokines to reeducate TILs, improving their potency and function in vivo. Carl June and colleagues have described the development of a next-generation K562-based aAPC platform capable of expressing multiple gene inserts, including human lymphocyte antigen (HLA)-A2; CD64 (the high-affinity Fc receptor), CD80, CD83, CD86, CD137L (4-1BBL), and CD252 (Ox40L); and a variety of T-cell supporting cytokines [71]. Cell-based aAPCs have proven to be more efficient at activating and expanding CD8+ CD28– T cells, and antigen-specific T cells, than the magnetic bead-based aAPC [71].
TIL therapy is only possible for a fraction of patients. To generate TIL, a tumor mass must first be resected, which is not always possible. Additionally, that tumor mass must contain TIL, and those TIL must be responsive to the existing ex vivo expansion protocols. For many patients, these limitations make TIL therapy impossible. One strategy to make adoptive therapy available to a larger patient population involves engineering polyclonal T cells to redirect their specificity towards tumor antigens. This can be accomplished by transducing lymphocytes with a cloned T-cell receptor (TCR) of high affinity to tumor-associated epitopes. In this case, the cloned heterodimeric TCR is transduced to mixed peripheral blood T cells isolated from the patient, creating a large population of bispecific T cells, which are polyclonal with respect to their original TCR, but potentially monoclonal for the cloned TCR [72].
A second strategy to generate novel tumor-targeted T cells is to transduce the polyclonal population with receptors that recognize antigens in an MHC-unrestricted fashion. These so-called chimeric antigen receptors (CARs) are fusion genes encoding an extracellular domain that specifically binds to tumor epitopes through a single-chain variable fragment (scFv) linked to intracellular signaling modules (such as the CD3 zeta chain, TCRz) that mediate T-cell activation [72–74]. The scFv contains the V H and V L chains of an antitumor antibody joined by a peptide linker of about 15 residues in length, and it confers the parental antibody’s specificity to the transduced T cells. In principle, universal targeting vectors can be constructed, because the scFvs bind to native cell surface epitopes and bypass the requirement for MHC restriction [75, 76]. Thus, in comparison to TCRs, CARs have two major advantages: (a) their HLA-independent recognition of antigen, which makes them broadly applicable regardless of the subject’s HLA and regardless of the level of HLA expression on tumor cells, and (b) their signaling, which redirects T-cell cytotoxicity and permits T-cell proliferation and survival upon repeat antigen exposure. A potential drawback stems from their potential immunogenicity, if scFv are nonhuman. This can be averted by using human scFv.
Pros
There is evidence that TIL-based adoptive therapy is an important opportunity in ovarian cancer. In the early 1990s, ovarian cancers were found to yield reactive TILs after IL-2 culturing in vitro [77, 78]. Moreover, in pilot clinical trials, patients who received adjuvant therapy with adoptive transfer of tumor-derived lymphocytes expanded ex vivo with IL-2, following surgical debulking and frontline chemotherapy, showed a survival advantage [33, 34]. Stage III EOC patients treated with consolidation adoptive transfer of expanded TILs after completion of cisplatin-based frontline chemotherapy (n = 13) had a 3-year overall survival rate of 100 %, while that of a control group of patients (n = 10) receiving only chemotherapy was 67.5 % (p < 0.01). The 3-year disease-free survival rate of the patients in the TIL group and in the control group was 82.1 and 54.5 %, respectively. While these results can be limited by the lack of randomization, they nevertheless support the feasibility of adoptive therapy for ovarian cancer [33].
TCR-based engineering represents a potentially powerful strategy for ovarian cancer therapy as TCRs that recognize HLA-A2-restricted epitopes from known ovarian cancer antigens such as NY-ESO-1 and p53 are available for clinical testing as well [79–82]. Optimization through selection of naturally occurring or recombinant high-affinity receptors, engineering to prevent recombination with endogenous TCR, and the use of lentiviral vectors developed in the June lab with transfection efficiency above 90 % are poised to improve this approach significantly [83].
Adoptive transfer of T cells engineered to express chimeric receptors is also expected to be useful for ovarian cancer patients once the tools are refined. Some of the CARs investigated in vitro and in vivo target ovarian cancer antigens including FBP [84, 85], MUC-1 [86], HER-2, and mesothelin [87].
Cons
So far, there has only been a single study of adoptive transfer of CAR T cells in ovarian cancer [88]. Patients received autologous T cells which had been transduced with an FRα-specific CAR. While this study demonstrated safety, the results were disappointing. There were no clinically evident tumor responses—most likely due to low expression of the transgenic CAR and poor persistence of the transferred T cells [88]. However, strategies to address these issues are being developed. For instance, T-cell persistence can be dramatically improved by using human scFv and by adding costimulatory signaling capabilities to the intracytoplasmic domain of CARs. Indeed, one issue needing to be addressed with CARs is that signaling through the cytosolic domain of the usual scFv-TCRz construct does not fully replicate the multichain TCR signaling complex. This can be solved by incorporating additional signaling modules in the cytoplasmic domain of the chimeric receptor. The value of such innovations was recently demonstrated in a mouse xenograft model. Similar to the unsuccessful clinical trial, T cells were transduced with a CAR targeting FRα. However, in this study, the signaling domain of costimulatory molecule CD137 was added to the CAR’s intracellular tail. When transferred into mice, these CAR T cells demonstrated enhanced in vivo persistence and tumor infiltration and achieved tumor regression superior to that seen in mice treated with T cells lacking the CD137 signaling domain [89].
Nonspecific Immunomodulation
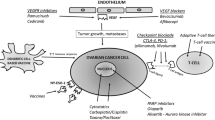
Given the limitations of immunotherapy, there is a reason to hope that modulating immune checkpoints (Fig. 29.1) by activation of effector cells, depletion of Tregs, or activation of professional APCs could substantially improve the therapeutic efficacy of vaccines or adoptively transferred T cells. Certain chemotherapy regimens promote antitumor immunity through each of these mechanisms. Additionally, a number of nonspecific immunotherapies, including immunomodulatory cytokines, Toll-like receptor (TLR) agonists, and functional antibodies, are being developed to achieve these goals. Many of these nonspecific therapies may prove to be valuable adjuvants to more targeted immunotherapies, including vaccination and adoptive T-cell therapy.

Modulation of immune checkpoints can lead to improved therapeutic efficacy of vaccines or adoptively transferred T cells
Pros
The immunomodulatory effects of chemotherapy can be broadly grouped into three mechanisms. First, chemotherapy-induced tumor cell death can result in in situ vaccination. Drugs such as doxorubicin, idarubicin, mitoxantrone, and oxaliplatin induce immunogenic tumor cell death, which facilitates tumor antigen uptake by professional antigen-presenting cells and subsequent antigen presentation to antitumor T cells. Second, some chemotherapy drugs can also induce direct activation of antigen-presenting cells. Since the 1980s, it has been recognized that cyclophosphamide administered at standard dose prior to cancer vaccines significantly enhanced immunotherapy. However, the mechanism of this phenomenon was initially unclear [90]. A recent study in the mouse reported that a myelosuppressive dose of cyclophosphamide induces rebound myelopoiesis and leads to the emergence of tumor-infiltrating DCs that secrete more IL-12 and less IL-10 and are fully capable of priming T-cell responses [91]. In addition, metronomic or low-dose, non-myelotoxic administration of paclitaxel, doxorubicin, vincristine, and other drugs can cause activation and maturation of DCs, including increased IL-12 secretion, a critical factor required for T-cell priming. Signaling via STAT4 and Rho GTPases may account for these effects [92]. The third mechanism by which chemotherapy achieves immunomodulation is through suppression of immune inhibitory cells. For instance, oral administration of metronomic cyclophosphamide was shown to induce a profound and selective reduction of circulating CD4+CD25+ regulatory T cells and restored T and NK effector functions in end-stage cancer patients [93]. Cyclophosphamide may also have additional effects contributing to restoration of the immune response; it can enhance IFN-γ production by splenocytes in a mouse model [94]. Conventional paclitaxel therapy also caused a significant decline in both numbers and activity of Treg, enhancing CD4+ and CD8+ activity systemically in patients with non-small cell lung cancer [95]. The mechanisms behind each of these immunomodulatory mechanisms are quite complex, and our understanding is still in its infancy. But effects appear to be dependent on drug type, dose, and schedule as well as the immune cell type.
