
Abstract
Classical galactosaemia is a rare disorder of carbohydrate metabolism caused by galactose-1-phosphate uridyltransferase (GALT) deficiency (EC 2.7.7.12). The disease is life threatening if left untreated in neonates and the only available treatment option is a long-term galactose restricted diet. While this is lifesaving in the neonate, complications persist in treated individuals, and the cause of these, despite early initiation of treatment, and shared GALT genotypes remain poorly understood. Systemic abnormal glycosylation has been proposed to contribute substantially to the ongoing pathophysiology. The gross N-glycosylation assembly defects observed in the untreated neonate correct over time with treatment. However, N-glycosylation processing defects persist in treated children and adults.
Congenital disorders of glycosylation (CDG) are a large group of over 100 inherited disorders affecting largely N- and O-glycosylation.
In this review, we compare the clinical features observed in galactosaemia with a number of predominant CDG conditions.
We also summarize the N-glycosylation abnormalities, which we have described in galactosaemia adult and paediatric patients, using an automated high-throughput HILIC-UPLC analysis of galactose incorporation into serum IgG with analysis of the corresponding N-glycan gene expression patterns and the affected pathways.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Classical Galactosaemia
- Galactose-1-phosphate Uridyltransferase
- Galactosaemic Patients
- Galactose Supplementation
- Galactose Diet
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Inborn errors of metabolism to include the primary Congenital Disorders of Glycosylation (CDG) and galactosaemia can provide vast information regarding disordered metabolic pathways involving glycosylation and potentially modifiable steps (Brinkman et al. 2006; Morava et al. 2015; Sun et al. 2015).
The number of CDG has increased dramatically over the last few years. Over 100 disorders are now described with on-going characterization of new subsets (Rymen and Jaeken 2014; Scott et al. 2014; Cartault et al. 2015; Freeze et al. 2015). While different CDG have well characterized defects in glycosylation, disorders such as galactosaemia, often termed secondary disorders of glycosylation, are less well defined (Morava et al. 2015). An understanding of the shared disturbed metabolic pathways could lead to improved understanding of the pathophysiology of these disorders of glycosylation and possibly improve therapeutic approaches.
Galactosaemia is a group of rare autosomal recessive carbohydrate metabolism disorders caused by deficiency of enzymes involved in the metabolism of the aldose monosaccharide galactose (Fridovich-Keil and Walter 2008). The most severe type of galactosaemia, Classical galactosaemia (OMIM #230400) (subsequently referred to as galactosaemia in this review), is caused by profound deficiency of the galactose-1-phosphate uridyltransferase (GALT) enzyme (EC 2.7.7.12). Following galactose intake in the affected neonate, there is a toxic build-up of intermediates of galactose metabolism. Strict dietary restriction of galactose is lifesaving in the neonate, but mild to severe long-term complications persist, including significant cognitive impairment and infertility in females, regardless of genotype or age at onset of treatment (Schweitzer et al. 1993; Fridovich-Keil and Walter 2008; Krabbi et al. 2011; Coss et al. 2013; Timson 2015). The cause of this pathophysiology is currently under review.
The toxic build-up of galactose intermediates coupled with deficiency of pathway product is proposed to contribute to the development of these complications. These intermediates can result in competitive inhibition of glycosyltransferases (Lai et al. 2003). A shortage of end-product UDP-hexose sugars could also lead to disruption of glycosylation in the posttranslational modification (PTM) of proteins and lipids (Ng et al. 1989; Ornstein et al. 1992).
Dysregulation of a number of genes and pathways has been observed in galactosaemia (Coman et al. 2010; Coss et al. 2014; Maratha et al. 2016), along with abnormal glycosylation profiles of glycoproteins in both treated and untreated patients (Charlwood et al. 1998; Quintana et al. 2009; Coman et al. 2010; Berry 2011).
Early stage perturbations of glycosylation, gene expression and inositol signaling during prenatal galactose intoxication, in combination with long-term galactose restriction and individual endogenous galactose production, likely have a substantial role in determining the long-term complications seen in galactosaemia (Berry et al. 2004; Huidekoper et al. 2005; Coss et al. 2014; Schadewaldt et al. 2014; Maratha et al. 2016). Understanding the role of glycosylation in the development of these complications is essential.
Congenital Disorders of Glycosylation
CDG are a large group of mainly autosomal recessive inherited disorders affecting the glycan synthesis. These can be divided into the following major categories: disorders of protein N-glycosylation or O-glycosylation, disorders of lipid and glycosylphosphatidylinositol (GPI) anchor glycosylation and disorders of multiple glycosylation pathways (Freeze 2006; Hennet 2012), with variable symptomatic severity. Almost all organs are affected with a particular impact on nervous development, immune, hepatic and gastrointestinal systems (Freeze and Aebi 2005; Freeze et al. 2015).
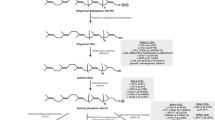
The majority of CDG disorders are caused by defects in the N-glycosylation pathway in which N-glycans are attached to arginine on proteins. N-glycan synthesis starts in the endoplasmic reticulum (ER), where the assembled product is attached. The processing of N-glycans into complex and hybrid structures continues in the Golgi apparatus. CDG-type I (CDG-I) abnormalities result from an abnormality of N-glycan assembly in the ER and CDG-type II (CDG-II) abnormalities result from abnormalities in N-glycan processing after transfer to the protein in the ER or steps occurring in the Golgi apparatus (Freeze 2013; Freeze et al. 2015), as depicted in Fig. 1.

