
Abstract
For thousands of years people were delivered helplessly to various kinds of infections, which often reached epidemic proportions and have cost the lives of millions of people. This is precisely the age since mankind has been thinking of infectious diseases and the question of their causes. However, due to a lack of knowledge, the search for strategies to fight, heal, and prevent the spread of communicable diseases was unsuccessful for a long time. It was not until the discovery of the healing effects of (antibiotic producing) molds, the first microscopic observations of microorganisms in the seventeenth century, the refutation of the abiogenesis theory, and the dissolution of the question “What is the nature of infectious diseases?” that the first milestones within the history of antibiotics research were set. Then new discoveries accelerated rapidly: Bacteria could be isolated and cultured and were identified as possible agents of diseases as well as producers of bioactive metabolites. At the same time the first synthetic antibiotics were developed and shortly thereafter, thousands of synthetic substances as well as millions of soil borne bacteria and fungi were screened for bioactivity within numerous microbial laboratories of pharmaceutical companies. New antibiotic classes with different targets were discovered as on assembly line production. With the beginning of the twentieth century, many of the diseases which reached epidemic proportions at the time—e.g., cholera, syphilis, plague, tuberculosis, or typhoid fever, just to name a few, could be combatted with new discovered antibiotics. It should be considered that hundred years ago the market launch of new antibiotics was significantly faster and less complicated than today (where it takes 10–12 years in average between the discovery of a new antibiotic until the launch). After the first euphoria it was quickly realized that bacteria are able to develop, acquire, and spread numerous resistance mechanisms. Whenever a new antibiotic reached the market it did not take long until scientists observed the first resistant germs. Since the marketing of the first antibiotic there is a neck-on-neck race between scientists who discover natural or develop semisynthetic and synthetic bioactive molecules and bacteria, which have developed resistance mechanisms. The emphasis of this chapter is to give an overview of the history of antibiotics research. The situation within the pre-antibiotic era as well as in the early antibiotic era will be described until the Golden Age of Antibiotics will conclude this time travel. The most important antibiotic classes, information about their discovery, activity spectrum, mode of action, resistance mechanisms, and current application will be presented.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Definition of the Term Antibiotic
In 1947, S. A. Waksman defined the term “antibiotic” as follows: “An antibiotic is a chemical substance, produced by micro-organisms, which has the capacity to inhibit the growth of and even to destroy bacteria and other micro-organisms” (Waksman 1947). Today, “antibiotic” has multiple meanings: (I) an organic chemical of natural or synthetic origin that inhibits or kills pathogenic bacteria; (II) any antimicrobial substance, or, (III) in the Waksman tradition, limited to antimicrobial substances of microbial origin. Anyway, all of these definitions coexist in literature and for a detailed discussion of “What is an antibiotic?” I refer to Bentley and Bennett (2003). Within this review the term antibiotic is used as any antimicrobial substance, independent if it is of natural, semisynthetic, or synthetic origin.
2 From the Early Beginning to the Golden Age of Antibiotics Research
2.1 The Pre-antibiotic Era
The discovery and subsequent large-scale production of antibiotics in the early twentieth century was one of the most important achievements in the history of medicine. This new wonder drugs, in addition to comprehensive knowledge about pathogens and improved hygiene measures, took away the fear of many infectious diseases and extremely increased quality and expectancy of life. While in 1900 contagious diseases were the most common cause of death in the USA, in 2000 only a negligible percentage died of infectious diseases (US National Center for Health Statistics). However, nowadays in many countries of the Third World the situation is comparable to 1900, whereas in industrial countries infectious diseases are mainly a problem of immune-suppressed people (HIV or cancer patients) or patients infected with multi-resistant pathogens. In the days when nothing was known about the path of infections, infection prevention, antibiotics, and vaccination, for thousands of years mankind was tortured by huge epidemics like syphilis, smallpox, malaria, typhus, yellow fever, leprosy, tuberculosis, Spanish Influenza, cholera, and plague, to name just a few.
The disastrous situation of people in cases of pandemics during the pre-antibiotic era should briefly be described exemplarily for plague: Plague is caused by the bacterium Yersinia pestis, which was named in honor of its discoverer Alexandre Émile Yersin (1863–1943). Yersin was a Swiss-born bacteriologist and as a member of the French Colonial Health Service he was sent to Hong Kong in 1894 to investigate the outbreak of bubonic plague (Hawgood 2008). He was the first who isolated the causative agent of pestilence from dissected buboes. Overall, Y. pestis was responsible for at least three pandemics in history: the Justinian plague which caused almost 100 million deaths, the “Black Death”—period in the fourteenth century during which tens of millions of people in Europe died (Haensch et al. 2010), and the outbreak between 1895 and 1930 with about 12 million victims, mostly in India (Kool 2005). Plague has not been eradicated yet and without antibiotic therapy the mortality rate is between 50 and 90 % (Lippi and Conti 2002). Furthermore nowadays pest is still a great public health and infection control threat. An aerosol of the agent could be used by terrorists as a biological weapon, which would cause the human transmissible, contagious pneumonic plague (Inglesby et al. 2000), in comparison to bubonic plague which is exclusively transmitted by bites of infected rodent fleas.
However, in the Middle Ages, bacteria and viruses as pathogenic agents, prophylactic measures as vaccines or even effective drugs as antibiotics were completely unknown. Bad air, miasms, inconvenient planetary constellations, or an imbalance between the four elements fire, earth, water and air and the four body juices blood, mucus, yellow bile, and black bile (Hippocratic Doctrine of the Four Juices; Hart 2001) have been held responsible of any kind of illnesses. Concerning serious infection diseases, doctors and physicians were unable to provide valuable assistance for suffering patients. In the case of plague infections, for example, they recommended energy sapping bloodlettings, diets with herbs, sometimes mixed with meat of vipers, open north windows, and if everything else fails, praying. Christian clerics and scholars of the occident interpreted such catastrophes as a “sign of God’s wrath” and a “deserved punishment of mankind’s sins.” While the Black Death raged most furiously around the year 1350, the desperation of people was such big that curious phenomena like the “flagellant movements” became very popular: Increasing numbers of followers joined desperate penitent religious groups which swarmed through the land and whose members whipped themselves to bleed. In this way they hoped to avert God’s anger from humankind (Würth 2012). Nonetheless, physicians’ advices for post exposure prophylaxis like wearing masks, avoiding close contact to, or separation of, infected persons were quiet useful from today’s infectious biology point of view. The government of Milano had even ordered walling of pest-infected persons in their houses, but probably these extreme measures were exceptional cases (Bulst 1977).
Thousands of years ago, since at least 1500 before Christ, at least healing effects of mushrooms, beer yeast, and molds are known to be valuable in treatment of infected wounds. These microorganisms were gladly used for medical applications, even if the practitioners were not able to explain the phenomenon of the effect (Duckett 1999). It was reported about ancient physicians in Egypt, Persian, and Greek, for example, that they treated patients with compresses and tonics made from herbs, molds, and organic compounds for more than 1000 years ago. Also bioactive compounds of plants were medically used as seen in the example of the first systematic chemotherapy, reported in the seventeenth century: Indigenous people of South America used a powder made of the cinchona tree bark (Cinchona pubescens (a, Fig. 1) or C. officinalis) against fever and later on for prevention and treatment of malaria, which has been introduced to South America by Spanish conquistadors. Later on (in 1820), the most active agent quinine, an alkaloid with antipyretic activity, was isolated by the French chemists Pierre-Joseph Pelletier and Joseph-Bienaimé Caventou (Rezende 2006) (1, Fig. 1) and synthesized by the American chemists Woodward and Doering in 1945. As reported by Jäger (2011), today quinine and other antimalaria agents in the bark’s extract of Cinchona spp. are still of medical importance, because the evolution of Plasmodium falciparum strains which are resistant to synthetic drugs (mainly chloroquine) led to rise in the malaria incidence in Asia and Africa.