Pleiotropic immune activation can also be achieved with cytokines and Toll-like receptor agonist therapy. Type I and II interferons and IL-2 are the most extensively studied cytokines for tumor therapy.
IFN-γ has been shown to have direct antiproliferative activity on ovarian cancer cells in vitro, which proved to be synergistic with cisplatin and doxorubicin [96–98]. In vitro and in vivo, IFN-γ upregulates HLA class I and class II molecules and antigen presentation in ovarian tumor cells [99], a requisite for recognition by T cells. In fact, HLA class I expression by the tumor correlates with the intensity of T-cell infiltration [100], a predictor of longer survival. Furthermore, IFN-γ has antiangiogenic effects [101].
Interleukin-2 (IL-2) promotes expansion and enhances the cytotoxicity of effector immune cells [102]. In addition, IL-2 can restore T-cell function following suppression by negative regulatory receptors such as PD-1 (see below). Because ovarian cancer patients exhibit spontaneous antitumor immune response, IL-2 therapy may be a rational approach to activate preexisting immunity or enhance immunomodulatory therapy. Intraperitoneal IL-2 was used in a phase I/II study in 41 patients with laparotomy-confirmed persistent or recurrent ovarian cancer. Weekly IL-2 infusion of 24 h duration was relatively well tolerated and demonstrated evidence of long-term efficacy in a modest number of patients. The toxicities of systemic IL-2 are significant; however, the peritoneal delivery method appeared to reduce the number and severity of the toxicities until the concentration in the intraperitoneal infusion reached the point where serum IL-2 became detectable. The appearance of systemic toxicity such as hypotension and thrombocytopenia, as well as locoregional dose-limiting toxicity (catheter infection), was associated with the highest doses. Twenty percent of patients had a negative third look, i.e., exhibited pathologic evidence of complete response and no residual disease at repeat abdominal exploration [29]. Recently, the therapeutic potential of several additional cytokines has been of increasing interest. IL-7, IL-15, IL-18, and IL-21 provide possible alternatives to IL-2. However, their function and clinical use are still under investigation [103–112].
Like cytokines, TLR agonists have multifaceted stimulatory effects on the immune system. TLR triggering induces DC maturation, which leads to the upregulation of costimulatory molecules, including CD40, CD80, and CD86, and secretion of immunomodulatory cytokines and chemokines. In addition, TLRs can directly stimulate the proliferation of CD4+ and CD8+ T cells as well as reverse the suppressive function of Treg cells [113–115]. Several clinical trials have demonstrated that administration of agonists for TLRs 3, 4, 7, and 9 can enhance activity of cancer vaccines in the context of non-small cell lung cancer [116], non-Hodgkins lymphoma [117, 118], glioblastoma [119], superficial basal cell carcinoma [120], and melanoma [121–124]. Adding TLR 3, 4, 7, or 9 ligands was shown to activate CD8+ cytotoxic T cells with increased IFN-alpha production and promote a stimulatory cytokine milieu at the tumor microenvironment [125, 126].
The use of antibodies to block T-cell inhibitory receptors such as CTLA-4 and PD-1 can lead to sustained activation and proliferation of tumor-specific T cells, preventing anergy or exhaustion and thereby allowing the development of an effective tumor-specific immune response. The majority of clinical data to date have emerged from studies in patients with melanoma [127], where CTLA-4 blockade has yielded objective responses. In a small study of ovarian cancer patients, one patient experienced a durable objective radiographic response. Multiple infusions of anti-CTLA-4 antibody every 3–5 months maintained disease control over 4 years [31]. The toxicities of CTLA-4 treatment showed similar pattern compared with those shown in melanoma patients, namely, grade I, rash in most of the patients (8/9); grade I or II, constitutional symptoms in 33 % (3/9) and sweet’s syndrome in 22 % (1/9); and grade III, diarrhea in 22 % of the patients(2/9). Tumor regression correlated with the CD8+/Treg ratio, suggesting that other forms of therapy that target Treg depletion may provide a highly effective form of treatment when combined with the tumor vaccine and CTLA-4 antibody arsenal [31].
Another way to enhance antitumor T-cell activity is through blockade of the PD-1 pathway. PD-1, expressed on activated T cells, binds PD-L1 and PD-L2 ligands. PD-L2 is restricted to professional antigen-presenting cells, while PD-L1 is expressed on many tissues. Importantly, ovarian carcinoma cells as well as tumor-infiltrating tolerogenic DCs and myeloid-derived suppressor cells express PD-L1 [128, 129], and expression levels correlate with disease course. Constitutive expression of PD-L1 by tumors conferred resistance to immunotherapy in mice [130], while antibodies blocking PD-L1 or PD-1 profoundly enhanced the efficacy of immunotherapy [130, 131]. A phase I study using PD-1 blocking antibody showed the antibody to be safe and well tolerated in patients with hematologic malignancies. Clinical benefit was observed in 33 % of the patients, with one complete remission [132].
Antibodies targeting the IL-2 receptor alpha chain (also known as CD25) can be used to deplete Tregs. In mouse models, the use of anti-CD25 monoclonal antibody before vaccination led to complete tumor rejection and establishment of long-lasting tumor immunity with no autoimmune complications [133, 134]. Daclizumab, which is an FDA-approved humanized IgG1-kappa mAb that binds specifically to CD25 [135], has been used in autoimmune disorders [136, 137], acute graft-versus-host disease [138], and in cancer patients with CD25+ T-cell malignancies [139]. The advantage of daclizumab is that it is well tolerated and has a half-life of 20 days [140]. In a recent study, daclizumab was used in a single dose of 1 mg/m2 prior to hTERT peptide vaccine for metastatic breast cancer. Total CD4+CD25+ and CD4+CD25+FoxP3+ cells remained suppressed for several weeks after a single infusion. Importantly, administration of anti-CD25 antibody was compatible with effective vaccination [141].
The main mechanism of immune stimulation by CD40 agonists (including recombinant CD40 ligand and agonistic anti-CD40 antibodies) is activation of CD40-expressing DCs, resulting in increased survival, upregulation of costimulatory molecules, and secretion of critical cytokines for T-cell priming, such as IL-12. In vitro human cell studies have also been conducted to evaluate whether recombinant CD40L is able to stimulate maturation of DCs derived from ovarian cancer patients. In one study, autologous DCs from ten ovarian cancer patients were pulsed with killed primary tumors as a source of tumor antigens. DCs were then cultured in the presence of TNF, TRANCE (tumor necrosis factor-related activation-induced cytokine), and CD40L to induce maturation. These mature whole lysate-pulsed DCs were able to stimulate CD8+ T cells that secreted IFN-γ in responses to ovarian tumor antigens. Similar results were also obtained in another study where DCs derived from ovarian cancer patients who were in remission were first loaded with HOCl-SKOV-3 tumor lysate and subsequently matured with activating anti-CD40 antibody [142]. In this study, mature DCs were able to stimulate both CD8+ and CD4+ antitumor T-cell responses. All these results highly suggested a potential benefit of using CD40L or anti-CD40 activating antibody as an adjuvant in DC-based whole tumor cell immunotherapy. Additional value of administering CD40 agonists in vivo is provided by the fact that ovarian cancers, like many tumors, express the CD40 receptor [143–146] and respond to CD40 ligation with apoptosis and growth inhibition in vitro and in vivo [145, 147, 148].
Cons
The usefulness of IL-2, although FDA approved for treatment of melanoma and renal cell carcinoma, has several limitations. Alone or in the context of adoptive immunotherapy, IL-2 is used at MTD, which induces a systemic inflammatory response with significant morbidity including multiple organ toxicities, most significantly the heart, lungs, kidneys, and central nervous system. Another manifestation of IL-2 toxicity is capillary leak syndrome, resulting in a hypovolemic state and fluid accumulation in the extravascular space [149]. Additionally, IL-2 is essential for the peripheral homeostasis of CD4+CD25+Foxp3+ Treg cells, and it is now known that IL-2 is also an important activator of Treg suppressive activity in vivo [150].