Schematic representation of: (a) N-glycan assembly in rER (Congenital Disorders of Glycosylation-type I, CDG-I) and (b) N-glycan processing in Golgi apparatus (Congenital Disorders of Glycosylation-type II, CDG-II). Blue arrows for ALG9 (Alpha-1, 2-mannosyltransferase), MGAT1 and MGAT3 represent respective gene expression pattern observed in our recent study (Maratha et al. 2016)
Approximately 1% of the human genome encodes genes involved in glycosylation and over half of all proteins are N-glycosylated (Freeze 1998; Pivac et al. 2011). Glycoproteins are central to many key biological systems such as cell–cell signaling, and are important in coagulation, immunity, fertility, etc. (Zoldos et al. 2010).
Galactosaemia has been reported as a secondary disorder of glycosylation, displaying characteristics of both CDG-I and CDG-II defects with both glycan assembly and processing defects observed (Charlwood et al. 1998; Sturiale et al. 2005; Quintana et al. 2009; Coman et al. 2010; Coss et al. 2012).
CDG and galactosaemia share multiple clinical characteristics. Tables 1 and 2 summarize a number of CDG syndromes (I and II) with symptoms which may also be observed in galactosaemia including neurological involvement, coagulopathies and liver disease. For example, PMM2-CDG, the most common subtype of CDG, is an N-glycan assembly defect caused by lack of phosphomannomutase 2 (PMM2) which converts mannose-6-phosphate to mannose-1-phosphate. The enzymatic deficiency results in reduced GDP-mannose required for the synthesis of the lipid-linked oligosaccharide (LLO) precursor. The symptoms observed are commonly intellectual disability, hypotonia, cerebellar dysfunction, polyneuropathy and stroke-like episodes (Dinopoulos et al. 2007).
The key clinical adverse outcomes seen in galactosaemia include intellectual disabilities, speech abnormalities and primary ovarian insufficiency (POI) in females. Intellectual disability occurs in at least 50% of affected individuals (Waggoner et al. 1990; Schweitzer et al. 1993; Shield et al. 2000; Doyle et al. 2010; Waisbren et al. 2012; Coss et al. 2013; Rubio-Agusti et al. 2013). Abnormal myelination was first documented in galactosaemia in 1971 (Haberland et al. 1971; Lebea and Pretorius 2005). There is considerable variability in IQs documented between galactosaemia patients with scores ranging from very low to well above average.
The presence of speech and language abnormalities (commonly verbal dyspraxia) is well documented in galactosaemia. One well-described entity is Childhood Apraxia of Speech (CAS). Overall, speech and language disorders are estimated to affect at least 25% of individuals with galactosaemia, commonly presenting in childhood (Schweitzer et al. 1993; Potter et al. 2008, 2013; Timmers et al. 2012; Waisbren et al. 2012; Coss et al. 2013), with pathophysiological correlates studied in fMRI brain studies (Timmers et al. 2015).
Over 80% of galactosaemia females suffer from POI (91% in Irish female patients) (Waggoner et al. 1990; Sanders et al. 2009; Coss et al. 2013). The clinical presentation varies from primary amenorrhea to delayed pubertal development followed by irregular menses or secondary amenorrhea. This results in infertility or sub-fertility as a predominant feature in galactosaemia females of childbearing age (Rubio-Gozalbo et al. 2010).
The pathophysiology for this presentation is still unknown. Mechanisms proposed include prenatal toxicity with galactose and metabolites possibly causing premature follicular apoptosis/atresia, abnormal cell signaling and hormone/receptor glycosylation abnormalities. Hypoglycosylation of follicle stimulating hormone (FSH) could theoretically alter its function and lead to POI in galactosaemia. However biochemical tests have shown inconclusive results (Prestoz et al. 1997; Gubbels et al. 2011).
Male galactosaemia patients seem to be less affected and have successfully fathered offspring, reaching puberty spontaneously although the age of onset can be delayed (Rubio-Gozalbo et al. 2010). PMM2-CDG patients also virilize normally through puberty but with occurrences of decreased testicular volume and increased serum FSH concentrations (de Zegher and Jaeken 1995).
Altered leptin signaling secondary to glycosylation abnormalities may be contributory (Knerr et al. 2013). Leptin is a key energy and fat storage regulator (Kratzsch et al. 2002) and abnormal leptin signaling due to hypoglycosylation has also been considered in abnormal fat distribution in PMM2-CDG (Wolthuis et al. 2013).
Osteopenia is another long-term complication associated with galactosaemia. Osteopenia and other skeletal abnormalities are common clinical findings in CDG (Coman et al. 2008; Rimella-Le-Huu et al. 2008). It has been suggested that hypoglycosylation of noncollagenous bone proteins may be responsible for decreased bone mass and increased osteocalcin levels in PMM2-CDG patients (Barone et al. 2002).
Decreased bone mineral density has been consistently observed in galactosaemia patients (Rubio-Gozalbo et al. 2002; Panis et al. 2004; Waisbren et al. 2012; Batey et al. 2013; Coss et al. 2013; Doulgeraki et al. 2014). Decreased bone metabolism has been suggested as the mechanism of reduced bone mineral density in galactosaemia as well as abnormal galactosylation of the collagen matrix (Kaufman et al. 1993).
Reduced levels of insulin-like growth factor 1 (IGF-1), carboxylated osteocalcin, N-terminal telopeptide and C-terminal telopeptide have been reported in serum samples from galactosaemia patients (Panis et al. 2004; Fridovich-Keil and Walter 2008).