Chemical structure of quinine 1 and photograph of Cinchona pubescens (a) (photo kindly provided by Dr. Heinke Jäger)
However, without the discovery and description of bacteria (and later on also viruses, both are agents of many infectious diseases), the successful story of antibiotics could not have been written. Although it has long been assumed that creatures exist which are indistinguishable with the naked eye, the first important milestone in the history of microbiology was set by Antonie van Leeuwenhoek (1632–1723), a tradesman and “amateur” scientist. Using his handcrafted microscopes he could visualize microstructures of various objects. Without any university education, he was the first who observed, described, and drew bacteria, sperms, and erythrocytes. His illustrations impressed with precision and attention to detail and gave a hitherto unknown insight to micro objects.
Also in the seventeenth century, the healing effects of molds were described by John Parkington, an English apothecary. He recommended in his book Theatrum Botanicum (Zimdahl 2015) to use these microorganisms for treatment of infections. Then, for about 200 years, there were no major advances with regard to “infection research.” However, in the nineteenth century, more and more scientists and physicians became interested in molds with antibacterial activity: Theodor Billroth (1829–1894), a German-Austrian surgeon, performed experiments with bacterial cultures and moulds to research their role in “accidental wound diseases.” He described, that occasionally when Penicillium grew in a culture, bacteria failed to grow. He observed the antibiotic activity of the fungi, but he could not draw the right conclusions, because he suggested that Penicillium could have “modified the medium” so as to render it unsuitable for bacterial growth (Majno and Joris 1979). In addition, Billroth searched for the causes of sepsis. Therefore he investigated microbial blood and pus preparations and described “Coccobacteria septica,” later called staphylococci and streptococci (Klein et al. 2012). In 1870 Sir John Scott Burdon-Sanderson (1828–1905), an English physiologist, also mentioned that culture fluid covered with mould inhibits bacterial growth (Williams 2013). At this time bacteria were already well known and described as contaminants in milk, wine, and silkworms by Louis Pasteur, as well as in blood by Billroth, but their role as agents of human diseases had not yet been proven. In 1875, another English physician, John Tyndall (1820–1893), conducted contamination experiments with air-exposed tubes of broth to investigate contamination by bacteria from the air. He also confirmed the aforementioned effect that in tubes with Penicillium growth bacteria could not be observed. Tyndall concluded that there was a battle between the bacteria and the mould, and “in every case where the mould was thick and coherent, the bacteria died or became dormant and fell to the bottom as a sediment” (Tyndall 1877). He observed that Penicillium was able to inhibit the growth of bacteria falling into the tubes from the air. However, as mentioned above in Tyndall’s times, the importance of bacteria as human pathogens was still not proven and therefore he also failed to assess the dimension of this observation (Friedman 1998). A few years later Joseph Lister (1827–1912), an English surgeon, also suspected bacteria to be responsible for infections. He was pioneering the development of antiseptic surgery by using chemical antiseptics, such as phenol, to kill bacteria on operating equipment and on wounds (De la Bédoyère 2005). He also cultivated a mould, described as Penicillium glaucum, together with bacteria in urine and observed that the bacteria did not grow in presence of the mold (Rubin 2007). In contrast to his predecessors, he postulated an antimicrobial activity and proved this theory in 1884, when he cured an abscess of a nurse with P. glaucum soaked tissues. Unfortunately Lister did not publish his results and was not able to produce sufficient amounts of extract or even isolate the active substance, later known as penicillin.
In the second part of the nineteenth century two main questions tasked the scientists: “Does spontaneous generation (abiogenesis) exist?” and “What is the nature of infectious diseases?” (Madigan et al. 2000). According to the theory of abiogenesis, bacteria, which could be observed under the microscope in numerous amounts from samples of rotten, but not from fresh food, develop spontaneously from dead material—but exclusively after contact with fresh air. Therefore the supporters of abiogenesis agreed that air is necessary for microbial development, but they could not explain the underlying cause. The myth was definitely refuted by the French chemist Louis Pasteur (1822–1895), who performed experiments to prove that without contamination microbial growth is not possible. He showed that in a sterile broth microbes could not develop spontaneously, but only after air contact. And he further detected that the same microbes, which grow in large amounts in air-exposed broth, can be found in small amounts in the air. Preliminary work had been carried out by the Italian priest Lazzaro Spallanzani (1729–1799) who already proved in the middle of the eighteenth century that microbial growth did not develop in boiled and afterwards hermetically sealed broth (Klein et al. 2012). As a consequence of the refuted germ theory numerous efficient sterilization techniques have been developed (Madigan et al. 2000). Furthermore, Pasteur discovered the principles of vaccination (anthrax, fowl cholera, rabies), lactic acid and alcoholic fermentation, and pasteurization.
The challenge to elucidate the “nature of infectious diseases” was met by Robert Koch (1843–1910). Long before Koch’s time, priest physicians in ancient India and China already presumed that pathogens could be transmitted between living beings. They tried to prevent smallpox infections by immunization of healthy persons with smallpox material from ill persons (immunization by variolation; Klein et al. 2012). In the sixteenth century, people also assumed that “something” could be transmitted from ill to healthy persons which therefore came down with the same illness. Consequently some diseases were classified as contagious, because they are able to spread within a population. Ignaz Semmelweis (1818–1865), an Austrian-Hungarian gynecologist found out that puerperal fever could be transmitted from deceased women in childbed to healthy pregnant woman. As the transmission vehicle he identified unwashed hands of physicians who carried out vaginal examination of birthing mothers after they autopsied women who died on puerperal fever. Thereupon Semmelweis ordered a consequently disinfection of hands with chlorine water before vaginal examinations and thus secured a place in history as a pioneer of infection prophylaxis and later on called “Mother’s Savior” (Klein et al. 2012). Joseph Lister, who also provided some evidence that bacteria could be responsible for infections, successfully introduced antiseptic measures in surgery. After a comprehensive literature research, Jakob Henle (1809–1885) came to the conclusion that during infection a specific infection material is transferred from ill to healthy persons (Klein et al. 2012). Koch was the first who successfully proved this theory and the correlation between a causative agent (Bacillus anthracis) and a disease (anthrax; Madigan et al. 2000). Later, in 1877, his colleague Pasteur observed that cultures of the anthrax bacilli, when contaminated with molds, became inhibited. Further great achievements of Koch included the development of solid media for bacterial cultivation and separation, the implementation of microphotography and, in 1876 the discovery of B. anthracis spores. Hereby he could describe the previously incompletely explained infection chain of anthrax. Using this example, he postulated four criteria to establish a causative relationship between a specific microbe and a disease (Henle-Koch postulates), which are still valid. Koch also identified the specific causative agents of cholera (Vibrio cholerae) and tuberculosis (Mycobacterium tuberculosis). When he started his work on tuberculosis in 1881, consumption was responsible for one-seventh of the reported deaths (Madigan et al. 2000). He succeeded in both, staining M. tuberculosis in tissue and cultivating the demanding germ, but staining and cultivating was challenging: The mycobacterial surface is rich in lipids (mycolic acid), acid resistant and therefore not accessible to traditional staining techniques. Koch invented a dye (alkaline methylene blue and Bismarck-brown) which served as a precursor of the Ziehl–Neelsen dye, a standard dye in many microbiological laboratories today. For the isolation and enrichment of mycobacteria, Koch used coagulated blood serum and thus created the basis for realistic experiments of antibiotics evaluation in vitro where the test conditions should mimic the situation in vivo as much as possible. Next, he infected guinea pigs with pure cultures of M. tuberculosis and proved that his formerly established four postulates could be fulfilled. As appreciation of his work about tuberculosis he was awarded the 1905 Novel Prize.
Now, where bacteria as the possible agents of diseases were identified, the discovery and isolation of the first antibiotic was just a blink away. About 30 years before Alexander Fleming falsely passed into history as the discoverer of penicillin (see also Karwehl and Stadler 2016), Ernest Duchesne (1874–1912), a French medical officer, noticed the antibacterial activity of molds (Duckett 1999), as so many before him. However, he went one step further and tried to identify the specific cause of the inhibition. During his work in a military hospital he observed Arabian hostlers, who stored their saddles in a dark, moisture chamber to support mould growth. They explained that saddles treated by this method supported wound healing of saddle sores. As mentioned above, the wound healing effect of molds was not new. Duchesne started a couple of precise experiments. He infected guinea pigs with Escherichia coli (formerly named “Bacillus coli communis”) or Salmonella typhi (formerly named “Eberth’s bacilli”), the latter of which is the agent of typhoid fever, and treated them with a suspension of the isolated mould. To his surprise all medicated animals survived. He also investigated interactions between bacteria and antibiotic producing fungi and was the first who discussed a therapeutic application of these antibiotic producing microorganisms. Duchesne described his results in the thesis “Contribution to the study of vital competition between microorganisms: antagonism between moulds and microbes” (Duchesne 1897). His thesis was unfortunately not accepted by the Institute Pasteur. Later on, in 1940, his work was confirmed by Chain, Florey, and coworkers, who identified the antibiotic produced by a Penicillium sp. and its effect against Gram-positive and Gram-negative bacteria (Chain et al. 1940; see chapter “Beta lactam antibiotics : penicillin, ampicillin, methicillin” elsewhere in this review). Last but not least Ernest Duchesne was honored posthumously by the Académie nationale de Médecine in 1949, four years after Fleming and colleagues had received the Nobel Prize for the rediscovery of the antibiotic effect of penicillin.
2.2 The Early Antibiotic Era
Anyway, the first antibiotic was described by the Italian medical scientist Bartolomeo Gosio (1863–1944). In 1893 Gosio explored the question why many people of Southern Europe and Southern USA, who lived in impoverished conditions, came down with pellagra. The diet among this social class mainly consisted of corn, which was suspected to be fungi contaminated. It was assumed that the consumption of this contaminated corn causes pellagra (Sydenstricker 1958; Bentley 2000; Nord 2010). The clinical symptoms of pellagra include photosensitive dermatitis, diarrhea, dementia, and death. Gosio isolated and cultivated Penicillium brevi-compactum from the corn and obtained a crystalline product within a filtrate. He found out that this substance showed antibiotic activity against Bacillus anthracis, the causative agent of anthrax. Therefore Gosio was the first one ever described a natural antibiotic (Gosio 1893; Gosio 1896). Gosio’s discovery and therefore a possible use for medical application was forgotten until two American scientists, Carl Alsberg and Otis Fisher Black resynthesized the substance in 1912 and named it mycophenolic acid (Bentley 2000; for more information and structure of mycophenolic acid see Fig. 1 in Karwehl and Stadler 2016). However, in 1937 the cause of pellagra was identified as a nicotinic acid deficiency by Conrad Elvehjem (Burris et al. 1990).
2.3 Salvarsan
The activity of the first synthetic antibiotic , the arsenic derivative arsphenamine, (Dioxy-diamino-arsenobenzol-dihydrochloride; Ehrlich and Bertheim 1912), trade name Salvarsan, was discovered by Paul Ehrlich (1854–1915), a German physician in 1909 in cooperation with the chemist Alfred Bertheim (1879–1914; synthesis) and the bacteriologist Sahachiro Hata (1873–1938; biological testing). Syphilis is a chronic, venereal lethal disease caused by the spirochaete Treponema pallidum, which was discovered by Fritz Schaudinn (1871–1906) and Erich Hoffmann (1868–1959) in 1905. At Ehrlich’s time syphilis was a rampant, worldwide affliction. It is assumed that T. pallidum was introduced to Spain in 1493 by returnees of Columbus’ discovery trip. They imported the pathogen from Haiti to Spain from where syphilis was later on spread rapidly fast by mercenaries from Napoli throughout Europe (Berger et al. 2012). Initially, this disease was treated with guaiac-bark, a cure adopted from the Indios. Later on, till the end of the nineteenth century, syphilis was usually treated with inorganic mercury salts, which has often resulted in fatal heavy metal poisoning with a simultaneous low efficacy. Around the year 1900 Ehrlich searched for dyes with specific affinity to bacteria but not to human or animal tissue. He argued that certain chemoreceptors on parasites, microorganisms, and cancer cells would be different from analogous structures in host tissues (Drews 2000). The idea was to find toxic substances, like arsenic, with these discriminatory properties for medical purposes against infectious diseases (“magic bullet”; Madigan et al. 2000). He tested a large number of aniline dyes and found out that methylene blue stains parasitic protozoa like plasmodia. Afterwards he administered methylene blue to two patients with milder symptoms of malaria and noted that the fever attacks disappeared after a few days of methylene blue administration and at the latest after eight days Plasmodium falciparum could not be detected in the patient’s blood anymore (Guttmann and Ehrlich 1891). Even if this experiment had no statistical significance, herewith Ehrlich can be given credit of having laid the foundation stone of modern chemotherapy. He was the first one to report that a synthetic drug could be used successfully to treat a specific disease, although the dye showed much less effect than the traditional used quinine.
In connection with his research about syphilis, which was still incurable at his time, Ehrlich took special note of a study published by Thomas (1905), who described aminophenyl arsenic acid, commercial name atoxyl, to be effective in the treatment of sleeping sickness. This disease is caused by the parasitic protozoan Trypanosoma brucei and was the greatest cause of death in Africa at that time. Atoxyl has been synthesized and published by Pierre Béchamp (1816–1908) in 1863 (Béchamp 1863). Ehrlich started an extensive screen of several hundred organoarsenic derivatives of atoxyl which were tested by his colleague Hata in vivo in syphilis-infected rabbits (Ehrlich and Hata 1910; 2, Fig. 2).

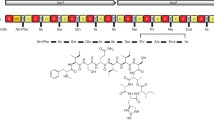
Atoxyl; sodium;(4-aminophenyl)-hydroxyarsinate 2
In 1909 compound no. 606 turned out to be the big hit: the arsenic derivative arsphenamine was also called “Ehrlich 606” and later on sold as Salvarsan (Fig. 3) (Ehrlich 1910). Salvarsan consists of about 30 % arsenic, showed “parasitotropic” properties but lacked “organotropic” properties (Bosch and Rosich 2008). With Salvarsan the scientists pioneered in 1910 the field of targeted therapies, the breakthrough treatment for Treponema, other spirochetes and also trypanosomiasis. This new drug selectively binds the pathogens and the arsenate kills them—the perfect “magic bullet.” Salvarsan is generally viewed as the first modern chemotherapeutic agent because it was discovered as a result of a rational screening program (Bennett 2015). From November 1910, the pharmaceutical company Hoechst (Frankfurt) was producing 12,000–14,000 ampoules Salvarsan per day for clinical trials and therapy worldwide. By treatment with Salvarsan two-thirds of syphilis-infected people could be cured up to 1928, although it had serious side effects like hypersensitivity problems due to arsenic poisoning.

Information sheet of Salvarsan (kindly provided by Prof. Dr. Christoph Friedrich, Bildarchiv des Instituts für Geschichte der Pharmazie der Universität Marburg)
Ehrlich continued to evaluate further synthesized compounds and in 1914 an enhanced version of Salvarsan, neoarsphenamine, marked as Neosalvarsan, was developed (4, Fig. 4; Williams 2009). Increased solubility and less toxicity due to a lower arsenical content (19 %) were the main advantages of Neosalvarsan. Unfortunately this improved product was also associated with side effects like nausea and vomiting. A further problem of both compounds was the need to store them in sealed vials under nitrogen atmosphere to prevent oxidation (Bosch and Rosich 2008). However, Salvarsan/Neosalvarsan was the drug of choice until penicillin became available in 1940. Ehrlich assigned the structure of Salvarsan (3, Fig. 4) with an As = As double bond. In 2005, an extensive mass spectral analysis by Lloyd and coworkers identified the structure of Salvarsan to have As–As single bonds and is a mixture consisting of cyclo-As3 and cyclo-As5 species (5 and 6, Fig. 4; Lloyd et al. 2005). Neosalvarsan also had an overall narrow spectrum activity (Aminov 2010).