Many clinical trials have demonstrated the efficacy of type I interferon therapy in the treatment of hematologic malignancies [151–153], melanoma [154–158], and renal cell carcinoma [159–161]. In contrast, trials in ovarian carcinoma were less encouraging. Intraperitoneal recombinant IFN-α alone or combined with cisplatin as salvage therapy for persistent ovarian cancer after primary chemotherapy has shown clinical efficacy in small volume disease [162, 163], but there was no significant effect in a cohort of patients with recurrent, platinum-resistant disease [164]. A large randomized, phase III trial (n = 300) conducted in patients with epithelial ovarian cancer concluded that INF-a2a as maintenance therapy following surgery and/or chemotherapy is not effective alone [165].
Conflicting results from trials involving IFN-γ administration highlight the difficulty in designing immunomodulation therapies. In one instance, a threefold prolongation of progression-free survival was observed in a phase III multicenter study from Europe with subcutaneous administration of rhIFN-γ combined with MTD cisplatin and cyclophosphamide chemotherapy, with minimal added toxicity [166]. However, in a subsequent randomized phase III trial conducted in the USA, addition of subcutaneous rhIFN-γ to carboplatin and paclitaxel did not improve survival [167]. Although one cannot exclude that racial and other demographic differences may account for opposite results, these data may indicate that the choice of chemotherapy drugs is in fact critical in combinatorial approaches with immunotherapy. Indeed, whereas cyclophosphamide has potent immunomodulatory effects on suppressive Tregs, high-dose steroids, which are necessarily given with paclitaxel to prevent acute hypersensitivity reactions, are immunosuppressive and induce Treg in the setting of antigen presentation.
Similarly, the use of TLR agonists in the clinic requires careful preclinical evaluation. For example, in the absence of specific cell-mediated antitumor immunity, nonspecific activation of inflammation might in fact promote tumor growth rather than reducing it [168]. TLR4 agonists were shown to promote tumor cell survival, tumor growth, and paclitaxel resistance in a proportion of ovarian cancer cells [169, 170].
Meanwhile, agonistic anti-CD40 antibody is best used in combination with vaccines or TLR agonists [171, 172]. This is because, when used alone, it can accelerate the deletion of tumor-specific cytotoxic lymphocytes [173].
Conclusions
In the past decade, we have witnessed important advances in the development of immunotherapies for gynecologic cancers. First, ovarian cancers are now seen as potentially immunogenic tumors, a characterization formerly reserved only for melanoma and renal cell cancer. Second, the a priori notion that chemotherapy drugs antagonize immune mechanisms altogether was challenged by evidence that select chemotherapy drugs commonly used to treat gynecologic cancers have important immunomodulatory effects. This has opened the door to explore interactions of these drugs with natural antitumor immunity. Third, several mechanisms of tumor immune escape, accounting for failure of immunotherapy, have been deciphered, and the importance of combinatorial immunotherapy targeting both adaptive and innate effector and suppressor mechanisms has been proven. Fourth, this decade has produced novel and potent bona fide stimulants of innate and adaptive immunity. The next decade will be the time to test and optimize these combinations to maximize efficacy and decrease toxicity. Rational combinations of agents will require understanding of their precise mechanism of action in order to select combinations yielding positive interactions.
Future Directions
Evidence now convincingly shows that ovarian cancers are immunogenic tumors. The dramatic advances in laboratory technology and clinical procedures in cellular immunotherapy, along with the development of powerful immunomodulatory antibodies, create new opportunities in ovarian cancer therapeutics. The challenge for the next decade will be to test rational combinations that offer maximal clinical benefit at the lowest cost.
Selection of appropriate patients for clinical trial participation will also be quite influential. Additional biomarkers are needed to maximize selection of patients who may benefit from immunotherapy. Evidence to date indicates that many ovarian cancer patients display a spontaneous antitumor immune response. These patients may be best suited for vaccine therapy or TIL-based therapy as they are the most likely to harbor a natural repertoire of tumor-reactive T cells with tumor rejecting potential that can be expanded in vivo or ex vivo. In addition, patients whose tumors exhibit intraepithelial T cells may be most likely to respond to immunotherapy as the tumor microenvironment is already conducive to T-cell homing and engraftment. Finally, more work will be necessary to develop strategies to integrate immunotherapy with current standard of care. We have previously demonstrated that patients with advanced ovarian cancer whose tumors exhibit low frequency of intraepithelial CD8+ T cells or high Ki67 expression are more likely to draw benefit from aggressive surgical cytoreduction, while debulking did not significantly affect the survival of patients with brisk CD8+ T cells or low Ki67 expression [17]. It is possible that immunotherapy with adoptive transfer of TILs and/or vaccine plus immunomodulation could be a rational adjuvant therapy for patients with intraepithelial T cells following conventional debulking surgery and chemotherapy. Based on the observation that VEGF antibody blockade enhances T-cell infiltration in tumors and that its efficacy depends on antitumor CD8 T-cell response [174], it is possible that patients with intraepithelial T cells may also respond better to bevacizumab or other VEGF inhibitors. On the other hand, our data suggest that maximal debulking efforts should be undertaken in tumors with low T cells and it is possible that these patients are not the best candidates for adjuvant immunotherapy that exploits natural antitumor immune response. Personalized adoptive therapy with engineered T cells redirected against known tumor epitopes might be the most efficient approach to adjuvant immunotherapy in patients with low level of naturally occurring TILs. Careful preclinical evaluation in well-characterized animal models will be necessary to evaluate combinations before undertaking clinical studies. However, the major challenge facing the field at present is to conduct randomized clinical trials demonstrating sufficient clinical benefit to justify the logistics and expense of customized cellular therapies. A positive outcome from immunotherapy trials in terms of effective therapy, extension of progression free, and overall survival would represent a major advancement for patients with advanced ovarian cancer.
Concluding Comments
-
Appropriate diagnostic methods are needed to identify patients suitable for immunotherapy.
-
Novel pharmacodynamics biomarkers need to be implemented to provide proper metrics for effectiveness of immune therapies.
-
New strategies need to be developed to integrate immunotherapy with standard of care or targeted therapies to offer long-term and durable benefit.
-
Formulation of effective, scalable, and reproducibly manufactured vaccines, T-cell products and immunomodulatory agents will be necessary to make these therapies commercially viable.
-
Randomized clinical trials need to be conducted to demonstrate sufficient clinical benefit in order to justify the expense of immunotherapies and the integration of them into standard of care.
References
Schlienger K, Chu CS, Woo EY, Rivers PM, Toll AJ, Hudson B, et al. TRANCE- and CD40 ligand-matured dendritic cells reveal MHC class I-restricted T cells specific for autologous tumor in late-stage ovarian cancer patients. Clin Cancer Res. 2003;9(4):1517–27. PubMed PMID: 12684428. Epub 2003/04/10.
Goodell V, Salazar LG, Urban N, Drescher CW, Gray H, Swensen RE, et al. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J Clin Oncol. 2006;24(5):762–8. PubMed PMID: 16391298.
Hayashi K, Yonamine K, Masuko-Hongo K, Iida T, Yamamoto K, Nishioka K, et al. Clonal expansion of T cells that are specific for autologous ovarian tumor among tumor-infiltrating T cells in humans. Gynecol Oncol. 1999;74(1):86–92. PubMed PMID: 10385556.
Halapi E, Yamamoto Y, Juhlin C, Jeddi-Tehrani M, Grunewald J, Andersson R, et al. Restricted T cell receptor V-beta and J-beta usage in T cells from interleukin-2-cultured lymphocytes of ovarian and renal carcinomas. Cancer Immunol Immunother. 1993;36(3):191–7. PubMed PMID: 8439980. Epub 1993/01/01.
Fisk B, Blevins TL, Wharton JT, Ioannides CG. Identification of an immunodominant peptide of HER-2/neu protooncogene recognized by ovarian tumor-specific cytotoxic T lymphocyte lines. J Exp Med. 1995;181(6):2109–17. PubMed PMID: 7539040. Pubmed Central PMCID: 2192068. Epub 1995/06/01.
Kooi S, Freedman RS, Rodriguez-Villanueva J, Platsoucas CD. Cytokine production by T-cell lines derived from tumor-infiltrating lymphocytes from patients with ovarian carcinoma: tumor-specific immune responses and inhibition of antigen-independent cytokine production by ovarian tumor cells. Lymphokine Cytokine Res. 1993;12(6):429–37. PubMed PMID: 8123759. Epub 1993/12/01.