Serum IgG N-Glycosylation Abnormalities in Galactosaemia
As discussed earlier, substantial N-glycan abnormalities have been demonstrated in serum transferrin IEF patterns in galactosaemia (Charlwood et al. 1998; Sturiale et al. 2005; Quintana et al. 2009). UDP-galactose substrate deficiency is one of the proposed contributing pathophysiological mechanisms (Gibson et al. 1995; Lai et al. 2003; Parkinson et al. 2013; Jumbo-Lucioni et al. 2014).
We have documented hypoglycosylation and gross N-glycan assembly defects in the whole serum of untreated neonates with galactosaemia similar to what has been observed in CDG-I defects (Coman et al. 2010; Coss et al. 2014). While the N-glycan assembly defects resolve within the first 6 months of life with galactose restriction, it is apparent that, after this initial effect, N-glycan processing defects persist, even in young children (Coss et al. 2014).
We also performed a study of N-glycan processing defects in 10 treated galactosaemia adults on a restricted galactose diet in comparison to matched controls using serum IgG analysis (the most abundant circulating N-glycan glycoprotein), analyzed by NP-HPLC to monitor the effects and potential benefits of galactose supplementation with galactosylation of IgG used as a specific biomarker of dietary galactose tolerance. We demonstrated an increase in non-galactosylated (G0) and monogalactosylated (G1) structures with decreased digalactosylated structures (G2) in diet-restricted galactosaemia patients, indicating continued N-glycan processing defects despite treatment (Coss et al. 2012). Five subjects followed a moderate galactose liberalization trial over 16 weeks. Their IgG N-glycan profiles showed consistent individual alterations in response to diet liberalization with improvement of profiles for three of the five subjects at a galactose intake of 1000 mg/day.
We recently also published a study of 13 children with galactosaemia which indicated that a moderate increase in galactose intake may be well tolerated in children and may improve glycosylation (Knerr et al. 2015).
We previously also identified that children with galactosaemia had lower serum leptin levels than normal controls, expressed as SDS for gender and pubertal age (Knerr et al. 2013). In the above diet relaxation study, there was no statistical significant difference noted in serum leptin levels in the patient control group and the diet relaxation groups at the baseline point. However, patients in the galactose supplementation group had, as a trend, slightly higher leptin levels at the end of the study than patient controls (p < 0.05), but within the normal range.
We have now established a rapid automated robotic hydrophilic interaction ultra-performance liquid chromatography N-glycan analysis for the measurement of IgG N-glycan galactose incorporation applied to adult galactosaemia patients which has demonstrated significant differences between the G0/G1 and G0/G2 incorporation ratios and controls (Stockmann et al. 2016).
This analysis of IgG glycosylation has also recently been applied to the CDG condition MAN1B1-CDG using this methodology (Saldova et al. 2015).
To further identify the specific N-glycosylation steps that are affected in galactosaemia, we performed further glycan subset analysis in the IgG glycosylation study of 40 galactosaemia treated patients compared to controls. In this work, we identified a significant increase in core fucosylated neutral glycans and a significant decrease in core fucosylated, afucosylated bisected glycans and N-linked mannose-5 glycans in circulating serum IgG N-glycans (Maratha et al. 2016). Figure 1, amended from this study, illustrates the steps in N-glycan synthesis, which may be affected in this pathway.
Abnormal Gene Expression and Cell Signaling in Galactosaemia
In a pilot microarray study of T-lymphocyte RNA expression from four galactosaemia patients (Coman et al. 2010), we identified extensive dysregulation of genes affecting many signaling pathways including MAP kinase, regulation of actin cytoskeleton, ubiquitin mediated proteolysis, inositol signaling, inflammatory pathways and glycan biosynthesis pathways (Coman et al. 2010), and we subsequently validated dysregulation of a number of N-linked glycosylation biosynthesis genes linked to CDG-1 and CDG-II, e.g. ALG (1, 2, 8 and 9) in a larger study (Coss et al. 2014).
We also confirmed and noted the dysregulation of the genes ANXA1 and ALG9 (Alpha-1,2-mannosyltransferase) (which also responded to differing levels of galactose exposure), in cultured galactosaemia patient fibroblast cells (Coss et al. 2014).
We have subsequently studied the expression of a number of these genes and other related relevant N-glycan biosynthesis genes in peripheral blood mononuclear cells from affected galactosaemia adult patients. We noted significant dysregulation of two key N-glycan biosynthesis genes ALG9, which was up-regulated (p < 0.001), and MGAT1, which was down-regulated (p < 0.01) with additional dysregulation of the genes FUT8, and MGAT3 (Maratha et al. 2016). The site of action of these genes is illustrated in Fig. 1.
The ALG9 (Alpha-1,2-mannosyltransferase) gene product is involved in the addition of the seventh and ninth mannose sugar to the growing N-glycan, essential for the formation of the initial oligosaccharide chain. It has been proposed that the interaction of ALG9 with ALG12 is required for the ultimate formation of the disaccharide glycan, which influences further downstream processing of N-glycans, indicating a potential regulatory role for the ALG9 gene in glycosylation (Coss et al. 2014). The clinical phenotype for ALG9-CDG has recently been expanded (see Table 1) (AlSubhi et al. 2016).
The decreased expression of the MGAT1 gene also has significant pathological correlates. The MGAT1 gene encodes GlcNAc transferase I (Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase), which adds GlcNAc to high-mannose sites, an essential early step in producing all branched complex and hybrid N-glycans.
Inactivation of the MGAT1 gene in mice was shown to impair oogenesis, and mouse MGAT1 knockouts were unviable (Shi et al. 2004).