Salvarsan, arsphenamine; structure as assumed by Ehrlich with an As-As double bond 3; Neosalvarsan 4, 5 and 6 mixture of the trimer and pentamer of Salvarsan as elucidated by Lloyd et al. (2005)
Already in 1908 Paul Ehrlich was awarded the Nobel Prize in Physiology or Medicine together with Ilya Ilyich Mechnikov “in recognition of their work on immunity.” The mode of action of Salvarsan is still unknown.
3 The Golden Age of Antibiotic Discovery: From Sulfa Drugs to Quinolones
3.1 Sulfa Drugs: Prontosil
The discovery of antibiotic activity of the synthetic sulfa drug class by Paul Gerhard Domagk (1895–1964) in the 1930th marked a further milestone in the history of antibiotics . Domagk, a German physician, already practiced in treating war wounds, was tasked with the identification of antibacterial azo dyes. These dyes were tested in large scale in vitro and in vivo (mice, rats, rabbits, guinea pigs) in his institute at Bayer. At Domagk’s time Bayer was a part of IG Farben, a huge German chemical trust of six companies: BASF, Bayer, Hoechst, Agfa, Cassella, and Kalle founded in winter 1925/26 (Spoerer and Streb 2013). The screening program was inspired by Ehrlich’s success to combat syphilis, as described above. After World War I, research and development concerning antibiotics have been intensified substantially as a result of the horrendous battle injuries suffered by the armies of the war, which were frequently magnified by subsequent bacterial infection of the wounds. However, also poorly nourished civilian populations were highly susceptible to bacterial infections and birthing women were endangered acutely to die from childbed fever (Bentley 2009). Initial comprehensive screening efforts of hundreds of dyes, synthesized by chemists at the Hoechst Company failed to produce a useful antibacterial agent (Fig. 5).

Examples of some azo dyes (from left to right): Sudan IV (staining of lipids), Evans Blue (dye for immunofluorescence), methyl red (pH-indicator), Sudan black (staining of lipids), Sudan green, and basic aniline dye methylene blue (stains parasitic protozoa like plasmodia)
Probably the main problem of these studies was the lack of reliable tests for antibacterial activity (Rubin 2007). Domagk developed an ingenious method to test the survival rate of mice previously infected with a highly virulent strain of hemolytic Streptococcus. He confirmed the reliability of his test system by using known antibacterial substances and those without activity. On the basis of preliminary studies a large number of sulfonamide-containing dyes were synthesized by the chemists Fritz Mietzsch (1896–1956) and Josef Klarer (1898–1953) at Bayer and subsequently tested by Domagk. In 1932 a red colored dye turned out to be highly effective in protecting mice from a lethal dose of Streptococci. For this substance, sulfamidochrysoidine, Domagk proclaimed a 100 % success rate, as assessed by the mouse protection assay, when administered prior to a challenge with the potentially lethal microorganisms (Rubin 2007). Interestingly, this dye, trade name Prontosil (7, Fig. 6), was highly active in infected mice but nearly ineffective in vitro (Domagk 1935). Later it was found out by Tréfouël and colleagues that sulfamidochrysoidine is a precursor and is degraded by enzymes within the body to the active drug sulfanilamide (8, Fig. 6; Tréfouël 1935). Sulfanilamide was already described by the Austrian chemist Paul Gelmo (1879–1961) in 1908 (patent registration by Heinrich Hörlein, Bayer AG, in 1909). He had synthesized and characterized the substance for his doctoral dissertation, but unfortunately the potent therapeutic properties of sulfanilamide went unrecognized until the above-mentioned rediscovery.

Prodrug Prontosil; 4-[(2,4-diaminophenyl)diazenyl] benzenesulfonamide) 7; Active form sulfanilamide (4-aminobenzenesulfonamide) 8
Prontosil was marketed from 1935 on, and immediately deaths as a result of pneumonia, childbed fever and meningitis decreased drastically. With Prontosil an effective drug against Gram-positive cocci (mainly Streptococcus) was discovered and Domagk awarded the 1939 Nobel Prize for the discovery of the antibiotic effect of Prontosil.
The target of sulfonamides, and the basis for their selectivity, is the enzyme dihydropteroate synthase (DHPS) in the folic acid pathway. Mammalian cells are not dependent on endogenous synthesis of folic acid and generally lack DHPS (Sköld 2000); therefore sulfonamides have a bacteriostatic effect on many Gram-positive and on some Gram-negative bacteria, too. As a result of the enormously profitable marketing of Prontosil, the search and development of new sulfa drugs boomed in the late 1930 and many thousands of derivatives with sulfanilamide structure have been created in the following years. Today sulfonamides are infrequently used, in part due to widespread resistance (Sköld 2000, which were first observed in 1942 (Lewis 2013). Resistances against sulfanilamide-derivatives are commonly based on pathogen’s capability to use external folic acid sources. Nowadays, sulfa drugs are mainly given in combination with trimethoprim, which inhibits a later folic acid synthesis step (dihydrofolate reductase inhibitor). In combination with trimethoprim sulfa drugs show a bactericide effect.
In 1940, despite the discovery of quinine for malaria, Salvarsan for syphilis, and the sulfa drugs for mainly Gram-positive cocci-infections, most agents of infectious diseases could still not be treated.
3.2 Beta Lactam Antibiotics: Penicillin, Ampicillin, Methicillin
“It was noticed that around a large colony of a contaminating mold the staphylococcus colonies became transparent and were obviously undergoing lysis.” This observation, published by Alexander Fleming (1881–1955) in 1929, is often called the “birth of the antibiotic era” (Fleming 1929). However, Fleming was neither able to produce appreciable amounts nor to elucidate the structure of penicillin and therefore large-scale production and structure elucidation took a further 10 years. Howard Walter Florey (1898–1968) and Ernst Boris Chain (1906–1979) elucidated the structure of penicillin in 1939 (for structure of penicillin G, the first penicillin used in therapy see Karwehl and Stadler 2016). In March 1940 Chain isolated a small quantity of a substance that contained only 1 % of the active compound. Subsequently Norman Heatley (1911–2004) enhanced the purification process and since 1939 large-scale production of penicillin was possible. It is worth mentioning that the antibacterial properties of mold had been known from ancient times, and that many scientists before Fleming had come upon the similar observations regarding the antimicrobial activity of Penicillium, as already described in detail by Duchesne in 1897 (see previous section of this review). The introduction of penicillin in therapy in 1941 was the next big milestone of this successful story (Chain et al. 1940) (c, Fig. 7). In 1945 the Nobel Prize in Physiology or Medicine was awarded Fleming, Chain and Florey “for the discovery of penicillin and its curative effect in various infectious diseases” and at the end of the Second World War penicillin was available for US military and civil population. It was the first antibiotic capable of killing Gram-positive bacteria including the pathogens that caused gonorrhea, syphilis, and puerperal infections.