Peoples GE, Goedegebuure PS, Smith R, Linehan DC, Yoshino I, Eberlein TJ. Breast and ovarian cancer-specific cytotoxic T lymphocytes recognize the same HER2/neu-derived peptide. Proc Natl Acad Sci U S A. 1995;92(2):432–6. PubMed PMID: 7831305.
Peoples GE, Anderson BW, Fisk B, Kudelka AP, Wharton JT, Ioannides CG. Ovarian cancer-associated lymphocyte recognition of folate binding protein peptides. Ann Surg Oncol. 1998;5(8):743–50. PubMed PMID: 9869522.
Dadmarz RD, Ordoubadi A, Mixon A, Thompson CO, Barracchini KC, Hijazi YM, et al. Tumor-infiltrating lymphocytes from human ovarian cancer patients recognize autologous tumor in an MHC class II-restricted fashion. Cancer J Sci Am. 1996;2(5):263–72. PubMed PMID: 9166543. Epub 1996/10/01.
Santin AD, Bellone S, Ravaggi A, Pecorelli S, Cannon MJ, Parham GP. Induction of ovarian tumor-specific CD8+ cytotoxic T lymphocytes by acid-eluted peptide-pulsed autologous dendritic cells. Obstet Gynecol. 2000;96(3):422–30. PubMed PMID: 10960637. Epub 2000/08/29.
Peoples GE, Schoof DD, Andrews JV, Goedegebuure PS, Eberlein TJ. T-cell recognition of ovarian cancer. Surgery. 1993;114(2):227–34. PubMed PMID: 8342128.
Albert ML, Darnell JC, Bender A, Francisco LM, Bhardwaj N, Darnell RB. Tumor-specific killer cells in paraneoplastic cerebellar degeneration. Nat Med. 1998;4(11):1321–4. PubMed PMID: 9809559. Epub 1998/11/11.
Lanitis E, Poussin M, Hagemann IS, Coukos G, Sandaltzopoulos R, Scholler N, et al. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol Ther. 2012;20(3):633–43.
Odunsi K, Matsuzaki J, Karbach J, Neumann A, Mhawech-Fauceglia P, Miller A, et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc Natl Acad Sci U S A. 2012;109(15):5797–802. PubMed PMID: 22454499. Pubmed Central PMCID: 3326498. Epub 2012/03/29.
Chu CS, Kim SH, June CH, Coukos G. Immunotherapy opportunities in ovarian cancer. Expert Rev Anticancer Ther. 2008;8(2):243–57. PubMed PMID: 18279065.
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–13. PubMed PMID: 12529460. Epub 2003/01/17.
Adams SF, Levine DA, Cadungog MG, Hammond R, Facciabene A, Olvera N, et al. Intraepithelial T cells and tumor proliferation: impact on the benefit from surgical cytoreduction in advanced serous ovarian cancer. Cancer. 2009;115(13):2891–902. PubMed PMID: 19472394. Epub 2009/05/28.
Clarke B, Tinker AV, Lee C, Subramanian S, van de Rijn M, Turbin D, et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type and BRCA1 loss. Mod Pathol. 2009;22(3):393–402.
Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(9):3360–5. PubMed PMID: 17360651.
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43. PubMed PMID: 16344461. Pubmed Central PMCID: 1311741. Epub 2005/12/14.
Shah CA, Allison KH, Garcia RL, Gray HJ, Goff BA, Swisher EM. Intratumoral T cells, tumor-associated macrophages, and regulatory T cells: association with p53 mutations, circulating tumor DNA and survival in women with ovarian cancer. Gynecol Oncol. 2008;109(2):215–9. PubMed PMID: 18314181.
Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61(12):4766–72. PubMed PMID: 11406550. Epub 2001/06/19.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. PubMed PMID: 15322536. Epub 2004/08/24.
Kryczek I, Wei S, Zhu G, Myers L, Mottram P, Cheng P, et al. Relationship between B7-H4, regulatory T cells, and patient outcome in human ovarian carcinoma. Cancer Res. 2007;67(18):8900–5. PubMed PMID: 17875732.
Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36. PubMed PMID: 18157142.
Kandalaft LE, Facciabene A, Buckanovich RJ, Coukos G. Endothelin B receptor, a new target in cancer immune therapy. Clin Cancer Res. 2009;15(14):4521–8. PubMed PMID: 19567593. Epub 2009/07/02.
Yoshihara K, Tsunoda T, Shigemizu D, Fujiwara H, Hatae M, Masuzaki H, et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin Cancer Res. 2012;18(5):1374–85. PubMed PMID: 22241791. Epub 2012/01/14.
Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21(2):233–40. PubMed PMID: 19304471. Epub 2009/03/24.
Edwards RP, Gooding W, Lembersky BC, Colonello K, Hammond R, Paradise C, et al. Comparison of toxicity and survival following intraperitoneal recombinant interleukin-2 for persistent ovarian cancer after platinum: twenty-four-hour versus 7-day infusion. J Clin Oncol. 1997;15(11):3399–407. PubMed PMID: 9363872.
Vlad AM, Budiu RA, Lenzner DE, Wang Y, Thaller JA, Colonello K, et al. A phase II trial of intraperitoneal interleukin-2 in patients with platinum-resistant or platinum-refractory ovarian cancer. Cancer Immunol Immunother. 2010;59(2):293–301. PubMed PMID: 19690855. Epub 2009/08/20.
Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A. 2008;105(8):3005–10. PubMed PMID: 18287062. Epub 2008/02/22.
Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A. 2003;100(8):4712–7. PubMed PMID: 12682289.
Fujita K, Ikarashi H, Takakuwa K, Kodama S, Tokunaga A, Takahashi T, et al. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995;1(5):501–7. PubMed PMID: 9816009.
Aoki Y, Takakuwa K, Kodama S, Tanaka K, Takahashi M, Tokunaga A, et al. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991;51(7):1934–9. PubMed PMID: 2004379.
Hung CF, Wu TC, Monie A, Roden R. Antigen-specific immunotherapy of cervical and ovarian cancer. Immunol Rev. 2008;222:43–69. PubMed PMID: 18363994. Pubmed Central PMCID: 2692865. Epub 2008/03/28.
Odunsi K, Sabbatini P. Harnessing the immune system for ovarian cancer therapy. Am J Reprod Immunol. 2008;59(1):62–74. PubMed PMID: 18154597. Epub 2007/12/25.
Sabbatini P, Odunsi K. Immunologic approaches to ovarian cancer treatment. J Clin Oncol. 2007;25(20):2884–93. PubMed PMID: 17617519. Epub 2007/07/10.
Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–15. PubMed PMID: 15340416.
Sabbatini P, Spriggs D, Aghajanian C, Hensley M, Tew W, Konner J, et al. Consolidation strategies in ovarian cancer: observations for future clinical trials. Gynecol Oncol. 2010;116(1):66–71. PubMed PMID: 19836827. Epub 2009/10/20.
Reinartz S, Kohler S, Schlebusch H, Krista K, Giffels P, Renke K, et al. Vaccination of patients with advanced ovarian carcinoma with the anti-idiotype ACA125: immunological response and survival (phase Ib/II). Clin Cancer Res. 2004;10(5):1580–7. PubMed PMID: 15014007.
Gulley JL, Arlen PM, Tsang KY, Yokokawa J, Palena C, Poole DJ, et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin Cancer Res. 2008;14(10):3060–9. PubMed PMID: 18483372. Pubmed Central PMCID: 2673097. Epub 2008/05/17.
Disis ML, Goodell V, Schiffman K, Knutson KL. Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. J Clin Immunol. 2004;24(5):571–8. PubMed PMID: 15359116.
Chianese-Bullock KA, Irvin Jr WP, Petroni GR, Murphy C, Smolkin M, Olson WC, et al. A multipeptide vaccine is safe and elicits T-cell responses in participants with advanced stage ovarian cancer. J Immunother. 2008;31(4):420–30. PubMed PMID: 18391753. Epub 2008/04/09.
Tsuda N, Mochizuki K, Harada M, Sukehiro A, Kawano K, Yamada A, et al. Vaccination with predesignated or evidence-based peptides for patients with recurrent gynecologic cancers. J Immunother (1997). 2004;27(1):60–72. PubMed PMID: 14676634.