Galactosaemia and CDG: Dietary Treatment Approaches
Our studies of IgG N-glycosylation with varying effects of galactose exposure in galactosaemia adults and children have indicated the presence of significant interindividual tolerance of exogenous galactose in galactosaemia patients. There are reports on individuals who have relaxed the diet at an early age with good outcomes (Lee et al. 2003; Panis et al. 2006). It appears that some affected individuals with galactosaemia may have more ability to utilize alternative, accessory pathways to metabolize galactose and its metabolites than others and may tolerate moderate amounts of exogenous dietary galactose (Coss et al. 2014). This may be influenced by epigenetic regulation (Lauc and Zoldos 2009). As an illustrative example, the over-expression of human UDP-glucose pyrophosphorylase (hUGP2), an ‘accessory pathway’ enzyme, using both galactose-1-phosphate and glucose-1-phosphate as substrates, rescued GALT-deficient yeast cells from galactose toxicity (Lai and Elsas 2000). The UGP2 reaction may not be relevant under normal physiological conditions as high toxic levels of galactose-1-phosphate seem to be required (Lai et al. 2003), as glucose-1-phosphate is the preferred substrate (Leslie et al. 2005). As referred earlier, it is possible that some patients may have the ability to generate more UDP-galactose, using excess galactose-1-phosphate as a substrate (Lai and Elsas 2000; Fridovich-Keil and Walter 2008).
Considering this variability in accessory pathways of galactose metabolism and linked glycosylation, we propose that the clinical outcomes observed in galactosaemia are multifactorial, influenced by prenatal toxicity and postnatal variation in accessory glycosylation pathways (Coss et al. 2012; Knerr et al. 2015).
Also, while the severe restriction of dietary galactose in the affected newborn is life saving and largely reverses the N-glycan assembly defect, our studies suggest that over restriction of galactose in the long-term may contribute to ongoing N-glycan processing defects, evident in all the galactosaemia patients whom we have studied to date (Coman et al. 2010; Coss et al. 2012; Knerr et al. 2015; Stockmann et al. 2016).
The manipulation of exogenous provided sugar substrates in CDG is informative. At least four subtypes of CDG have been treated with dietary modulation of sugars: MPI-CDG, SLC55C1-CDG, PGM1-CDG (Hendriksz et al. 2001; Harms et al. 2002; Penel-Capelle et al. 2003; de Lonlay and Seta 2009) and SLC35A2-CDG (Ng et al. 2013; Dorre et al. 2015).
SLC55C1-CDG is caused by a decreased affinity of the GDP-fucose transporters resulting in decreased fucose, resulting in immunological defects and severe psychomotor delay (Goreta et al. 2012). Supplementation with oral fucose challenges the defective transporters leading to clinical improvements in some patients, with correction of immunological dysfunction and psychomotor improvement (Marquardt et al. 1999; Jaeken 2010).
PGM1-CDG phosphoglucomutase 1 deficiency (E.C 5.4.2.2) is caused by disruption of the glucose metabolism pathway whereby phosphoglucomutase catalyzes the bidirectional transfer of phosphate from position 1 to 6 on glucose. Deficiency of this enzyme, now characterized clinically by hypoglycaemia, liver disease, cardiomyopathy, short stature, cleft palate and normal intelligence, has previously been associated with a primary muscle disease, Glycogen Storage Disease, XIV (Morava 2014). There are a limited number of patients reported in the literature; one of the first reported suffered from exercise-induced rhabdomyolysis and muscular glycogenosis (Stojkovic et al. 2009; Timal et al. 2012; Morava 2014). The disruption of this pathway (caused by reduced PGM1) results in dysregulation of glycolysis and disruption of the galactose metabolism pathway. It has been suggested that the build-up of glucose-1-phosphate competes with galactose-1-phosphate for the UDP-glucose pyrophosphorylase enzyme. This drives the product of the pyrophosphorylase pathway towards UDP-glucose, reducing the level of UDP-galactose (Perez et al. 2013). If this is the dysregulated pathway of PGM1-CDG, then it would indicate there is some biologically relevant level of UDP-galactose produced from the UDP-glucose pyrophosphorylase pathway, which has direct relevance for galactosaemia. A study of PGM1-CDG patients treated with a combination of d-galactose and complex carbohydrate supplementation improved serum transferrin hypoglycosylation and ameliorated clinical symptoms. This study indicated increased levels of activated UDP-galactose in the treated patients which improved glycosylation (Morava 2014).
In addition, Ng et al. in 2013 reported a disorder of the X-linked gene UDP-galactose transporter SLC35A2. This disorder leads to galactose-deficient glycoproteins as measured by N-glycans from whole serum using MALDI-TOF. This showed increased levels of hypogalactosylated glycans, particularly biantennary species (Ng et al. 2013). Interestingly, in 3 affected children, the neonatal profile improved and normalized during the first few years of life. In a recently reported child with this transporter deficiency, dietary galactose supplementation resulted in nearly complete normalization of the abnormal transferrin glycosylation pattern (Dorre et al. 2015).
The beneficial effect of galactose supplementation for PGM1-CDG and SLC35A2 deficiency suggests the physiological need for supplementary exogenous galactose in the presence of UDP galactose limited bioavailability. This is of possible relevance to galactosaemia.
Conclusion
There are many biochemical and clinical similarities between galactosaemia and CDG syndromes. Early (prenatal and perinatal) dysregulation of glycosylation in galactosaemia must be a major determinant of both neurological/cognitive and reproductive deficits, while ongoing abnormalities in glycosylation and associated gene dysregulation and associated cell signaling abnormalities may also have relevant pathophysiological consequences.