Penicillium chrysogenum Q176 on agar a; Light microscopic image of a conidiophore of P. chrysogenum b (both pictures are kindly provided by Prof. Dr. Ulrich Kück and Dr. Julia Böhm, Ruhr-University Bochum); Label of Penicillin containing antiseptic powder c (kindly provided by Prof. Dr. Christoph Friedrich, Bildarchiv des Instituts für Geschichte der Pharmazie der Universität Marburg)
A great contribution to the history of antibiotic research was Fleming’s development of a new screening method. He revolutionized the search for bioactive compounds from microorganisms by spreading soil or soil dilutions on agar plates, inoculated with pathogenic bacteria. Subsequently he searched for inhibition zones in lawns of these pathogens. This procedure saved time, money and required much less resources than any testing in animal disease models. As a result, this method became widely used in mass screenings for antibiotic producing microorganisms by many researchers in academia and industry (Aminov 2010). As also mentioned by Karwehl and Stadler (2016), modern taxonomic studies revealed that the name of the fungus from which penicillin was first obtained (“Penicillium notatum” and later “P. chrysogenum” Fig. 7a and b), is actually P. rubens (Houbraken et al. 2011).
Penicillin and derivatives belong to the class of β-lactam antibiotics , which further include cephalosporins, carbapenems, and monobactams. Characteristic for this group is a β-lactam ring within the molecular structure. Most β-lactam antibiotics are potent inhibitors of cell wall biosynthesis. The first β-lactam antibiotic penicillin G was mainly effective against Gram-positive bacteria, because the cell wall of most Gram-negatives is impermeable for this agent. Subsequently developed semisynthetic penicillins (methicillin, oxacillin, ampicillin, carbenicillin) show a broad-spectrum activity and also inhibit several Gram-negative pathogens. Many bacteria, like staphylococci or E. coli for example, are able to produce β-lactamases; enzymes which destroy the β-lactam ring and therefore render the antibiotic ineffective. Today, β-lactam antibiotics are often given in combination with β-lactamase inhibitors (clavulanic acid, sulbactam). As cited by Aminov (2010), Rollo and coworkers investigated possible resistance emergence under laboratory conditions in 1952 and predicted that: “Syphilis has now been treated with arsenicals for about 40 years without any indications of an increased incidence of arsenic-resistant infections, and this work gives grounds for hoping that the widespread use of penicillin will equally not result in an increasing incidence of infections resistant to penicillin” (Rollo et al. 1952). While this is true for Treponema pallidium as reported by Cha et al. (2004), this is not the case for many other pathogenic bacteria, including the Enterobacteriaceae, which have become resistant not only to the original penicillin but also to semisynthetic penicillins, cephalosporins, and newer carbapenemes (Kumarasamy et al. 2010). Fleming was among the first who cautioned about the potential resistance to penicillin and already noted in 1929, “that the growth of E. coli and a number of other bacteria belonging to the coli-typhoid group was not inhibited by penicillin.” He explained this observation that the dosage was too little or too shortly given. A few years later, Abraham and Chain made an extract of E. coli and found a substance that destroyed the growth-inhibiting property of penicillin. They published these results in 1940 (Abraham and Chain 1940) and in 1945 the first resistances against penicillin in therapy were already observed (Lewis 2013). This was the beginning of an armament race with uncertain outcomes between scientists, who develop new antibiotics and microorganism, which already have, develop, or acquire and, still worse, spread diverse resistant mechanisms by conjugation, transformation, and transduction of the encoding genes. Usually resistance develops within two to three years after the introduction of a new antibiotic treatment (Davies 2006). This is nowhere more apparent than in the steady evolution of beta-lactamases by point mutation under the selective pressure of successive introductions of new beta-lactamase-resistant penicillins, cephalosporins, carbapenems and monobactams (Jacoby and Bush 2005). Between 2010 and 2014 two novel cephalosporins (ceftaroline fosamil; ceftobiprole) for treatment of acute bacterial skin/skin structure infections and community-acquired pneumonia as well as a cephalosporin-beta-lactamase inhibitor combination (ceftolozane-tazobactam), reached the market (Hesterkamp 2015).
The semisynthetic penicillin-derivate ampicillin (para-aminobenzyl penicillin), was first described by Brewer and Jonson (1953) and later on patented by the US chemist John Clark Sheehan. Ampicillin is a broad-spectrum antibiotic. It shows the same activity like penicillin but in addition ampicillin acts against a couple of Gram-negative pathogens like Enterococcus faecalis, Escherichia coli, Haemophilus influenza, and Proteus mirabilis. Ampicillin is still used in therapy.
Methicillin (celbenin), also a semisynthetic penicillin-derivate, was developed by Beecham, a British pharmaceutical company in 1959 (later SmithKlineBeecham [SKB] and now Glaxo). It has a narrow spectrum activity but was, together with oxacillin, the first beta-lactamase-resistant substance of this antibiotic class and therefore it seemed to be a very promising drug in the early 1960. Methicillin was used in therapy against beta-lactamase-producing pathogens such as Staphylococcus aureus that would otherwise be resistant to most penicillins. However, already in 1961 first resistances against methicillin were reported from staphylococci (Barber 1961) and today methicillin is no longer in clinical use. The resistance of methicillin-resistant S. a ureus strains (MRSA) is based on a modified penicillin-binding protein (PBP2a). Because in the past resistance tests have been conducted with methicillin as lead-antibiotic, the designation “methicillin-resistant” was deduced for strains which show acquired resistance against all beta-lactam-antibiotics (penicillins, cephalosporins, carbapenems). Later on oxacillin was used as test antibiotic and led to oxacillin-resistant S. a ureus strains (ORSA).
In 1945, an Italian physician, Guiseppe Brotzu, isolated the ascomycete Acremonium chrysogenum from Sardinian coastal seawater. Brotzu detected an antibiotic effects of extracts generated from this fungus and later, in 1955, the structure of the active compound, cephalosporin C, was elucidated (Newton and Abraham 1955). For more information of the history of cephalosporins see Bloemendal and Kück (2014) and Karwehl and Stadler (2016).
3.3 The Discovery of Soil Inhabiting Bacteria as Reliable Sources for New Antibiotics
The discovery and successful marketing of penicillin as well as World War II, which suggested the need for new agents to control infectious diseases and epidemics certain to arise, has led to intensive search for new synthetic and natural antibiotics around the 1940. One main focus was set on the isolation and screen of microorganisms from the environment for antimicrobial activity. Many drug companies established departments of microbiology and fermentation units, and there were only a few large Pharma companies that did not participate in the search for new antibiotics (Drews 2000).
3.4 Tyrothricin/Gramicidin
In 1939, the American microbiologist René Dubos (1901–1982) isolated tyrothricin, a mixture of linear and cyclic polypeptide antibiotics, from the soil bacterium Bacillus brevis. He found out that tyrothricin decomposed the capsule of Pneumococcus bacteria, which were the major cause of pneumonia since the late nineteenth century down to the present day. The main constituent of tyrothricin is alkaline tyrocidine, in addition to lipophilic gramicidins A, B, and C. Tyrothricin was the first commercially available antibiotic (Dubos and Hotchkiss 1942; Dubos et al. 1942), but the current application is limited to skin infections and infections of mouth and pharynx.
Also from B. brevis Rollin D. Hotchkiss and Dubos isolated gramicidin D (Hotchkiss and Dubos 1940 a, b), a heterogeneous mixture of antibiotic compounds (gramicidin A, B, and C) which are synthesized via the nonribosomal pathway by nonribosomal peptide-synthetases. Gramicidin D (D from Dubos) is a membrane channel forming linear pentadecapeptide antibiotic, which is active against Gram-positive bacteria. In 1944 Georgyi F. Gause and Mariya G. Brazhnikova detected the antibacterial gramicidin S (Soviet Gramicidin) also in supernatants of B. brevis. This cyclic decapeptide was produced by a strain from Russian soil (Gause and Brazhnikova 1944).
In the early 1940 the biochemist Selman Abraham Waksman (1888–1973), along with Albert Schatz (1922–2005) and Elizabeth Bugie (1920–2001) also searched for soil borne microorganisms that, among other test germs, would kill or inhibit the growth of penicillin-resistant bacteria. The main focus was set on drugs to combat M. tuberculosis, the causative agent of tuberculosis. M. tuberculosis is insensitive against Salvarsan, Prontosil and penicillins and was responsible for the death of every fourth adult in Europe and USA around 1900. Waksman and colleagues made the pioneering discovery that in particular bacteria of the genus Streptomyces (group actinobacteria) are highly promising candidates for the production of new antibiotics . He and his students screened soil bacteria for bioactivity and searched for growth inhibition zones surrounding single colonies of isolated soil microbes on agar plates, based on Fleming’s method. Subsequently, they tested the inhibition on specifically targeted pathogenic bacteria.
3.5 Actinomycin
The first promising substance isolated under this initial screening program was found in 1940: Actinomycin, a polypeptide, was also the first antibiotic isolated from an actinobacterium: Streptomyces antibioticus subsp. antibioticus (formerly named Actinomyces antibioticus) (Fig. 8, a–c) (Waksman and Woodruff 1940).

Streptomyces antibioticus subsp. antibioticus agar plates media ISP 5, ISP 2, ISP 3, ISP 4 a; Spore chain morphology and spore surface in SEM C × 7500 b. S. antibioticus subsp. antibioticus agar plates media 5006, 5265, 5315 c. Pictures were kindly provided by PD Dr. Joachim Wink
The substance is active against a broad range of bacteria and even showed promise of attacking a tuberculosis strain, but it proved too toxic for antibacterial therapeutic use in humans (Waksman and Woodruff 1941). Actinomycin D was also the first antibiotic with anticancer activity and is nowadays still in use in antitumor therapy. It binds to DNA duplexes, thereby interfering with the action of enzymes engaged in replication and transcription. Based on its mode of action, actinomycin has also become an important tool in molecular and cell biology (Hollstein 1973).
3.6 Bacitracin
Bacitracin, a broad-spectrum polypeptide antibiotic complex, inhibits mainly Gram-positive pathogens like streptococci and staphylococci and the anaerobic germ Clostridium difficile by preventing the bacterial cell wall synthesis. It was originally isolated from Bacillus subtilis and Bacillus licheniformis and described by Johnson et al. (1945). It inhibits peptidoglycan synthesis in Gram-positive bacteria by binding to a lipid pyrophosphate carrier that transports cell wall precursors to the growing cell wall (Husain 2004). Commercial bacitracin is a mixture of different related polypeptides with the main compound bacitracin B. Due to its systemic toxicity, bacitracin is mainly used local against wound infections, burn injuries and skin grafts. Four major bacitracin resistance mechanisms have been detected until 2012 and are described in detail in Charlebois et al. (2012).
3.7 Aminoglycosides: Streptomycin, Neomycin, Kanamycin, Gentamicin
The next promising hit within the screening program was the first described antibiotic of the aminoglycoside class: Streptomycin. This compound was isolated by Schatz et al. (1944) from Streptomyces anulatus subsp. griseus (previously named S. griseus) (Fig. 9a–d). Streptomycin and many other aminoglycosides are high potent broad-spectrum antibiotics and act primarily by impairing bacterial protein synthesis through binding to the 30S ribosomal subunit. This antibiotic class is still in clinical use, albeit mostly in combination therapy or in topical application because of their insufficient pharmaceutical properties and severe side effects. Streptomycin was also the first anti-tuberculoticum used in therapy (1946) and furthermore it showed activity against several other diseases that could not be cured by penicillin. The history of aminoglycosides thereafter is marked by the successive introduction of a series of milestone compounds (kanamycin, gentamicin, and tobramycin) which definitively established the usefulness of this class of antibiotics for the treatment of Gram-negative bacillary infections (Mingeot-Leclercq et al. 1999; Shaw et al. 1993).

Streptomyces anulatus subsp. griseus agar plates media ISP 1 + 3 a; Spore chains and spore surface (SEM × 7500) b Streptomycin ampoule c. This image was kindly provided by Prof. Dr. Christoph Friedrich, Bildarchiv des Instituts für Geschichte der Pharmazie der Universität Marburg; Streptomyces anulatus subsp. griseus agar plates media ISP 2 + 4 d. Pictures 1, 2, and 4 were kindly provided by PD Dr. Joachim Wink
For the co-discovery of streptomycin Waksman was awarded the Nobel Prize in 1952. Resistances against aminoglycosides are based on alteration of the ribosomal binding sites (streptomycin only), decreased uptake and/or accumulation of the drug in bacteria and the bacterial expression of enzymes which modify the antibiotic and thereby inactivate it (Davies and Wright 1997). Today, out of the various microorganisms screened, species of Streptomyces are still the most important producers of naturally occurring antibiotics, producing approximately two-thirds of all known antibiotics (Baltz 1998; Weber et al. 2003). However, no new drugs have been obtained from these approaches since the mid-1980s and it is even very difficult to find novel carbon skeletons in Streptomyces, suggesting that the genus has been exhaustively explored.
In the 1940, a large number of antibiotics with activities against Gram-negatives, including rickettsiae, and Gram-positives, including mycobacteria, were isolated from various species of the genus Streptomyces. Therefore the focus was set on the actinomycetes as potential producers of antimicrobial agents that might possess promising chemotherapeutic properties. The following characteristics of new antibiotics were particularly important: (1) High activity against Gram-negative bacteria and mycobacteria; (2) antibiotic action against streptomycin-resistant bacteria; (3) low toxicity to animals; (4) other desirable properties, such as activity against rickettsiae, viruses, tumors and phages (fide Waksman et al. 1949 Footnote 1).
Within this scope, neomycin, a new aminoglycoside broad-spectrum antibiotic active against streptomycin-resistant bacteria, including M. tuberculosis was isolated from Streptomyces fradiae and described by Waksman and Lechevalier (1949). Neomycin shows broad-spectrum activity as other antibiotics of the aminoglycoside group. Resistance against neomycin is conferred by aminoglycoside 3′-phosphotransferase genes, which inactivate neomycin by phosphorylation. There is a cross-resistance of neomycin to kanamycin and partially also to gentamicin.
In the 1950s another important broad-spectrum aminoglycoside antibiotic, kanamycin, a mixture of derivatives A, B and C, was isolated from Streptomyces kanamyceticus, a strain cultivated from Japanese soil (Takeuchi et al. 1957). The antibacterial spectrum includes mycobacteria, many Gram-positive and most Gram-negative pathogens. However, due to a couple of serious side effects, today kanamycin is mainly used for local therapy of eye infections (in the form of eye drops) and as a reserve antibiotic. Resistance mechanisms are as described for neomycin.
Gentamicin (genticin), a mixture of similar aminoglycosides, is produced by Micromonospora purpurea and was discovered in 1963 in the laboratories of the Schering Corporation, Bloomington, New Jersey (No authors listed, 1967). Gentamicin has a similar activity spectrum to related antibiotics such as neomycin and kanamycin (exerting both a bacteriostatic and bactericidal effect), but a rather greater activity: Almost all enterobacteria, including species of Aerobacter, Escherichia, Klebsiella, Salmonella, Shigella, Proteus, and also some Pseudomonas aeruginosa-strains are sensitive to gentamicin. Among Gram-positives, staphylococci are the most sensitives. Due to strong side effects (nephrotoxic reactions, ototoxicity) nowadays gentamicin is only used as an emergency antibiotic.
Back to the 1940s, the screening of fermentation broths of actinomycetes yielded a variety of antibiotics that were relatively quickly developed for clinical use. The antibiotic activity was detected, as described above, by agar diffusion assays in which fermentation samples were applied to filter paper discs that were placed on an agar plate inoculated with a bacterial test germ (Silver 2012). After this screening method had been successfully established in many laboratories, one hit followed another.
3.8 Chloramphenicol
A new antibiotic with broad-spectrum activity against various Gram-positive and Gram-negative pathogens, anaerobes, spirochetes, rickettsiae, chlamydiae and mycoplasma, was chloramphenicol (9, Fig. 10), isolated by John Ehrlich (Ehrlich et al. 1947) and colleagues from Streptomyces venezuelae (Ehrlich et al. 1948). The biological activity of chloramphenicol was described by Smith et al. (1947) and Gottlieb et al. (1948). The drug was introduced into clinical practice in 1949 (trade name Chloromycetin). Chloramphenicol also became the first antibiotic accessible by total synthesis (Controulis et al. 1949).