Chu CS, Boyer J, Coukos G, Rubin SC, Morgan MA, Bendig DL. Autologous dendritic cell (IDD-6) vaccination as consolidation for advanced ovarian cancer. In: SGO annual meeting on women’s cancer. Tampa; 2008.
Hernando JJ, Park TW, Kubler K, Offergeld R, Schlebusch H, Bauknecht T. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: clinical and immunological evaluation of a phase I trial. Cancer Immunol Immunother. 2002;51(1):45–52. PubMed PMID: 11845259. Epub 2002/02/15.
Gong J, Nikrui N, Chen D, Koido S, Wu Z, Tanaka Y, et al. Fusions of human ovarian carcinoma cells with autologous or allogeneic dendritic cells induce antitumor immunity. J Immunol. 2000;165(3):1705–11. PubMed PMID: 10903782.
Ioannides CG, Platsoucas CD, Freedman RS. Immunological effects of tumor vaccines: II. T cell responses directed against cellular antigens in the viral oncolysates. In Vivo. 1990;4(1):17–24. PubMed PMID: 2103838.
Ioannides CG, Platsoucas CD, Patenia R, Kim YP, Bowen JM, Morris M, et al. T-cell functions in ovarian cancer patients treated with viral oncolysates: I. Increased helper activity to immunoglobulins production. Anticancer Res. 1990;10(3):645–53. PubMed PMID: 2142392.
Schirrmacher V. Clinical trials of antitumor vaccination with an autologous tumor cell vaccine modified by virus infection: improvement of patient survival based on improved antitumor immune memory. Cancer Immunol Immunother. 2005;54(6):587–98. PubMed PMID: 15526097.
Benencia F, Courreges MC, Conejo-Garcia JR, Mohammed-Hadley A, Coukos G. Direct vaccination with tumor cells killed with ICP4-deficient HSVd120 elicits effective antitumor immunity. Cancer Biol Ther. 2006;5(7):867–74. PubMed PMID: 16861891.
Benencia F, Courreges MC, Coukos G. Whole tumor antigen vaccination using dendritic cells: comparison of RNA electroporation and pulsing with UV-irradiated tumor cells. J Transl Med. 2008;6:21. PubMed PMID: 18445282.
Benencia F, Courreges MC, Fraser NW, Coukos G. Herpes virus oncolytic therapy reverses tumor immune dysfunction and facilitates tumor antigen presentation. Cancer Biol Ther. 2008;7(8):1194–205. PubMed PMID: 18458533.
Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30(5):413–21.
Odunsi K, Qian F, Matsuzaki J, Mhawech-Fauceglia P, Andrews C, Hoffman EW, et al. Vaccination with an NY-ESO-1 peptide of HLA class I/II specificities induces integrated humoral and T cell responses in ovarian cancer. Proc Natl Acad Sci U S A. 2007;104(31):12837–42. PubMed PMID: 17652518. Pubmed Central PMCID: 1937553. Epub 2007/07/27.
Neller MA, López JA, Schmidt CW. Antigens for cancer immunotherapy. Semin Immunol. 2008;20(5):286–95.
Cannon MJ, O’Brien TJ. Cellular immunotherapy for ovarian cancer. Expert Opin Biol Ther. 2009;9(6):677–88. PubMed PMID: 19456205. Epub 2009/05/22.
van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–7. PubMed PMID: 1840703. Epub 1991/12/13.
Pardoll DM. Spinning molecular immunology into successful immunotherapy. Nat Rev Immunol. 2002;2(4):227–38. PubMed PMID: 12001994.
Gulley JL. Therapeutic vaccines: the ultimate personalized therapy? Human Vaccin Immunother. 2013;9(1):219–21. PubMed PMID: 22995839.
June CH. Adoptive T, cell therapy for cancer in the clinic. J Clin Invest. 2007;117(6):1466–76. PubMed PMID: 17549249. Epub 2007/06/06.
Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–57. PubMed PMID: 15800326.
Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99(25):16168–73. PubMed PMID: 12427970. Epub 2002/11/13.
Perret R, Ronchese F. Memory T cells in cancer immunotherapy: which CD8 T-cell population provides the best protection against tumours? Tissue Antigens. 2008;72(3):187–94. PubMed PMID: 18627571. Epub 2008/07/17.
Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–12. PubMed PMID: 16203864. Pubmed Central PMCID: 1397916. Epub 2005/10/06.
Huang J, Kerstann KW, Ahmadzadeh M, Li YF, El-Gamil M, Rosenberg SA, et al. Modulation by IL-2 of CD70 and CD27 expression on CD8+ T cells: importance for the therapeutic effectiveness of cell transfer immunotherapy. J Immunol. 2006;176(12):7726–35. PubMed PMID: 16751420. Epub 2006/06/06.
Powell Jr DJ, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105(1):241–50. PubMed PMID: 15345595. Epub 2004/09/04.
Shen X, Zhou J, Hathcock KS, Robbins P, Powell Jr DJ, Rosenberg SA, et al. Persistence of tumor infiltrating lymphocytes in adoptive immunotherapy correlates with telomere length. J Immunother (1997). 2007;30(1):123–9. PubMed PMID: 17198091. Epub 2007/01/02.
Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175(10):7046–52. PubMed PMID: 16272366. Epub 2005/11/08.
Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009;106(41):17469–74. PubMed PMID: 19805141. Epub 2009/10/07.
Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15(5):981–8. PubMed PMID: 17375070.
Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3(1):35–45. PubMed PMID: 12509765. Epub 2003/01/02.
Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96(2):467–74. PubMed PMID: 10887107. Epub 2000/07/11.
Brocker T, Karjalainen K. Adoptive tumor immunity mediated by lymphocytes bearing modified antigen-specific receptors. Adv Immunol. 1998;68:257–69. PubMed PMID: 9505091.
Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–8. PubMed PMID: 2513569. Pubmed Central PMCID: 298636. Epub 1989/12/01.
Pinthus JH, Waks T, Kaufman-Francis K, Schindler DG, Harmelin A, Kanety H, et al. Immuno-gene therapy of established prostate tumors using chimeric receptor-redirected human lymphocytes. Cancer Res. 2003;63(10):2470–6. PubMed PMID: 12750268.
Freedman RS, Edwards CL, Kavanagh JJ, Kudelka AP, Katz RL, Carrasco CH, et al. Intraperitoneal adoptive immunotherapy of ovarian carcinoma with tumor-infiltrating lymphocytes and low-dose recombinant interleukin-2: a pilot trial. J Immunother Emphasis Tumor Immunol. 1994;16(3):198–210. PubMed PMID: 7834119.
Ioannides CG, Den Otter W. Concepts in immunotherapy of cancer: introduction. In Vivo. 1991;5(6):551–2. PubMed PMID: 1810436.
Theoret MR, Cohen CJ, Nahvi AV, Ngo LT, Suri KB, Powell Jr DJ, et al. Relationship of p53 overexpression on cancers and recognition by anti-p53 T cell receptor-transduced T cells. Hum Gene Ther. 2008;19(11):1219–32. PubMed PMID: 19848582. Epub 2009/10/24.
Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity. 2009;30(5):656–65. PubMed PMID: 19464988. Epub 2009/05/26.
Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, et al. Characterization of genetically modified T-cell receptors that recognize the CEA:691–699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res. 2009;15(1):169–80.
Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. 2008;180(9):6116–31. PubMed PMID: 18424733. Epub 2008/04/22.
June CH, Blazar BR, Riley JL. Engineering lymphocyte subsets: tools, trials and tribulations. Nat Rev Immunol. 2009;9(10):704–16. PubMed PMID: 19859065. Epub 2009/10/28.
Wang G, Chopra RK, Royal RE, Yang JC, Rosenberg SA, Hwu P. A T cell-independent antitumor response in mice with bone marrow cells retrovirally transduced with an antibody/Fc-gamma chain chimeric receptor gene recognizing a human ovarian cancer antigen. Nat Med. 1998;4(2):168–72. PubMed PMID: 9461189.
Parker LL, Do MT, Westwood JA, Wunderlich JR, Dudley ME, Rosenberg SA, et al. Expansion and characterization of T cells transduced with a chimeric receptor against ovarian cancer. Hum Gene Ther. 2000;11(17):2377–87. PubMed PMID: 11096442.
Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180(7):4901–9. PubMed PMID: 18354214. Epub 2008/03/21.
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–5. PubMed PMID: 19211796. Pubmed Central PMCID: 2651342. Epub 2009/02/13.
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–15. PubMed PMID: 17062687.
Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011;71(13):4617–27. PubMed PMID: 21546571. Epub 2011/05/07.
Berd D, Mastrangelo MJ. Active immunotherapy of human melanoma exploiting the immunopotentiating effects of cyclophosphamide. Cancer Invest. 1988;6(3):337–49. PubMed PMID: 3167614. Epub 1988/01/01.
Radojcic V, Bezak KB, Skarica M, Pletneva MA, Yoshimura K, Schulick RD, et al. Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer Immunol Immunother. 2009. PubMed PMID: 19590872. Epub 2009/07/11.
Shurin GV, Tourkova IL, Kaneno R, Shurin MR. Chemotherapeutic agents in noncytotoxic concentrations increase antigen presentation by dendritic cells via an IL-12-dependent mechanism. J Immunol. 2009;183(1):137–44. PubMed PMID: 19535620. Epub 2009/06/19.
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007;56(5):641–8. PubMed PMID: 16960692. Epub 2006/09/09.
Schiavoni G, Mattei F, Di Pucchio T, Santini SM, Bracci L, Belardelli F, et al. Cyclophosphamide induces type I interferon and augments the number of CD44(hi) T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood. 2000;95(6):2024–30. PubMed PMID: 10706870.
Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, et al. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin Immunol. 2008;129(2):219–29. PubMed PMID: 18771959. Epub 2008/09/06.
Nehme A, Julia AM, Jozan S, Chevreau C, Bugat R, Canal P. Modulation of cisplatin cytotoxicity by human recombinant interferon-gamma in human ovarian cancer cell lines. Eur J Cancer. 1994;30A(4):520–5. PubMed PMID: 8018412. Epub 1994/01/01.
Melichar B, Hu W, Patenia R, Melicharova K, Gallardo ST, Freedman R. rIFN-gamma-mediated growth suppression of platinum-sensitive and -resistant ovarian tumor cell lines not dependent upon arginase inhibition. J Transl Med. 2003;1(1):5. PubMed PMID: 14572312.
Wall L, Burke F, Smyth JF, Balkwill F. The anti-proliferative activity of interferon-gamma on ovarian cancer: in vitro and in vivo. Gynecol Oncol. 2003;88(1 Pt 2):S149–51. PubMed PMID: 12586108.
Freedman RS, Kudelka AP, Kavanagh JJ, Verschraegen C, Edwards CL, Nash M, et al. Clinical and biological effects of intraperitoneal injections of recombinant interferon-gamma and recombinant interleukin 2 with or without tumor-infiltrating lymphocytes in patients with ovarian or peritoneal carcinoma. Clin Cancer Res. 2000;6(6):2268–78. PubMed PMID: 10873077.
Kooi S, Zhang HZ, Patenia R, Edwards CL, Platsoucas CD, Freedman RS. HLA class I expression on human ovarian carcinoma cells correlates with T-cell infiltration in vivo and T-cell expansion in vitro in low concentrations of recombinant interleukin-2. Cell Immunol. 1996;174(2):116–28. PubMed PMID: 8954611.
Duda DG, Sunamura M, Lozonschi L, Kodama T, Egawa S, Matsumoto G, et al. Direct in vitro evidence and in vivo analysis of the antiangiogenesis effects of interleukin 12. Cancer Res. 2000;60(4):1111–6. PubMed PMID: 10706132. Epub 2000/03/08.
Ohta M, Mitomi T, Kimura M, Habu S, Katsuki M. Anomalies in transgenic mice carrying the human interleukin-2 gene. Tokai J Exp Clin Med. 1990;15(4):307–15. PubMed PMID: 2130538.
Capitini CM, Chisti AA, Mackall CL. Modulating T-cell homeostasis with IL-7: preclinical and clinical studies. J Intern Med. 2009;266(2):141–53. PubMed PMID: 19623690. Epub 2009/07/23.
Ribas A. Update on immunotherapy for melanoma. J Natl Compr Canc Netw. 2006;4(7):687–94. PubMed PMID: 16884670. Epub 2006/08/04.
Andersson A, Yang SC, Huang M, Zhu L, Kar UK, Batra RK, et al. IL-7 promotes CXCR3 ligand-dependent T cell antitumor reactivity in lung cancer. J Immunol. 2009;182(11):6951–8. PubMed PMID: 19454692. Epub 2009/05/21.
Sharma S, Wang J, Huang M, Paul RW, Lee P, McBride WH, et al. Interleukin-7 gene transfer in non-small-cell lung cancer decreases tumor proliferation, modifies cell surface molecule expression, and enhances antitumor reactivity. Cancer Gene Ther. 1996;3(5):302–13. PubMed PMID: 8894249. Epub 1996/09/01.
Shanmugham LN, Petrarca C, Frydas S, Donelan J, Castellani ML, Boucher W, et al. IL-15 an immunoregulatory and anti-cancer cytokine. Recent advances. J Exp Clin Cancer Res. 2006;25(4):529–36. PubMed PMID: 17310844. Epub 2007/02/22.
Brandt K, Singh PB, Bulfone-Paus S, Ruckert R. Interleukin-21: a new modulator of immunity, infection, and cancer. Cytokine Growth Factor Rev. 2007;18(3–4):223–32. PubMed PMID: 17509926. Epub 2007/05/19.
Thompson JA, Curti BD, Redman BG, Bhatia S, Weber JS, Agarwala SS, et al. Phase I study of recombinant interleukin-21 in patients with metastatic melanoma and renal cell carcinoma. J Clin Oncol. 2008;26(12):2034–9. PubMed PMID: 18347008. Epub 2008/03/19.
Carroll RG, Carpenito C, Shan X, Danet-Desnoyers G, Liu R, Jiang S, et al. Distinct effects of IL-18 on the engraftment and function of human effector CD8 T cells and regulatory T cells. PLoS One. 2008;3(9):e3289. PubMed PMID: 18818761. Pubmed Central PMCID: 2538560. Epub 2008/09/27.
Robertson MJ, Mier JW, Logan T, Atkins M, Koon H, Koch KM, et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin Cancer Res. 2006;12(14 Pt 1):4265–73. PubMed PMID: 16857801.
Robertson MJ, Kirkwood JM, Logan TF, Koch KM, Kathman S, Kirby LC, et al. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. 2008;14(11):3462–9. PubMed PMID: 18519778.
Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, et al. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science. 2005;309(5739):1380–4. PubMed PMID: 16123302.
Crellin NK, Garcia RV, Hadisfar O, Allan SE, Steiner TS, Levings MK. Human CD4+ T cells express TLR5 and its ligand flagellin enhances the suppressive capacity and expression of FOXP3 in CD4+CD25+ T regulatory cells. J Immunol. 2005;175(12):8051–9. PubMed PMID: 16339542. Epub 2005/12/13.
Tabiasco J, Devevre E, Rufer N, Salaun B, Cerottini JC, Speiser D, et al. Human effector CD8+ T lymphocytes express TLR3 as a functional coreceptor. J Immunol. 2006;177(12):8708–13. PubMed PMID: 17142772. Epub 2006/12/05.
Manegold C, Gravenor D, Woytowitz D, Mezger J, Hirsh V, Albert G, et al. Randomized phase II trial of a toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combination with first-line taxane plus platinum chemotherapy for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26(24):3979–86. PubMed PMID: 18711188. Epub 2008/08/20.
Link BK, Ballas ZK, Weisdorf D, Wooldridge JE, Bossler AD, Shannon M, et al. Oligodeoxynucleotide CpG 7909 delivered as intravenous infusion demonstrates immunologic modulation in patients with previously treated non-Hodgkin lymphoma. J Immunother. 2006;29(5):558–68. PubMed PMID: 16971811. Epub 2006/09/15.
Leonard JP, Link BK, Emmanouilides C, Gregory SA, Weisdorf D, Andrey J, et al. Phase I trial of toll-like receptor 9 agonist PF-3512676 with and following rituximab in patients with recurrent indolent and aggressive non Hodgkin’s lymphoma. Clin Cancer Res. 2007;13(20):6168–74. PubMed PMID: 17947483. Epub 2007/10/20.