The persistence of aberrant glycosylation and disruption of CDG-related genes in long-term treated galactosaemia patients suggest that this is a major area in galactosaemia research. This requires further investigation which may offer new biomarkers to monitor affected individuals and enhance our understanding of this and related conditions.
References
AlSubhi S, AlHashem A, AlAzami A et al (2016) Further delineation of the ALG9-CDG phenotype. JIMD Rep. doi:10.1007/8904-2015-504
Barone R, Pavone V, Pennisi P, Fiumara A, Fiore CE (2002) Assessment of skeletal status in patients with congenital disorder of glycosylation type IA. Int J Tissue React 24:23–28
Batey LA, Welt CK, Rohr F et al (2013) Skeletal health in adult patients with classic galactosemia. Osteoporos Int 24:501–509
Berry GT (2011) Is prenatal myo-inositol deficiency a mechanism of CNS injury in galactosemia? J Inherit Metab Dis 34:345–355
Berry GT, Moate PJ, Reynolds RA et al (2004) The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab 81:22–30
Brinkman RR, Dube MP, Rouleau GA, Orr AC, Samuels ME (2006) Human monogenic disorders - a source of novel drug targets. Nat Rev Genet 7:249–260
Cartault F, Munier P, Jacquemont ML et al (2015) Expanding the clinical spectrum of B4GALT7 deficiency: homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur J Hum Genet 23:49–53
Chantret I, Dupré T, Delenda C et al (2002) Congenital disorders of glycosylation type Ig is defined by a deficiency in dolichyl-P-mannose:Man7GlcNAc2-PP-dolichyl mannosyltransferase. J Biol Chem 277:25815–25822
Chantret I, Dancourt J, Dupré T et al (2003) A deficiency in dolichyl-P-glucose:Glc1Man9GlcNAc2-PP-dolichyl alpha3-glucosyltransferase defines a new subtype of congenital disorders of glycosylation. J Biol Chem 278:9962–9971
Charlwood J, Clayton P, Keir G, Mian N, Winchester B (1998) Defective galactosylation of serum transferrin in galactosemia. Glycobiology 8:351–357
Coman D, Irving M, Kannu P, Jaeken J, Savarirayan R (2008) The skeletal manifestations of the congenital disorders of glycosylation. Clin Genet 73:507–515
Coman DJ, Murray DW, Byrne JC et al (2010) Galactosemia, a single gene disorder with epigenetic consequences. Pediatr Res 67:286–292
Coss KP, Byrne JC, Coman DJ et al (2012) IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol Genet Metab 105:212–220
Coss KP, Doran PP, Owoeye C et al (2013) Classical Galactosaemia in Ireland: incidence, complications and outcomes of treatment. J Inherit Metab Dis 36:21–27
Coss KP, Treacy E, Cotter E et al (2014) Systemic gene dysregulation in classical Galactosaemia: Is there a central mechanism? Mol Genet Metab 113:177–187
de Lonlay P, Seta N (2009) The clinical spectrum of phosphomannose isomerase deficiency, with an evaluation of mannose treatment for CDG-Ib. Biochim Biophys Acta 1792:841–843
de Lonlay P, Seta N, Barrot S et al (2001) A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 38:14–19
de Zegher F, Jaeken J (1995) Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 37:395–401
Dinopoulos A, Mohamed I, Jones B, Rao S, Franz D, deGrauw T (2007) Radiologic and neurophysiologic aspects of stroke-like episodes in children with congenital disorder of glycosylation type Ia. Pediatrics 119:e768–e772
Dorre K, Olczak M, Wada Y et al (2015) A new case of UDP-galactose transporter deficiency (SLC35A2-CDG): molecular basis, clinical phenotype, and therapeutic approach. J Inherit Metab Dis 38:931–940
Doulgeraki A, Monopolis I, Deligianni D, Kalogerakou M, Schulpis KH (2014) Body composition in young patients with galactose metabolic disorders: a preliminary report. J Pediatr Endocrinol Metab 27:81–86
Doyle CM, Channon S, Orlowska D, Lee PJ (2010) The neuropsychological profile of galactosaemia. J Inherit Metab Dis 33:603–609
Drouin-Garraud V, Belgrand M, Grünewald S et al (2001) Neurological presentation of a congenital disorder of glycosylation CDG-Ia: implications for diagnosis and genetic counseling. Am J Med Genet 101:46–49
Dupré T, Vuillaumier-Barrot S, Chantret I et al (2010) Guanosine diphosphate-mannose:GlcNAc2-PP-dolichol mannosyltransferase deficiency (congenital disorders of glycosylation type Ik): five new patients and seven novel mutations. J Med Genet 47:729–735
Etzioni A, Frydman M, Pollack S et al (1992) Brief report: recurrent severe infections caused by a novel leukocyte adhesion deficiency. N Engl J Med 327:1789–1792
Frank CG, Grubenmann CE, Eyaid W, Berger EG, Aebi M, Hennet T (2004) Identification and functional analysis of a defect in the human ALG9 gene: definition of congenital disorder of glycosylation type IL. Am J Hum Genet 75:146–150
Freeze HH (1998) Disorders in protein glycosylation and potential therapy: tip of an iceberg? J Pediatr 133:593–600
Freeze HH (2006) Genetic defects in the human glycome. Nat Rev Genet 7:537–551
Freeze HH (2013) Understanding human glycosylation disorders: biochemistry leads the charge. J Biol Chem 288:6936–6945