Structure of chloramphenicol 9
Chloramphenicol had been the only antibiotic available that was consistently active against Salmonella species including Salmonella typhi but due to its high toxicity, today the compound is given only in cases of specific indication. Chloramphenicol acts as inhibitor of the protein synthesis in bacteria and binds reversibly to the 50S subunit. Here the transfer of amino acids to growing peptide chains is prevented by suppression of peptidyl transferase activity. Thus the drug inhibits peptide bond formation, ultimately leading to inhibition of protein formation.
3.9 Tetracyclines
Another important antibiotic class discovered from Streptomyces species are the tetracyclines. The first tetracyclines, aureomycin (chlortetracycline), discovered by Duggar (1948) and terramycin (oxytetracycline: 10, Fig. 11), described by Finlay et al. (1950) have been isolated from the fermenter broths of Streptomyces aureofaciens and S. rimosus, respectively. Tetracycline molecules act bacteriostatic by inhibition of protein biosynthesis and comprise a linear fused tetracyclic nucleus to which a variety of functional groups are attached.

Structure of terramycin 10
Commercially applied tetracycline molecules bind to the 30S ribosomal subunit and block the entry of the amino-acyl tRNA into the A site of the ribosome. Therefore they are broad-spectrum antibiotics and show activity against a wide range of Gram-positive and Gram-negative germs including pathogens such as chlamydiae, mycoplasmas, rickettsiae, and protozoan parasites.
The absence of major adverse side effects has led to extensive use of tetracyclines in human and animal infection therapy, but the emergence of microbial resistance has limited their effectiveness (Chopra and Roberts 2001). As referred by Chopra and Roberts (2001), Levy (1984) reported that until the mid-1950, the majority of commensal and pathogenic bacteria was sensitive against tetracyclines. Hughes and Datta (1983) found out that among 433 different strains of Enterobacteriaceae, collected between 1917 and 1954, only 2 % were resistant to tetracyclines. Four different mechanisms of tetracycline resistance have been identified so far: drug inactivation, active efflux, ribosomal protection, and alteration of the target. As the majority of tetracycline resistance genes are associated with mobile plasmids, transposons, conjugative transposons, and integrons, these genes can spread from species to species into a wide range of genera by conjugation (Chopra and Roberts 2001). The first tetracycline-resistant bacterium, Shigella dysenteriae, was isolated in 1953 (Falkow 1975), and just two years later the first multiple-drug resistant Shigella was isolated. Multi-drug resistances which include resistance against tetracyclines has been identified in an increasing number of Gram-negative and Gram-positive species. Goldstein et al. (1994) found out that at the beginning of the 1990s approximately 90 % of methicillin-resistant S. aureus, 70 % of Streptococcus agalactiae, 70 % of multiple-drug resistant Enterococcus faecalis, and 60 % of the multiple-drug resistant Streptococcus pneumoniae strains were also resistant against tetracycline. Until the 1970s, a couple of naturally tetracyclines as well as those from semisynthetic approaches reached the market (Chopra and Roberts 2001). Aureomycin, for example, has been synthesized since 1959. Despite the widespread of tetracycline resistances, this antibiotic class is still of great interest for antibiotic therapy. Today the focus is set on new derivatives which are under examination for potential introduction as clinical agents to circumvent existing tetracycline resistance mechanisms (McMurry and Levy 2000; Tymiak et al. 1993) and on tetracycline efflux pump inhibitors that could be used in conjunction with older tetracyclines to restore their activity (Nelson et al. 1993; Nelson and Levy 1999).
Another broad-spectrum tetracycline, chelocardin, is produced by Amycolatopsis sulphurea (former Nocardia sulphurea) and was first described by Oliver et al. (1962). The structure was elucidated by Mitscher et al. (1970). Chelocardins are regarded as structurally atypical tetracyclines with antibacterial activity (Oliva et al. 1992). The exact mode of action is still unknown. Like other existing natural product scaffolds, chelocardins have not been developed because their suboptimal pharmacological properties could not be addressed at the time. Lešnik et al. (2015) demonstrated that reviving such compounds through the application of biosynthetic engineering can deliver novel drug candidates. They introduced the carboxamido moiety of tetracyclines (an important structural feature for their bioactivity) into the chelocardins, and generated a broad-spectrum antibiotic lead with significantly improved activity, including all Gram-negative pathogens of the ESKAPE panel (Enterococcus faecium, S. aureus, Klebsiella pneumoniae, Acinetobacter baumannii, P. aeruginosa, Enterobacter spp.). As the molecular target site of chelocardin seems to be different from that of the regular tetracyclins, there is some hope that this molecule can soon be turned into a novel broad-spectrum antibiotic with which we can combat the newly arising multi-resistant Gram-negative bacteria.
3.10 Macrolides/Ketolides
The important class of macrolides was discovered by James M. McGuire, who isolated and described the first lead compound erythromycin (Ilotycin) from Streptomyces erythreus. The strain was isolated from a soil sample collected on a small Philippine island in 1952 (McGuire et al. 1952). Macrolides are macrocyclic polyketides that typically consist of a 12–16 chained macrocyclic lactone ring with a glycosidic linked amino sugar. Macrolide antibiotics bind to the 50S subunit of the bacterial ribosome and inhibit the translocase and therefore inhibit the protein synthesis. They act primarily bacteriostatic, mainly against metabolically active bacteria. The pathogen spectrum of macrolides includes Gram-positive cocci (staphylococci, streptococci) and rods, Gram-negative pathogens like Legionella pneumophila, chlamydia, Bordetella pertussis, spirochaetes, Haemophilus influenza, and cell wall lacking mycoplasmas.
Three resistance mechanisms against macrolide antibiotics have been reported: (I) mutation or modification of the target (methylation of 23S rRNA of the 50S subunit); (II) efflux of exclusively 14- and/or 15-membered macrolides by increasing the number of efflux pumps, and (III) enzymatic inactivation by plasmid-coding esterases, as described for staphylococci and streptococci (Höck 2012b). To date erythromycin is still the standard antibiotic against infections of the respiratory tract and of throat-nose-ear sections, against pertussis and for patients with allergy to penicillin.
To remove the acid instability of erythromycin, semisynthetic macrolides of the second generation have been developed (clarithromycin, azithromycin). When resistances against first- and second-generation macrolides appeared, the ketolides, an enhancement of macrolides, have been designed (Chellat et al. 2016). Ketolides are semisynthetic derivatives of the macrolide erythromycin A (Zhanel et al. 2002) and were first mentioned in literature by Griesgraber et al. (1996). The mode of action of ketolides is very similar to erythromycin and ketolides exhibit good activity against Gram-positive and some Gram-negative aerobes. They also have excellent activity against drug resistant S. pneumoniae, including some macrolide-resistant strains (Zhanel et al. 2002). Actually, telithromycin is the only ketolide that has so far made it to the market.
3.11 Glycopeptides
Also in the 1950s the glycopeptides (glycosylated, cyclic or polycyclic, nonribosomal peptides) produced by a diverse group of soil actinomycetes have been discovered. Glycopeptides inhibit a late stage in bacterial cell wall peptidoglycan synthesis of exclusively Gram-positive bacteria. The first described glycopeptide, vancomycin was described by McCormick et al. (1955) from Amycolatopsis orientalis, formerly Streptomyces orientalis. The producer was isolated from a soil sample collected in Borneo during a research program carried out by Eli Lilly. Vancomycin became available for clinical use as an anti-staphylococcal agent after the approval of the U.S. Food and Drug Administration (FDA) in 1958 (Jovetic et al. 2010), although its early use was somewhat limited by side effects.
The second glycopeptide antibiotic in clinical use is teicoplanin, which belongs to the same family as vancomycin. It was isolated from the fermentation broth of Actinoplanes teichomyceticus (Somma et al. 1984). Teicoplanin is a mixture of five components of very similar polarity including teichomycin, published by Parenti et al. (1978). Today, vancomycin and teicoplanin are used to treat serious Gram-positive bacterial infections that are resistant to other antibiotics like β-lactams (Kahne et al. 2005) and methicillin-resistant S. aureus infections. They serve as reserve antibiotics in the treatment of serious infections of oxacillin-resistant staphylococci and ampicillin-resistant enterococci as well as against enterocolitis caused by toxin producing Clostridium difficile.
The frequency of resistance to glycopeptide antibiotics has increased significantly over the past decades and multiple genera, including enterococci and staphylococci, have developed resistance to these drugs (Ferber 2003). Some bacteria show natural, intrinsic resistance against vancomycin and teicoplanin, for example enterococcal species and non-enterococcal organisms like Leuconostoc, Pediococcus, Lactobacillus, and Erysipelothrix (Nelson 1999).
Acquired resistances have manifested itself largely through the expression of genes encoding proteins that reprogram cell wall biosynthesis and, thus, evade the action of vancomycin and teicoplanin (Binda et al. 2014). Reduced sensitivity (only against vancomycin), resistance by overproduction of peptidoglycan, or change of the target by acquisition of resistance genes via horizontal gene transfer are possible resistance mechanisms (Höck 2012a).
Another glycopeptide antibiotic, ristocetin (ristomycin), was first described by Grundy et al. (1956/1957) from Amycolatopsis lurida. It was originally used to treat Gram-positive pathogenic infections in humans, particularly staphylococcal infections, but its use was soon discontinued, however, due to toxic side effects related to its ability to cause thrombocytopenia and platelet agglutination (Weiss et al. 1973).
Actually three new lipoglycopeptides, i.e., telavancin, dalbavancin, and oritavancin have been registered for infections due to S. aureus including MRSA (Hesterkamp 2015).
3.12 Streptogramins
Streptogramins, also discovered from soil inhabiting streptomycetes, were first mentioned in literature in 1953 by Charney et al. Streptogramins are natural cyclic peptides of different unique classes. Each member of the class is a combination of at least three structurally unrelated molecules (group A and group B). Group A streptogramins are polyunsaturated macrolactones, group B streptogramins are cyclic hexadepsipeptides (Khosla et al. 1999). Group A and group B streptogramins alone act bacteriostatic, together the effect is bactericidal. Both groups bind to bacterial ribosomes and inhibit the translation of messenger RNA at the elongation step (DiGiambattista et al. 1989). Their combination generates bactericidal activities and reduces the possibility of emergence of resistant strains. However, resistances against streptogramins have been described: The substances can be conferred by modification of the drug target (methylation of the 23S ribosomal RNA), resulting in resistance to group B but not group A streptogramins or by active efflux, which has been described in Staphylococcus epidermidis, affecting inter alia group B streptogramins (DiGiambattista et al. 1989). Cross-resistance between streptogramins, macrolides and lincosamides is common. A combination product of later developed derivatives showed strong activity against streptococci, staphylococci and pneumococci. Due to several side effects of streptogramins and better investigated monopreparations like linezolid and daptomycin, today streptogramins are no longer relevant in antibiotic therapy.
3.13 Lincosamide
The antibiotic class of lincosamides, including the natural product lincomycin (11, Fig. 12) and the semisynthetic analogue clindamycin (12, Fig. 12), was first characterized in the 1960s and is now used for treatment of a broad spectrum of infections. Lincosamides act bacteriostatically by binding to the 23S rRNA of the 50S subunit. They mimic the intermediate formed in the initial phase of the elongation cycle, thereby inhibiting the protein synthesis.