Carpentier A, Laigle-Donadey F, Zohar S, Capelle L, Behin A, Tibi A, et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006;8(1):60–6. PubMed PMID: 16443949. Epub 2006/01/31.
Stockfleth E, Trefzer U, Garcia-Bartels C, Wegner T, Schmook T, Sterry W. The use of toll-like receptor-7 agonist in the treatment of basal cell carcinoma: an overview. Br J Dermatol. 2003;149 Suppl 66:53–6. PubMed PMID: 14616352. Epub 2003/11/18.
Adams S, O’Neill DW, Nonaka D, Hardin E, Chiriboga L, Siu K, et al. Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using TLR7 agonist imiquimod as vaccine adjuvant. J Immunol. 2008;181(1):776–84. PubMed PMID: 18566444. Epub 2008/06/21.
Koido S, Hara E, Homma S, Torii A, Toyama Y, Kawahara H, et al. Dendritic cells fused with allogeneic colorectal cancer cell line present multiple colorectal cancer-specific antigens and induce antitumor immunity against autologous tumor cells. Clin Cancer Res. 2005;11(21):7891–900. PubMed PMID: 16278414. Epub 2005/11/10.
den Brok MH, Sutmuller RP, Nierkens S, Bennink EJ, Toonen LW, Figdor CG, et al. Synergy between in situ cryoablation and TLR9 stimulation results in a highly effective in vivo dendritic cell vaccine. Cancer Res. 2006;66(14):7285–92. PubMed PMID: 16849578. Epub 2006/07/20.
Lesimple T, Neidhard EM, Vignard V, Lefeuvre C, Adamski H, Labarriere N, et al. Immunologic and clinical effects of injecting mature peptide-loaded dendritic cells by intralymphatic and intranodal routes in metastatic melanoma patients. Clin Cancer Res. 2006;12(24):7380–8. PubMed PMID: 17189411. Epub 2006/12/26.
Hamdy S, Molavi O, Ma Z, Haddadi A, Alshamsan A, Gobti Z, et al. Co-delivery of cancer-associated antigen and toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine. 2008;26(39):5046–57. PubMed PMID: 18680779. Epub 2008/08/06.
Ramakrishna V, Vasilakos JP, Tario Jr JD, Berger MA, Wallace PK, Keler T. Toll-like receptor activation enhances cell-mediated immunity induced by an antibody vaccine targeting human dendritic cells. J Transl Med. 2007;5:5. PubMed PMID: 17254349. Epub 2007/01/27.
Fong L, Small EJ. Anti-cytotoxic T-lymphocyte antigen-4 antibody: the first in an emerging class of immunomodulatory antibodies for cancer treatment. J Clin Oncol. 2008;26(32):5275–83. PubMed PMID: 18838703. Epub 2008/10/08.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–7. PubMed PMID: 12704383.
Liu SM, Meng Q, Zhang QX, Wang SD, Liu ZJ, Zhang XF. Expression and significance of B7-H1 and its receptor PD-1 in human gastric carcinoma. Zhonghua Zhong Liu Za Zhi. 2008;30(3):192–5. PubMed PMID: 18756934. Epub 2008/09/02. chi.
Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65(3):1089–96. PubMed PMID: 15705911.
Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. 2007;56(5):739–45. PubMed PMID: 17195077. Epub 2006/12/30.
Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14(10):3044–51. PubMed PMID: 18483370. Epub 2008/05/17.
Benencia F, Coukos G. T regulatory cell depletion can boost DC-based vaccines. Cancer Biol Ther. 2005;28:4(6). PubMed PMID: 15917649.
Prasad SJ, Farrand KJ, Matthews SA, Chang JH, McHugh RS, Ronchese F. Dendritic cells loaded with stressed tumor cells elicit long-lasting protective tumor immunity in mice depleted of CD4+CD25+ regulatory T cells. J Immunol. 2005;174(1):90–8. PubMed PMID: 15611231.
Waldmann TA. Daclizumab (anti-Tac, Zenapax) in the treatment of leukemia/lymphoma. Oncogene. 2007;26(25):3699–703. PubMed PMID: 17530023. Epub 2007/05/29.
Kreijveld E, Koenen HJ, Klasen IS, Hilbrands LB, Joosten I. Following anti-CD25 treatment, a functional CD4+CD25+ regulatory T-cell pool is present in renal transplant recipients. Am J Transplant. 2007;7(1):249–55. PubMed PMID: 17109733. Epub 2006/11/18.
Nussenblatt RB, Fortin E, Schiffman R, Rizzo L, Smith J, Van Veldhuisen P, et al. Treatment of noninfectious intermediate and posterior uveitis with the humanized anti-Tac mAb: a phase I/II clinical trial. Proc Natl Acad Sci U S A. 1999;96(13):7462–6. PubMed PMID: 10377437. Epub 1999/06/23.
Przepiorka D, Kernan NA, Ippoliti C, Papadopoulos EB, Giralt S, Khouri I, et al. Daclizumab, a humanized anti-interleukin-2 receptor alpha chain antibody, for treatment of acute graft-versus-host disease. Blood. 2000;95(1):83–9. PubMed PMID: 10607689. Epub 1999/12/23.
Lehky TJ, Levin MC, Kubota R, Bamford RN, Flerlage AN, Soldan SS, et al. Reduction in HTLV-I proviral load and spontaneous lymphoproliferation in HTLV-I-associated myelopathy/tropical spastic paraparesis patients treated with humanized anti-Tac. Ann Neurol. 1998;44(6):942–7. PubMed PMID: 9851439. Epub 1998/12/16.
Vincenti F, Nashan B, Light S. Daclizumab: outcome of phase III trials and mechanism of action. Double Therapy and the Triple Therapy Study Groups. Transplant Proc. 1998;30(5):2155–8. PubMed PMID: 9723424. Epub 1998/09/02.
Rech AJ, Vonderheide RH. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann N Y Acad Sci. 2009;1174:99–106. PubMed PMID: 19769742. Epub 2009/09/23.
Chiang CL, Ledermann JA, Egla A, Elizabeth B, Katz DR, Chain BM. Oxidation of ovarian epithelial cancer cells by hypochlorous acid enhances immunogenicity and stimulates T cells that recognize autologous primary tumor. Clin Cancer Res. 2008;14(15):10.
Melichar B, Patenia R, Gallardo S, Melicharova K, Hu W, Freedman RS. Expression of CD40 and growth-inhibitory activity of CD40 ligand in ovarian cancer cell lines. Gynecol Oncol. 2007;104(3):707–13. PubMed PMID: 17166566. Epub 2006/12/15.
Hakkarainen T, Hemminki A, Pereboev AV, Barker SD, Asiedu CK, Strong TV, et al. CD40 is expressed on ovarian cancer cells and can be utilized for targeting adenoviruses. Clin Cancer Res. 2003;9(2):619–24. PubMed PMID: 12576427. Epub 2003/02/11.
Gallagher NJ, Eliopoulos AG, Agathangelo A, Oates J, Crocker J, Young LS. CD40 activation in epithelial ovarian carcinoma cells modulates growth, apoptosis, and cytokine secretion. Mol Pathol. 2002;55(2):110–20. PubMed PMID: 11950960. Pubmed Central PMCID: 1187159. Epub 2002/04/16.
Ciaravino G, Bhat M, Manbeian CA, Teng NN. Differential expression of CD40 and CD95 in ovarian carcinoma. Eur J Gynaecol Oncol. 2004;25(1):27–32. PubMed PMID: 15053058. Epub 2004/04/01.
Toutirais O, Gervais A, Cabillic F, Le Gallo M, Coudrais A, Leveque J, et al. Effects of CD40 binding on ovarian carcinoma cell growth and cytokine production in vitro. Clin Exp Immunol. 2007;149(2):372–7. PubMed PMID: 17565609. Pubmed Central PMCID: 1941941. Epub 2007/06/15.