Freeze HH, Aebi M (2005) Altered glycan structures: the molecular basis of congenital disorders of glycosylation. Curr Opin Struct Biol 15:490–498
Freeze HH, Eklund EA, Ng BG, Patterson MC (2015) Neurological aspects of human glycosylation disorders. Annu Rev Neurosci 38:105–125
Fridovich-Keil JL, Walter JH (2008) Galactosaemia Chapter 72 online metabolic and molecular bases of inherited diseases-OMMBID. In Valle DBA, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A et al (eds) Part 7: carbohydrates. McGraw Hill, New York
Gibson JB, Reynolds RA, Palmieri MJ et al (1995) Comparison of erythrocyte uridine sugar nucleotide levels in normals, classic galactosemics, and patients with other metabolic disorders. Metabolism 44:597–604
Goreta SS, Dabelic S, Dumic J (2012) Insights into complexity of congenital disorders of glycosylation. Biochem Med (Zagreb) 22:156–170
Gubbels CS, Thomas CM, Wodzig WK et al (2011) FSH isoform pattern in classic galactosemia. J Inherit Metab Dis 34:387–390
Haberland C, Perou M, Brunngraber EG, Hof H (1971) The neuropathology of galactosemia. A histopathological and biochemical study. J Neuropathol Exp Neurol 30:431–447
Harms HK, Zimmer KP, Kurnik K, Bertele-Harms RM, Weidinger S, Reiter K (2002) Oral mannose therapy persistently corrects the severe clinical symptoms and biochemical abnormalities of phosphomannose isomerase deficiency. Acta Paediatr 91:1065–1072
Hendriksz CJ, McClean P, Henderson MJ et al (2001) Successful treatment of carbohydrate deficient glycoprotein syndrome type 1b with oral mannose. Arch Dis Child 85:339–340
Hennet T (2012) Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim Biophys Acta 1820:1306–1317
Huidekoper HH, Bosch AM, van der Crabben SN, Sauerwein HP, Ackermans MT, Wijburg FA (2005) Short-term exogenous galactose supplementation does not influence rate of appearance of galactose in patients with classical galactosemia. Mol Genet Metab 84:265–272
Jaeken J (2010) Congenital disorders of glycosylation. Ann N Y Acad Sci 1214:190–198
Jumbo-Lucioni P, Parkinson W, Broadie K (2014) Overelaborated synaptic architecture and reduced synaptomatrix glycosylation in a Drosophila classic galactosemia disease model. Dis Model Mech 7:1365–1378
Kaufman FR, Loro ML, Azen C, Wenz E, Gilsanz V (1993) Effect of hypogonadism and deficient calcium intake on bone density in patients with galactosemia. J Pediatr 123:365–370
Knerr I, Coss KP, Doran PP et al (2013) Leptin levels in children and adults with classic galactosaemia. JIMD Rep 9:125–131
Knerr I, Coss KP, Kratzsch J et al (2015) Effects of temporary low-dose galactose supplements in children aged 5-12 y with classical galactosemia: a pilot study. Pediatr Res 78:272–279
Krabbi K, Uudelepp ML, Joost K, Zordania R, Ounap K (2011) Long-term complications in Estonian galactosemia patients with a less strict lactose-free diet and metabolic control. Mol Genet Metab 103:249–252
Kranz C, Denecke J, Lehle L et al (2004) Congenital disorder of glycosylation type Ik (CDG-Ik): a defect of mannosyltransferase I. Am J Hum Genet 74:545–551
Kranz C, Sun L, Eklund EA, Krasnewich D, Casey JR, Freeze HH (2007) CDG-Id in two siblings with partially different phenotypes. Am J Med Genet A 143A:1414–1420
Kratzsch J, Lammert A, Bottner A et al (2002) Circulating soluble leptin receptor and free leptin index during childhood, puberty, and adolescence. J Clin Endocrinol Metab 87:4587–4594
Lai K, Elsas LJ (2000) Overexpression of human UDP-glucose pyrophosphorylase rescues galactose-1-phosphate uridyltransferase-deficient yeast. Biochem Biophys Res Commun 271:392–400
Lai K, Langley SD, Khwaja FW, Schmitt EW, Elsas LJ (2003) GALT deficiency causes UDP-hexose deficit in human galactosemic cells. Glycobiology 13:285–294
Lauc G, Zoldos V (2009) Epigenetic regulation of glycosylation could be a mechanism used by complex organisms to compete with microbes on an evolutionary scale. Med Hypotheses 73:510–512
Lebea PJ, Pretorius PJ (2005) The molecular relationship between deficient UDP-galactose uridyl transferase (GALT) and ceramide galactosyltransferase (CGT) enzyme function: a possible cause for poor long-term prognosis in classic galactosemia. Med Hypotheses 65:1051–1057
Lee PJ, Lilburn M, Wendel U, Schadewaldt P (2003) A woman with untreated galactosaemia. Lancet 362:446
Leslie N, Yager C, Reynolds R, Segal S (2005) UDP-galactose pyrophosphorylase in mice with galactose-1-phosphate uridyltransferase deficiency. Mol Genet Metab 85:21–27
Lübke T, Marquardt T, von Figura K, Körner C (1999) A new type of carbohydrate-deficient glycoprotein syndrome due to a decreased import of GDP-fucose into the golgi. J Biol Chem 274:25986–25989
Maratha A, Stockmann H, Coss KP et al (2016) Classical galactosaemia: novel insights in IgG N-glycosylation and N-glycan biosynthesis. Eur J Hum Genet (Epub). doi.10.1038/1jhg.2015.254