Structures of lincomycin 11 and clindamycin 12
Lincomycin is produced by Streptomyces lincolnensis. The producer was originally isolated from a soil sample collected in Lincoln, Nebraska (Mason et al. 1962). Also already in 1962 it was found that the in vitro activity of lincomycin against Gram-positives was very similar to the macrolide erythromycin (Lewis et al. 1962). Macrolides and lincosamides are chemically distinct but share a similar mode of action: inhibition is limited to Gram-positive cocci (mainly staphylococci and streptococci) and bacilli, to Gram-negative cocci, and intracellular bacteria (chlamydia and rickettsia species). Gram-negative bacilli are generally resistant, with some exceptions (Leclercq 2002).
The later developed semisynthetic clindamycin is four to eight times more active than lincomycin against most Gram-positive organisms (Meyers et al. 1969), and also acts against anaerobic germs and toxoplasmas. Today, lincosamides make up an important class of antibiotics used against a wide range of pathogens, including MRSA (Morar et al. 2009).
3.14 Ansamycins—Rifamycin, Rifampicin
Rifamycins, substances of the ansamycin family, are a group of antibiotics which are naturally produced or chemically synthesized. The first rifamycin (rifomycin; 13, Fig. 13) was discovered and isolated in Italy in 1957 from Streptomyces mediterranei (Sensi et al. 1960) which has been renamed to Nocardia mediterranei, later to Amycolatopsis mediterranei, and finally to Amycolatopsis rifamycinica. The five rifamycin components are designated A, B, C, D, and E, whereby rifamycins O, S, and SV are derivatives of the inactive B component, and AG and X are derivatives of the O component. Rifamycin B, which chemical structure has been elucidated by Oppolzer et al. (1964), is the precursor of clinically used antibiotics that are effective against M. tuberculosis, the pathogenic agent of tuberculosis and M. leprae, the agent of leprosy. Greater potency was achieved through substitutions as exemplified by the semisynthetic derivatives rifamide, rifaximin, rifapentine, rifampicin (rifampin; 14, Fig. 13), and rifabutin (Carter 2009).

Structures of rifamycin B 13 and rifampicin 14
Rifampicin inhibits the RNA synthesis and acts bactericide on extra- and intracellular bacteria. In addition to mycobacteria, also staphylococci (including MRSA-strains), streptococci (including penicillin-resistant pneumococci), enterococci, Neisseria, Legionella, and other bacteria can be combated with rifampicin (Bange and Fille 2012). Due to resistance development caused by mutations of the RNA-polymerase gene (rpoB), normally a combination of different anti-tuberculosis drugs is given in combination: within the first two months: isoniazid, rifampicin, pyrazinamide and ethambutol; Up to the third month (stabilization phase) for at least four months: rifampicin and isoniazid (Bange et al. 2012). If a patient is infected with multi-drug resistant Mycobacteria (MDR-TB), the WHO recommends second-line or reserve drugs like kanamycin or linezolid, for example (WHO 2009).
3.15 (Fluoro) Quinolones: Nalidixic Acid, Ciprofloxacin
The synthetic quinolone antibiotics are arguably one of the most important classes of anti-infective agents. Their mode of action was new at the time of their discovery. They act bactericidal by inhibiting the bacterial enzyme DNA gyrase (topoisomerase II). DNA gyrase catalyses changes in the topology of DNA and can interconvert relaxed and supercoiled forms, introduce and remove catenanes and knots. Therefore these enzymes are essential in all bacteria to survival, but absent in higher eukaryotes, making them an attractive target for antibiotics (Collin et al. 2011).
When gyrase was discovered in E. coli in 1976 (Gellert et al. 1976), the two most important classes of gyrase inhibitors had already been described.
The first clinical quinolone, nalidixic acid (15, Fig. 14), was discovered accidentally as a by-product of the synthesis of the important antimalarial drug chloroquine. Nalidixic acid was patented by the Sterling Drug Company in 1963 (Nielsen et al.). Already in 1962 the antibacterial properties of nalidixic acid were described (Lesher et al. 1962; Bisacchi 2015). In 1964 the first clinical reports and launch by Sterling as a first-in-class antibacterial agent followed. Based on nalidixic acid the fluoroquinolones were developed. Within the search for more active derivatives of nalidixic acid, the chemist K. Grohe (Bayer Company) found a way to produce highly effective antibiotics (cycloaracylation):

Structure of nalidixic acid 15 and ciprofloxacin 16
The top product was the broad-spectrum fluoroquinolone ciprofloxacin (16, Fig. 14) (Grohe and Heitzer 1987). In comparison to nalidixic acid ciprofloxacin showed an enhanced activity and antibacterial spectrum. Out of more than 20.000 tested bacterial strains, 98.2 % were sensitive against this new drug. The substance was patented in 1981 (Grohe et al. 1981) and introduced to the market as ciprobay in 1987 (Neufeldt 2003). After their discovery, fluoroquinolones became one of the principal clinical weapons in the fight against bacterial infections. Up to now more than 10,000 quinolones have been synthesized.
At the beginning resistances against quinolones developed slowly, in comparison to other antibiotic classes, because quinolones act on two different targets, i.e., DNA gyrase and topoisomerase IV. However, their usefulness is now endangered by the rapid emergence of resistance which typically arises as a result of alterations in these target enzymes and of changes in drug entry and efflux (Jacoby 2005).
The only other class of gyrase inhibitors which has been introduced into clinical use are the aminocoumarins, with the most prominent drug novobiocin, produced by Streptomyces spheroides (later reclassified to Streptomyces niveus), and discovered in the 1950 (Heide 2014; Harris et al. 1955). Within the last two years, two promising DNA gyrase inhibitors reached the medical pipeline: Baumann et al. (2014) discovered three cystobactamids, which have been isolated from the soil borne myxobacterium Cystobacter sp.. Cystobactamids were highly active against various Gram-positive pathogens and one cystobactamid, designated 919-2, also efficiently inhibits the growth of E. coli and A. baumannii, confirming that the compounds can penetrate the outer membrane of Gram-negative bacteria (Baumann et al. 2014).
Albicidin, a peptide antibiotic with remarkable antibacterial activity against various Gram-positive and Gram-negative microorganisms, is naturally produced by the sugarcane pathogenic bacterium Xanthomonas albilineans and can also be produced through heterologous expression. However, the yield of albicidin by natural production as well as by heterologous expression was too low for providing sufficient material for bioactivity profiling and structure–activity studies. With the total synthesis of albicidin Kretz et al. (2015) succeeded in sufficient production of this promising substance, which is already known since the 1980s (Birch and Patil 1985). As the potential of quinolones is largely exhausted as a template for new type II topoisomerase inhibitors, cystobactamids and albicidin offer promising alternatives to generate novel antibiotics using medicinal chemistry and biosynthetic engineering.
3.16 Oxazolidinones
A further clinical important class of antibiotics was detected after the “Golden Age of Antibiotics ” and should also be mentioned within this chapter: The oxazolidinones, a new class of synthetic compounds against Gram-positives, were first described in 1978 in a patent (DuPont), primary as antibiotics against plant pathogens. In 1987 oxazolidinones were introduced to clinical tests in human medicine. The first commercial substance of this class, linezolid (17, Fig. 15), was introduced to the US market in 2000 (Stahlmann and Riecke 2004). As the name suggests, the most important structure element of oxazolidinones is the five member heterocyclic oxazolidinone ring with an acetamidomethyl group. Linezolid, like vancomycin, is a reserve antibiotic. While most of the widely known antibiotics (macrolides, chloramphenicol) inhibit bacterial protein synthesis at the peptide chain elongation stage, linezolid acts earlier by potent interaction with the 50S ribosomal subunit and therefore hinders the protein biosynthesis at the first step. Linezolid is active against all Gram-positives, aerobic as well as anaerobic growing germs and also against methicillin resistant S. aureus vancomycin resistant enterococci and acts mainly bacteriostatic. Gram-negatives, with the exception of Pasteurella, are resistant. Resistance to other protein synthesis inhibitors does not affect oxazolidinone activity; however rare development of oxazolidinone resistance cases, associated with 23S rRNA alterations during treatment, have been reported (Pandit et al. 2012).