Ghamande S, Hylander BL, Oflazoglu E, Lele S, Fanslow W, Repasky EA. Recombinant CD40 ligand therapy has significant antitumor effects on CD40-positive ovarian tumor xenografts grown in SCID mice and demonstrates an augmented effect with cisplatin. Cancer Res. 2001;61(20):7556–62. PubMed PMID: 11606394. Epub 2001/10/19.
Schwartz RN, Stover L, Dutcher J. Managing toxicities of high-dose interleukin-2. Oncology (Williston Park). 2002;16(11 Suppl 13):11–20. PubMed PMID: 12469935. Epub 2002/12/10.
Brandenburg S, Takahashi T, de la Rosa M, Janke M, Karsten G, Muzzulini T, et al. IL-2 induces in vivo suppression by CD4(+)CD25(+)Foxp3(+) regulatory T cells. Eur J Immunol. 2008;38(6):1643–53. PubMed PMID: 18493984. Epub 2008/05/22.
Allan NC, Richards SM, Shepherd PC. UK Medical Research Council randomised, multicentre trial of interferon-alpha n1 for chronic myeloid leukaemia: improved survival irrespective of cytogenetic response. The UK Medical Research Council’s Working Parties for Therapeutic Trials in Adult Leukaemia. Lancet. 1995;345(8962):1392–7. PubMed PMID: 7760609. Epub 1995/06/03.
Anger B, Porzsolt F, Leichtle R, Heinze B, Bartram C, Heimpel H. A phase I/II study of recombinant interferon alpha 2a and hydroxyurea for chronic myelocytic leukemia. Blut. 1989;58(6):275–8. PubMed PMID: 2736308. Epub 1989/06/01.
Foon KA, Roth MS, Bunn Jr PA. Alpha interferon treatment of low-grade B-cell non-Hodgkin’s lymphomas, cutaneous T-cell lymphomas, and chronic lymphocytic leukemia. Semin Oncol. 1986;13(3 Suppl 2):35–42. PubMed PMID: 3532334. Epub 1986/09/01.
Hersey P, Hasic E, MacDonald M, Edwards A, Spurling A, Coates AS, et al. Effects of recombinant leukocyte interferon (rIFN-alpha A) on tumour growth and immune responses in patients with metastatic melanoma. Br J Cancer. 1985;51(6):815–26. PubMed PMID: 3873953. Epub 1985/06/01.
Creagan ET, Ahmann DL, Green SJ, Long HJ, Frytak S, O’Fallon JR, et al. Phase II study of low-dose recombinant leukocyte A interferon in disseminated malignant melanoma. J Clin Oncol. 1984;2(9):1002–5. PubMed PMID: 6470751. Epub 1984/09/01.
Creagan ET, Ahmann DL, Green SJ, Long HJ, Rubin J, Schutt AJ, et al. Phase II study of recombinant leukocyte A interferon (rIFN-alpha A) in disseminated malignant melanoma. Cancer. 1984;54(12):2844–9. PubMed PMID: 6498762. Epub 1984/12/15.
Kirkwood JM, Ernstoff M. Melanoma: therapeutic options with recombinant interferons. Semin Oncol. 1985;12(4 Suppl 5):7–12. PubMed PMID: 2417333. Epub 1985/12/01.
Kirkwood JM, Ernstoff MS, Davis CA, Reiss M, Ferraresi R, Rudnick SA. Comparison of intramuscular and intravenous recombinant alpha-2 interferon in melanoma and other cancers. Ann Intern Med. 1985;103(1):32–6. PubMed PMID: 4003987. Epub 1985/07/01.
Quesada JR, Swanson DA, Gutterman JU. Phase II study of interferon alpha in metastatic renal-cell carcinoma: a progress report. J Clin Oncol. 1985;3(8):1086–92. PubMed PMID: 4020410. Epub 1985/08/01.
Levens W, Rubben H, Ingenhag W. Long-term interferon treatment in metastatic renal cell carcinoma. Eur Urol. 1989;16(5):378–81. PubMed PMID: 2776808. Epub 1989/01/01.
Rinehart JJ, Young D, Laforge J, Colborn D, Neidhart JA. Phase I/II trial of interferon-beta-serine in patients with renal cell carcinoma: immunological and biological effects. Cancer Res. 1987;47(9):2481–5. PubMed PMID: 3567933. Epub 1987/05/01.
Berek JS, Markman M, Stonebraker B, Lentz SS, Adelson MD, DeGeest K, et al. Intraperitoneal interferon-alpha in residual ovarian carcinoma: a phase II gynecologic oncology group study. Gynecol Oncol. 1999;75(1):10–4. PubMed PMID: 10502418.
Berek JS, Welander C, Schink JC, Grossberg H, Montz FJ, Zigelboim J. A phase I-II trial of intraperitoneal cisplatin and alpha-interferon in patients with persistent epithelial ovarian cancer. Gynecol Oncol. 1991;40(3):237–43. PubMed PMID: 2013446.
Markman M, Belinson J, Webster K, Zanotti K, Morrison B, Jacobs B, et al. Phase 2 trial of interferon-beta as second-line treatment of ovarian cancer, fallopian tube cancer, or primary carcinoma of the peritoneum. Oncology. 2004;66(5):343–6. PubMed PMID: 15331919.
Hall GD, Brown JM, Coleman RE, Stead M, Metcalf KS, Peel KR, et al. Maintenance treatment with interferon for advanced ovarian cancer: results of the Northern and Yorkshire gynaecology group randomised phase III study. Br J Cancer. 2004;91(4):621–6. PubMed PMID: 15305182. Pubmed Central PMCID: 2364769.
Windbichler GH, Hausmaninger H, Stummvoll W, Graf AH, Kainz C, Lahodny J, et al. Interferon-gamma in the first-line therapy of ovarian cancer: a randomized phase III trial. Br J Cancer. 2000;82(6):1138–44. PubMed PMID: 10735496.
Alberts DS, Marth C, Alvarez RD, Johnson G, Bidzinski M, Kardatzke DR, et al. Randomized phase 3 trial of interferon gamma-1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first-line treatment of advanced ovarian and primary peritoneal carcinomas: results from a prospectively designed analysis of progression-free survival. Gynecol Oncol. 2008;109(2):174–81. PubMed PMID: 18314182. Epub 2008/03/04.
Vakkila J, Lotze MT. Inflammation and necrosis promote tumour growth. Nat Rev Immunol. 2004;4(8):641–8. PubMed PMID: 15286730.
Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66(7):3859–68. PubMed PMID: 16585214. Epub 2006/04/06.
Kim KH, Xie Y, Tytler EM, Woessner R, Mor G, Alvero AB. KSP inhibitor ARRY-520 as a substitute for paclitaxel in type I ovarian cancer cells. J Transl Med. 2009;7:63. PubMed PMID: 19619321. Epub 2009/07/22.
Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229(1):152–72. PubMed PMID: 19426221. Epub 2009/05/12.
Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, et al. In situ stimulation of CD40 and toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. 2009;69(18):7329–37. PubMed PMID: 19738057. Pubmed Central PMCID: 2754806. Epub 2009/09/10.
Kedl RM, Jordan M, Potter T, Kappler J, Marrack P, Dow S. CD40 stimulation accelerates deletion of tumor-specific CD8(+) T cells in the absence of tumor-antigen vaccination. Proc Natl Acad Sci U S A. 2001;98(19):10811–6. PubMed PMID: 11526222. Pubmed Central PMCID: 58556. Epub 2001/08/30.
Manning EA, Ullman JG, Leatherman JM, Asquith JM, Hansen TR, Armstrong TD, et al. A vascular endothelial growth factor receptor-2 inhibitor enhances antitumor immunity through an immune-based mechanism. Clin Cancer Res. 2007;13(13):3951–9. PubMed PMID: 17606729. Epub 2007/07/04.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Kandalaft, L.E., Balint, K., Berek, J.S., Coukos, G. (2014). What Is the Future of Immunotherapy in Ovarian Cancer?. In: Ledermann, J., Creutzberg, C., Quinn, M. (eds) Controversies in the Management of Gynecological Cancers. Springer, London. https://doi.org/10.1007/978-0-85729-910-9_29
Download citation
DOI: https://doi.org/10.1007/978-0-85729-910-9_29
Published:
Publisher Name: Springer, London
Print ISBN: 978-0-85729-909-3
Online ISBN: 978-0-85729-910-9
eBook Packages: MedicineMedicine (R0)