Marquardt T, Denecke J (2003) Congenital disorders of glycosylation: review of their molecular bases, clinical presentations and specific therapies. Eur J Pediatr 162:359–379
Marquardt T, Luhn K, Srikrishna G, Freeze HH, Harms E, Vestweber D (1999) Correction of leukocyte adhesion deficiency type II with oral fucose. Blood 94:3976–3985
Matthijs G, Schollen E, Pardon E et al (1997) Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat Genet 16:88–92
Morava E (2014) Galactose supplementation in phosphoglucomutase-1 deficiency; review and outlook for a novel treatable CDG. Mol Genet Metab 112:275–279
Morava E, Zeevaert R, Korsch E et al (2007) A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur J Hum Genet 15:638–645
Morava E, Rahman S, Peters V, Baumgartner MR, Patterson M, Zschocke J (2015) Quo vadis: the re-definition of “inborn metabolic diseases”. J Inherit Metab Dis 38:1003–1006
Ng WG, Xu YK, Kaufman FR, Donnell GN (1989) Deficit of uridine diphosphate galactose in galactosaemia. J Inherit Metab Dis 12:257–266
Ng BG, Sharma V, Sun L et al (2011) Identification of the first COG-CDG patient of Indian origin. Mol Genet Metab 102:364–367
Ng BG, Buckingham KJ, Raymond K et al (2013) Mosaicism of the UDP-galactose transporter SLC35A2 causes a congenital disorder of glycosylation. Am J Hum Genet 92:632–636
Ornstein KS, McGuire EJ, Berry GT, Roth S, Segal S (1992) Abnormal galactosylation of complex carbohydrates in cultured fibroblasts from patients with galactose-1-phosphate uridyltransferase deficiency. Pediatr Res 31:508–511
Panis B, Forget PP, van Kroonenburgh MJ et al (2004) Bone metabolism in galactosemia. Bone 35:982–987
Panis B, Bakker JA, Sels JP, Spaapen LJ, van Loon LJ, Rubio-Gozalbo ME (2006) Untreated classical galactosemia patient with mild phenotype. Mol Genet Metab 89:277–279
Parkinson W, Dear ML, Rushton E, Broadie K (2013) N-glycosylation requirements in neuromuscular synaptogenesis. Development 140:4970–4981
Penel-Capelle D, Dobbelaere D, Jaeken J, Klein A, Cartigny M, Weill J (2003) Congenital disorder of glycosylation Ib (CDG-Ib) without gastrointestinal symptoms. J Inherit Metab Dis 26:83–85
Perez B, Medrano C, Ecay MJ et al (2013) A novel congenital disorder of glycosylation type without central nervous system involvement caused by mutations in the phosphoglucomutase 1 gene. J Inherit Metab Dis 36:535–542
Peters V, Penzien JM, Reiter G et al (2002) Congenital disorder of glycosylation IId (CDG-IId) -- a new entity: clinical presentation with Dandy-Walker malformation and myopathy. Neuropediatrics 33:27–32
Pivac N, Knezevic A, Gornik O et al (2011) Human plasma glycome in attention-deficit hyperactivity disorder and autism spectrum disorders. Mol Cell Proteomics 10:M110 004200
Potter NL, Lazarus JA, Johnson JM, Steiner RD, Shriberg LD (2008) Correlates of language impairment in children with galactosaemia. J Inherit Metab Dis 31:524–532
Potter NL, Nievergelt Y, Shriberg LD (2013) Motor and speech disorders in classic galactosemia. JIMD Rep 11:31–41
Prestoz LL, Couto AS, Shin YS, Petry KG (1997) Altered follicle stimulating hormone isoforms in female galactosaemia patients. Eur J Pediatr 156:116–120
Quintana E, Navarro-Sastre A, Hernandez-Perez JM et al (2009) Screening for congenital disorders of glycosylation (CDG): transferrin HPLC versus isoelectric focusing (IEF). Clin Biochem 42:408–415
Rafiq MA, Kuss AW, Puettmann L et al (2011) Mutations in the alpha 1,2-mannosidase gene, MAN1B1, cause autosomal-recessive intellectual disability. Am J Hum Genet 89:176–182
Rimella-Le-Huu A, Henry H, Kern I et al (2008) Congenital disorder of glycosylation type Id (CDG Id): phenotypic, biochemical and molecular characterization of a new patient. J Inherit Metab Dis
Rubio-Agusti I, Carecchio M, Bhatia KP et al (2013) Movement disorders in adult patients with classical galactosemia. Mov Disord 28:804–810
Rubio-Gozalbo M, Hamming S, van Kroonenburgh MJPG, Bakker J, Vermeer C, Forget P (2002) Bone mineral density in patients with classic galactosaemia. Arch Dis Child 87:57–60
Rubio-Gozalbo ME, Gubbels CS, Bakker JA, Menheere PP, Wodzig WK, Land JA (2010) Gonadal function in male and female patients with classic galactosemia. Hum Reprod Update 16:177–188
Rymen D, Jaeken J (2014) Skin manifestations in CDG. J Inherit Metab Dis 37:699–708
Saldova R, Stockmann H, O’Flaherty R, Lefeber DJ, Jaeken J, Rudd PM (2015) N-Glycosylation of serum IgG and total glycoproteins in MAN1B1 deficiency. J Proteome Res 14:4402–4412
Sanders RD, Spencer JB, Epstein MP et al (2009) Biomarkers of ovarian function in girls and women with classic galactosemia. Fertil Steril 92:344–351
Schadewaldt P, Kamalanathan L, Hammen HW, Kotzka J, Wendel U (2014) Endogenous galactose formation in galactose-1-phosphate uridyltransferase deficiency. Arch Physiol Biochem 120:228–239
Schweitzer S, Shin Y, Jakobs C, Brodehl J (1993) Long-term outcome in 134 patients with galactosaemia. Eur J Pediatr 152:36–43