Structure of linezolid 17
A second-generation oxazolidinone, tedizolid, has been registered for acute bacterial skin and skin structure infections by MRSA (Hesterkamp 2015).
Between 1940 and 1970 there was an explosion of antibiotic discoveries and their use in human medicine. Since 1970 the situation began to change and there has been a rapid drop in the introduction of new antibiotics and a sad departure by many large pharmaceutical companies from antibiotics as commercial products. In a comprehensive review Newman and Cragg (2016) summarized all new chemical entities and medical indications by source of compound between 1981 and 2014 and showed that natural products/natural product structures continue to play a dominant role in the discovery of leads for the development of drugs for the treatment of human diseases. But the Golden Age of antibiotics is a distant memory and the pipeline is almost empty (Cole 2014). Therefore, the actual situation is alarming: Dangerous rise in resistance to present antibiotics by pathogenic microbes, especially those nosocomial bacteria from the ESKAPE panel present an urgent need for the discovery of new antibiotics and chemical modifications of known antibiotic scaffolds.
For a current overview about “Natural products as sources of new drugs from 1981 to 2014” see Newman and Cragg (2016). For more information about “The clinical development and pipeline of antibiotics” see Hesterkamp (2015) and for “Strategies for the discovery and development of new antibiotics from natural products—three case studies” see Herrmann et al. (2016), the last two articles are within this EBook.
Notes
- 1.
This deviates from the modern definition of antibiotics, but is a literal citation of Waksman’s concept.
References
Abraham EP, Chain E (1940) An enzyme from bacteria able to destroy penicillin. Nature 146:837
Aminov RI (2010) A brief history of the antibiotic era: lessons learned and challenges for the future. Front Microbiol 1:134
Baltz RH (1998) Genetic manipulation of antibiotic producing Streptomyces. Trends Microbiol 2:76–83
Bange F-C, Fille M (2012) Antimykobakterielle therapie chapter 105. In: Suerbaum S, Hahn H, Burchard G-D, Kaufmann SHE, Schulz TF (eds) Medizinische Mikrobiologie und Infektiologie 7 Auflage Springer, Berlin
Bange F-C, Hahn H, Kaufmann SHE, Ulrichs T (2012) Mykobakterien. Chapter 41. In: Suerbaum S, Hahn H, Burchard G-D, Kaufmann SHE, Schulz TF (eds) Medizinische Mikrobiologie und Infektiologie 7 Auflage. Springer, Berlin
Barber M (1961) Methicillin-resistant staphylococci. J Clin Pathol 14:385–393
Baumann S, Herrmann J, Raju R, Steinmetz H, Mohr KI, Hüttel S, Harmrolfs K, Stadler M, Müller R (2014) Cystobactamids: myxobacterial topoisomerase inhibitors exhibiting potent antibacterial activity. Angew Chem Int Ed 53:1–6
Béchamp MA (1863) De l’action de la chaleur sur l’arseniate d’analine et de la formation d’un anilide de l’acide arsenique. Compt Rend 56:1172–1175
Bennett JW (2015) What is an antibiotic? In: Sánchez S, Demain AL (eds) Antibiotics-current innovations and future trends. Caister Academic Press
Bentley R (2000) Mycophenolic acid: a one hundred year odyssey from antibiotic to immunosuppressant. Chem Rev 100:3801–3826
Bentley R, Bennett JW (2003) What is an antibiotic? Revisited. Adv Appl Microbiol 52:303–331
Bentley R (2009) Different roads to discovery; Prontosil (hence sulfa drugs) and penicillin (hence b-lactams). J Ind Microbiol Biotechnol 36:775–786
Berger A, Fingerle V, Sing A (2012) Treponemen. Chapter 43. In: Suerbaum S, Hahn H, Burchard G-D, Kaufmann SHE, Schulz TF (2012) Medizinische Mikrobiologie und Infektiologie. 7. Auflage. Springer, Berlin
Binda E, Marinelli F, Marcone GL (2014) Old and new glycopeptide antibiotics: action and resistance. Antibiotics 3:572–594
Birch RG, Patil SS (1985) Preliminary characterization of an antibiotic produced by Xanthomonas albilineans which inhibits DNA synthesis in Escherichia coli. J Gen Microbiol 131:1069–1075
Bisacchi GS (2015) Origins of the Quinolone class of antibacterials: an expanded “Discovery Story”. J Med Chem 58:4874–4882
Bloemendal S, Kück U (2014) Cephalosporins. In: Martín JF, García-Estrada C, Zeilinger S (eds) Biosynthesis and molecular genetics of fungal secondary metabolites. Springer, New York, pp 43–64
Bosch F, Rosich L (2008) The contributions of Paul Ehrlich to pharmacology: a tribute on the occasion of the centenary of his Nobel Prize. Pharmacology 82:171–179
Brewer GA, Johnson MJ (1953) Activity and properties of para-aminobenzyl penicillin. Appl Microbiol 1:163–166
Bulst N (1977) Der Schwarze Tod. Demographische, wirtschaftliche und kulturgeschichtliche Aspekte der Pestkatastrophe von 1347–1352. Bilanz der neueren Forschung. Antrittsvorlesung an der Universität Heidelberg
Burris RH, Baumann A, Potter VR (1990) Conrad Arnold Elvehjem. Biographical memoir, Nat Acad Sc
Carter GT (2009) Natural products in drug discovery. In: Stromgaard K, Krogsgaard-Larsen P, Madsen U (eds.) Textbook of drug design and discovery, 4th edn. CRC Press Boca Raton, pp 89–106
Cha JY, Ishiwata A, Mobashery S (2004) A novel β-lactamase activity from a penicillin-binding protein of Treponema pallidum and why syphilis is still treatable with penicillin. J Biol Chem 279:14917–14921
Chain E, Florey HW, Gardner AD, Heatley NG, Jennings MA, Orr-Ewing J, Sanders AG (1940) Penicillin as a chemotherapeutic agent. Lancet ii: 226–228
Charlebois A, Jalbert LA, Harel J, Masson L, Archambault M (2012) Characterization of genes encoding for acquired bacitracin resistance. Clostridium perfringens PloS One 7(9). doi:10.1371
Charney J, Fisher WP, Curran C, Machlowitz RA, Tytell AA (1953) Streptogramin, a new antibiotic Antibiot Chemother 3: 1283–1286
Chellat MF, Raguz L, Riedl R (2016) Antibiotikaresistenzen gezielt überwinden. Angew Chem 128:2–32
Chopra I, Roberts M (2001) Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65:232–260
Cole ST (2014) Who will develop new antibacterial agents? Phil Trans R Soc B 369:20130430
Collin F, Karkare S, Maxwell A (2011) Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl Microbiol Biotechnol 92:479–497
Controulis J, Rebstock MC, Crooks HM (1949) Chloramphenicol (Chloromycetin). V. Synthesis. J Am Chem Soc 71:2463–2468
Davies J, Wright GD (1997) Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol 5:234–240
Davies J (2006) Where have all the antibiotics gone? Can J Infect Dis Med Microbiol 17:287–290
De la Bédoyère G (2005) The discovery of penicillin (milestones in modern science). Evans Brothers Ltd
Di Giambattista M, Chinali G, Cocito C (1989) The molecular basis of the inhibitory activities of type A and type B synergimycins and related antibiotics on ribosomes. J Antimicrob Chemother 24:485–507
Domagk G (1935) Ein Beitrag zur Chemotherapie der bakteriellen Infektionen. Deutsch Med Wochenschrift 61:250–253
Drews J (2000) Drug discovery: A historical perspective. Science 287:1960–1964
Dubos RJ, Hotchkiss RD (1942) Origin, nature and property of gramicidin and tyrocidine. Trans Studies Coll Phys Phila 4(10):11–19
Dubos RJ, Hotchkiss RD, Coburn AF (1942) The effect of gramicidin and tyrocidine on bacterial metabolism. J Biol Chem 146:421–426
Duchesne E (1897) Contribution à l’etude de la concurrence vitale chez les microorganismes. Antagonisme entre les moisissures et le microbes. Thèse. Lyon: Faculté de Médecine et de Pharmacie de Lyon
Duckett S (1999) Ernest Duchesne and the concept of fungal antibiotic therapy. Lancet 11(354):2068–2071
Duggar BM (1948) Aureomycin; a product of the continuing search for new antibiotics. Ann N Y Acad Sci 30:177–181
Ehrlich P (1910) Die Behandlung der Syphilis mit dem Ehrlichschen Präparat 606. Deutsche medizinische Wochenschrift: 1893–1896
Ehrlich P, Hata S (1910) Die Experimentelle Chemotherapie der Spirillosen. Springer, Berlin
Ehrlich P, Bertheim A (1912): Über das salzsaure 3.3-Diamino-4.4-dioxy-arsenobenzol und seine nächsten Verwandten. Berichte der deutschen chemischen Gesellschaft Bd 45(1):756–766
Ehrlich J, Bartz QR, Smith RM, Joslyn DA, Burkholder PR (1947) Chloromycetin, a new antibiotic from a soil actinomycete. Science 31:417
Ehrlich J, Gottlieb D, Burkholder PR, Anderson LE, Pridham TG (1948) Streptomyces venezuelae, N. sp., The source of chloromycetin. J Bacteriol 56:467–477
Falkow S (1975) Infectious multiple drug resistance. Pion Ltd London United Kingdom
Ferber D (2003) Triple-threat microbe gained powers from another bug. Science 302:1488
Finlay AC, Hobby GL, P’an SY, Regna PP, Routien JB, Seeley DB, Shull GM, Sobin BA, Vinson IAJW, Kane JH (1950) Terramycin, a new antibiotic. Science 27:85
Fleming A (1929) On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. Br J Exp Pathol 10(3):226–236
Gause GF, Brazhnikova MG (1944) Gramicidin S and its use in the treatment of infected wounds. Nature 154:703
Gellert M, Mizuuchi K, O’Dea MH, Nash HA (1976) DNA gyrase: an enzymethat introduces superhelical turns into DNA. Proc Natl Acad Sci USA 73:3872–3876
Gelmo P (1908) Über Sulfamide der p-Amidobenzolsulfonsäure. J prak Chem 77:369–382
Goldstein FW, Kitzis MD, Acar JF (1994) N, N-Dimethylglycylamido derivative of minocycline and 6-demethyl-6-deoxytetracycline, two new glycylcyclines highly effective against tetracycline-resistant Gram positive cocci. Antimicrob Agents Chemother 38: 2218–2220
Gosio B (1893) Contributo all’etiologia della pellagra; ricerche chimiche e batteriologische sulle alterazioni del mais. Giornale della Reale Accademia di Medicina di Torino 61:484–487
Gosio B (1896) Ricerdre batteriologische chirmide sulle alterazioni del mais. Rivista d’Ingienne e Sanita Publica 7:825–868
Gottlieb D, Bhattacharyya PK, Anderson HW, Carter HE (1948) Some properties of an antibiotic from a species of Streptomyces. J Bact 55:409–417
Griesgraber G, Or YS, Chu DTW, Nilius AM, Johnson PM, Flamm RK, Henry RF, Plattner JJ (1996) 3-Keto-11, 12-carbazate Derivatives of 6-O-Methylerythromycin A synthesis and in vitro activity. J Antibiot 49:465–477
Grohe K, Heitzer H (1987) Cycloaracylierung von Enaminen, I Synthese von 4-Chinolon-3-carbonsauren. Lieb Ann Chem 1:29–37
Grohe K, Zeiler HJ, Metzger K (1981) 1-cyclopropyl-6-fluor-1,4-dihydro-4-oxo-7-piperazino-chinolin-3-carbonsaeuren, Verfahren zu ihrer Herstellung sowie diese enthaltende antibakterielle Mittel. DE 3142854:A1
Grundy WE, Sinclair AC, Theriault RJ, Goldstein AW, Rickher CJ, Warren HB Jr, Oliver TJ, Sylvester JC (1956–1957) Ristocetin, microbiologic properties. Antibiot Annu 687–692
Guttmann P, Ehrlich P (1891) Über die Wirkung des Methylenblau bei Malaria. Berl Klin Wochenschr 28:953–956
Haensch S, Bianucci R, Signoli M et al (2010) Distinct clones of yersinia pestis caused the black death. Ed Nora J Besansky PLoS Pathogens 6(10):e1001134
Hart GD (2001) Descriptions of blood and blood disorders before the advent of laboratory studies. Histl Rev Brit J Haem 115:719–728
Harris DA, Reagan MA, Ruger M, Wallick H, Woodruff HB (1955) Discovery and antimicrobial properties of cathomycin, a new antibiotic produced by Streptomyces spheroides n. sp. Antibiot Annu 3:909–917
Hawgood BJ (2008) Alexandre Yersin (1863–1943): discoverer of the plague bacillus, explorer and agronomist. J Med Biog 16:167–172
Heide L (2014) New aminocoumarin antibiotics as gyrase inhibitors. Int J Med Microbiol 304:31–36