Scott K, Gadomski T, Kozicz T, Morava E (2014) Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis 37:609–617
Shi S, Williams SA, Seppo A et al (2004) Inactivation of the Mgat1 gene in oocytes impairs oogenesis, but embryos lacking complex and hybrid N-glycans develop and implant. Mol Cell Biol 24:9920–9929
Shield JP, Wadsworth EJ, MacDonald A et al (2000) The relationship of genotype to cognitive outcome in galactosaemia. Arch Dis Child 83:248–250
Sparks SE, Krasnewich DM (1993) Congenital disorders of glycosylation overview. In Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds) GeneReviews. University of Washington, Seattle, WA
Stockmann H, Coss KP, Rubio-Gozalbo ME et al (2016) IgG N-Glycosylation Galactose Incorporation Ratios for the Monitoring of Classical Galactosaemia. JIMD Rep. doi.10.1007/8904_2015_490
Stojkovic T, Vissing J, Petit F et al (2009) Muscle glycogenosis due to phosphoglucomutase 1 deficiency. N Engl J Med 361:425–427
Sturiale L, Barone R, Fiumara A et al (2005) Hypoglycosylation with increased fucosylation and branching of serum transferrin N-glycans in untreated galactosemia. Glycobiology 15:1268–1276
Sun J, Zhu K, Zheng W, Xu H (2015) A comparative study of disease genes and drug targets in the human protein interactome. BMC Bioinformatics 16 Suppl 5:S1
Tegtmeyer LC, Rust S, van Scherpenzeel M et al (2014) Multiple phenotypes in phosphoglucomutase 1 deficiency. N Engl J Med 370:533–542
Thiel C, Schwarz M, Hasilik M et al (2002) Deficiency of dolichyl-P-Man:Man7GlcNAc2-PP-dolichyl mannosyltransferase causes congenital disorder of glycosylation type Ig. Biochem J 367:195–201
Thiel C, Schwarz M, Peng J et al (2003) A new type of congenital disorders of glycosylation (CDG-Ii) provides new insights into the early steps of dolichol-linked oligosaccharide biosynthesis. J Biol Chem 278:22498–22505
Timal S, Hoischen A, Lehle L et al (2012) Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum Mol Genet 21:4151–4161
Timmers I, Jansma BM, Rubio-Gozalbo ME (2012) From mind to mouth: event related potentials of sentence production in classic galactosemia. PLoS One 7:e52826
Timmers I, van den Hurk J, Hofman PAM et al (2015) Affected functional networks associated with sentence production in classic galactosemia. Brain Res 1616:166–176
Timson DJ (2015) The molecular basis of galactosemia - Past, present and future. Gene. pii. S0378-1119 (15)00801-X
Van Geet C, Jaeken J (1993) A unique pattern of coagulation abnormalities in carbohydrate-deficient glycoprotein syndrome. Pediatr Res 33:540–541
Waggoner DD, Buist NR, Donnell GN (1990) Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis 13:802–818
Waisbren SE, Potter NL, Gordon CM et al (2012) The adult galactosemic phenotype. J Inherit Metab Dis 35:279–286
Weinstein M, Schollen E, Matthijs G et al (2005) CDG-IL: an infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am J Med Genet (A) 136:194–197
Wolthuis DF, van Asbeck EV, Kozicz T, Morava E (2013) Abnormal fat distribution in PMM2-CDG. Mol Genet Metab 110:411–413
Zeevaert R, Foulquier F, Cheillan D et al (2009) A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur J Med Genet 52:303–305
Zoldos V, Grgurevie S, Lauc G (2010) Epigenetic regulation of protein glycosylation. Biomol Concepts 1:253
Acknowledgments
The authors acknowledge funding from the Irish Health Research Board (HRB-HRA POR award), and (MRCG/ TSCUH CFFH HRB award) which have supported this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by: Jaak Jaeken
Appendices
Synopsis
An understanding of the link between galactosaemia and CDG, with a focus on abnormal N-glycosylation.
Compliance with Ethics Guidelines
This article does not contain any studies with human or animal subjects performed by any of the authors.
Ashwini Maratha and Eileen Treacy have been involved in the planning, conception, writing, drafting and reviewing this review. Hugh-Owen Colhoun assisted in writing this review. Ina Knerr, Karen Coss and Peter Doran have assisted in reviewing this review.
Conflict of Interest
The authors Ashwini Maratha, Hugh-Owen Colhoun, Ina Knerr, Karen Coss, Peter Doran and Eileen Treacy declare that they have no conflict of interest.
Rights and permissions
Copyright information
© 2016 SSIEM and Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Maratha, A., Colhoun, HO., Knerr, I., Coss, K.P., Doran, P., Treacy, E.P. (2016). Classical Galactosaemia and CDG, the N-Glycosylation Interface. A Review. In: Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V. (eds) JIMD Reports, Volume 34. JIMD Reports, vol 34. Springer, Berlin, Heidelberg. https://doi.org/10.1007/8904_2016_5
Download citation
DOI: https://doi.org/10.1007/8904_2016_5
Received:
Revised:
Accepted:
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-55585-9
Online ISBN: 978-3-662-55586-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)