Herrmann J, Lukežič T, Kling A, Baumann S, Hüttel S, Petković H, Müller R (2016) Strategies for the discovery and development of new antibiotics from natural products: three case studies. Curr Top Microbiol Immunol Springer EBook
Hesterkamp T (2015) Antibiotics clinical development and pipeline. Curr Top Microbiol Immunol. (Springer EBook). doi:10.1007/82_2015_451
Höck M (2012a) Glycopeptidantibiotika. Chapter 98. Suerbaum S, Hahn H, Burchard G-D, Kaufmann SHE, Schulz TF (eds) Medizinische Mikrobiologie und Infektiologie 7 Auflage. Springer, Berlin
Höck M (2012b) Makrolide. Chapter 102. Suerbaum S, Hahn H, Burchard G-D, Kaufmann SHE, Schulz TF (eds) Medizinische Mikrobiologie und Infektiologie 7. Auflage. Springer, Berlin
Hollstein U (1973) Actinomycin. chemistry and mechanism of action. Chem Rev 6:625–652
Hotchkiss RD, Dubos RJ (1940a) Fractionation of bactericidal agent from cultures of a soil bacillus. J Biol Chem 132:791–792
Hotchkiss RD, Dubos RJ (1940b) Chemical properties of bactericidal substances isolated from cultures of a soil bacillus. J Biol Chem 132:793–794
Houbraken J, Frisvad JC, Samson RA (2011) Fleming’s penicillin producing strain is not Penicillium chrysogenum but P. rubens. IMA Fungus 2(1):87–95
Hughes VM, Datta N (1983) Conjugative plasmids in bacteria of the ‘pre-antibiotic’ era. Nature 302:725–726
Husain MA (2004) Bacitracin, glycopeptide antibiotics, and the polymyxins. In: Craig CR, Stitzel RE (eds) Modern pharmacology with clinical applications 48:552
Inglesby TV, Dennis DT, Henderson DA et al (2000) Plague as a biological weapon: medical and public health management. Working Group on Civilian Biodefense JAMA 283:2281–2290
Jacoby GA (2005) Mechanisms of resistance to quinolones. Clin Inf Dis 41:120–126
Jacoby GA, Bush K (2005) Beta-lactam resistance in the 21st century. In: White DG, Alekshun MN, McDermott PF (eds) Frontiers in antimicrobial resistance. ASM Press, Washington, DC, pp 53–65
Jäger H (2011) Cinchona pubescens. In: Roloff, A., Weisgerber, H., Lang, U., Stimm, B. (Hrsg.): Enzyklopädie der Holzgewächse, Wiley-VCH, Weinheim, 58:1–14
Johnson BA, Anker H, Meleney FL (1945) Bacitracin: A new antibiotic produced by a member of the B. subtilis group. Science 12:376–377
Jovetic S, Zhu Y, Marcone GL, Marinelli F, Tramper J (2010) β-Lactam and glycopeptide antibiotics: first and last line of defense? Trends Biotechnol 28:596–604
Kahne, D Leimkuhler C, Lu W, Walsh C (2005) Glycopeptide and lipoglycopeptide antibiotics. Chem Rev 105:425–448
Karwehl S, Stadler M (2016) Exploitation of fungal biodiversity for discovery of novel antibiotics. In: Curr Top Microbiol Immunol, in press. doi:10.1007/82_2016_496
Khosla R, Verma DD, Kapur A, Aruna RV, Khanna N (1999) Streptogramins: a new class of antibiotics. Indian J Med Sci 53:111–119
Klein P, Falke D, Hahn H (2012) Ursprung der medizinischen Mikrobiologie. Chapter 2. In: Suerbaum S, Hahn H, Burchard G-D, Kaufmann SHE, Schulz TF (eds) Medizinische Mikrobiologie und Infektiologie. 7. Auflage. Springer, Berlin
Kool JL (2005) Risk of person-to-person transmission of pneumonic plague. Clin Infect Dis 40(8):1166–1172
Kretz J, Kerwat D, Schubert V, Grätz S, Pesic A, Semsary S, Cociancich S, Royer M, Süssmuth RD (2015) Total synthesis of albicidin: A lead structure from Xanthomonas albilineans for potent antibacterial gyrase inhibitors. Angew Chem Int Ed 54:1969–1973
Kumarasamy KK, Toleman MA, Walsh TR, Bagaria J, Butt F, Balakrishnan R, Chaudhary U, Doumith M, Giske CG, Irfan S, Krishnan P, Kumar AV, Maharjan S, Mushtaq S, Noorie T, Paterson DL, Pearson A, Perry C, Pike R, Rao B, Ray U, Sarma JB, Sharma M, Sheridan E, Thirunarayan MA, Turton J, Upadhyay S, Warner M, Welfare W, Livermore DM, Woodford N (2010) Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect Dis 10:597–602
Leclercq R (2002) Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications. Clin Infect Dis 34:482–492
Lesher GY, Froelich EJ, Gurett MD, Baily JH, Brundage RP (1962) 1,8-Naphthyridine derivatives. A new class of chemotherapeutic agents. J Med Pharm Chem 91:1063–1065
Lešnik U, Lukežič T, Podgoršek A, Horvat J, Polak T, Šala M, Jenko B, Harmrolfs K, Ocampo-Sosa A, Martínez-Martínez L, Herron PR, Fujs S, Kosec G, Hunter IS, Müller R (2015) Construction of a new class of tetracycline lead structures with potent antibacterial activity through biosynthetic engineering. Angew Chem Int Ed 54:3937–3940
Levy SB (1984) Resistance to the tetracyclines. In Bryan LE (ed) Antimicrobial drug resistance. Academic Press, pp 191–240
Lewis K (2013) Platforms for antibiotic discovery. Nat Rev Drug Discovery 12:371–387
Lewis C, Clapp HW, Grady JE (1962) In vitro and in vivo evaluation of lincomycin, a new antibiotic. Antimicrob Agents Chemother 2:570
Lippi D, Conti AA (2002) Plague, policy, saints and terrorists: a historical survey. J Infect 44(4):226–228
Lloyd NC, Morgan HW, Nicholson BK, Ronimus RS (2005) The composition of Ehrlich’s Salvarsan: resolution of a century-old debate. Angew Chem Int Ed 44:941–944
Madigan MT, Martinko JM, Parker J (2000) Brock Mikrobiologie. Spektrum Akademischer Verlag
Majno G, Joris I (1979) Billroth and Penicillium. Rev Infect Dis 5:880–884
Mason DJ, Dietz A, Deboer C (1962) Lincomycin-a new antibiotic. I. Discovery and biological properties AAC 2:554–559
McCormick MH, McGuire JM, Pittenger GE, Pittenger RC, Stark WM (1955–1956) Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot Annu 3:606–611
McGuire JM, Bunch RL, Anderson RC, Boaz HE, Flynn EH, Powell HM, Smith JW (1952) Ilotycin, a new antibiotic. Antibiot Chemother 2:281–283
McMurry LM, Levy SB (2000) Tetracycline resistance in Gram positive bacteria. 660–677. In: Fischetti VA, Novick RP, Ferretti JJ, Portnoy DA, and Rood JI (eds) Gram positive pathogens. American Society for Microbiology, Washington DC
Meyers BR, Kaplan K, Weinstein L (1969) Micro- biological and pharmacological behaviour of 7-chloro- lincomycin. Appl Microbiol 17:653
Mingeot-Leclercq M-P, Glupczynski Y, Tulkens PM (1999) Aminoglycosides: activity and resistance. AAC 43:727–737
Mitscher LA, Juvarkar JV, Rosenbrook W, Andres WW, Schenck JR, Egan RS (1970) Structure of chelocardin, a novel tetracycline antibiotic. J Am Chem Soc 92:6070–6071
Morar M, Bhullar K, Hughes DW, Junop M, Wright GD (2009) Structure and mechanism of the lincosamide antibiotic adenylyltransferase LinB. Structure 17:1649–1659
Nelson ML, Park BH, Andrew JS, Georgian VA, Thomas BC, Levy SB (1993) Inhibition of the tetracycline efflux antiport protein by 13-thio-substituted 5-hydroxy-6-deoxytetracyclines. J Med Chem 36:370–377
Nelson ML, Levy SB (1999) Reversal of tetracycline resistance mediated by different bacterial tetracycline resistance determinants by an inhibitor of the Tet(B) antiport protein. Antimicrob Agents Chemother 43:1719–1724
Nelson RRS (1999) Intrinsically vancomycin-resistant Gram-positive organisms: clinical relevance and implications for infection control. J Hosp Inf 42:275–282
Neufeldt S (2003) Chronologie Chemie: Entdecker und Entdeckungen Ed. Neufeldt S. 3. Auflage Wiley VCH
Newman DJ, Cragg GM (2016) Natural products as sources of new drugs from 1981 to 2014. J Nat Prod 79:629–661
Newton GGF, Abraham EP (1955) Cephalosporin C, a new antibiotic containing sulphur and D-aminoadipic acid. Nature 175:548
Nielsen ED, Hamilton PB, David Rosi D, Peruzzotti GP (1963) Microbiological oxidation of 7-methyl-1, 8-naphthyridines to 7-hydroxymethyl-1, 8-naphthyridines. US 3317401 A Sterling Drug Inc
No authors listed (1967) Gentamicin. Br Med J 21:158–159
Nord C (2010) Mycophenolic Acid—The road from an early antibiotic to a modern drug. Biologiskt aktiva naturprodukter I läkemedelsutvecklingen, pp 1–11
Oliva B, Gordon G, McNicholas P, Ellestad G, Chopra I (1992) Evidence that tetracycline analogs whose primary target is not the bacterial ribosome cause lysis of Escherichia coli. Antimicrob Agents Chemother 36:913–919
Oliver TJ, Ptokop JF, Bower RR, Otto RH (1962) Chelocardin, a new broad spectrum antibiotic. I. Discovery and biological properties. Antimicrob Agents Chemother 583–591
Oppolzer W, Prelog V, Sensi P (1964) Experientia 20:336–339
Pandit N, Singla RK, Shrivastava B (2012) Current updates on oxazolidinone and its significance. Int J Med Chem. doi:10.1155/2012/159285
Parenti F, Beretta G, Berti M, Arioli V (1978) Teicomycins, new antibiotics from Actinoplanes teicomyceticus nov. sp. I. Description of the producer strain, fermentation studies and biological properties. J Antibiot 31:276–283
Rezende L (2006) Chronology of science. Checkmark, New York
Rollo IM, Williamson J, Plackett RL (1952) Acquired resistance to penicillin and to neoarsphenamine in Spirochaeta recurrentis. Br J Pharmacol Chemother 7:33–41
Rubin RP (2007) A brief history of great discoveries in pharmacology. In celebration of the centennial anniversary of the founding of the american society of pharmacology and experimental therapeutics. Pharmacol Rev 59:289–359
Schatz A, Bugie E, Waksman S (1944) Streptomycin: A substance exhibiting antibiotic activity against gram positive and gram negative bacteria. Proc Exp Biol Med 55:66–69
Sensi P, Greco AM, Ballotta R (1960) Rifomycins. I. Isolation and properties of rifomycin B and rifomycin complex. Antibiot Annu 262–270
Shaw KJ, Rather PN, Hare RS, Miller GH (1993) Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 57:138–163
Silver LL (2012) Chapter 2 Rational Approaches to antibacterial discovery: pre-genomic directed and phenotypic screening, 33–75. In: Dougherty TJ, Pucci MJ (eds) Antibiotic discovery and development, Springer, Berlin
Sköld O (2000) Sulfonamide resistance: mechanisms and trends. Drug Resist Updat 3(3):155–160
Smith RM, Joslyn DA, Gruhzit OM, McLean W, Penner MA, Ehrlich J (1947) Chloromycetin: biological studies
Somma S, Gastaldo L, Corti A (1984) Teicoplanin, a new antibiotic from Actinoplanes teichomyceticus nov. sp. Antimicrob Agents Chemother 26:917–923
Spoerer M, Streb J (2013) Neue deutsche Wirtschaftsgeschichte des 20. Walter de Gruyter, Jahrhunderts
Stahlmann R, Riecke K (2004) Die Infektiologie. 5.3.14.1 Editors: Adam D, Doerr HW, Link H, Lode H (Hrsg.), Springer
Sydenstricker VP (1958) The History of Pellagra. Its Recognition as a Disorder of Nutrition and Its Conquest. Am J Clin Nutr 6(4):409–414
Takeuchi T, Hikiji T, Nitta K, Yamazaki S, Abe S, Takayama H, Umezawa H (1957) Biological studies on kanamycin. J Antibiot (Tokyo) 10:107–114
Thomas HW (1905) Some experiments in the treatment of trypanosomiasis. Brit Med J 1140
Tréfouël J and T, Nitti F, Bovet D (1935) Activité du p-aminophénylsulfamide sur l’infection streptococcique expérimentale de la souris et du lapin. C R Soc Biol 120(23):756
Tymiak AA, Aklonis C, Bolgar MS et al (1993) Novel tetracycline glycosides active against tetracycline-resistant bacteria. J Org Chem 58:535–537
Tyndall J (1877) The optical deportment of the atmosphere in relation to the phenomena of putrefaction and infection. Philos Trans R Soc Lond 166:27–74
Waksman SA, Woodruff HB (1940) Bacteriostatic and bacteriocidal substances produced by soil actinomycetes. Proc Soc Exp Biol 45:609–614
Waksman S, Woodruff HB (1941) Actinomyces Antibioticus, a new soil organism antagonistic to pathogenic and non-pathogenic bacteria. J Bacteriol 42:231–249
Waksman SA (1947) What is an antibiotic or an antibiotic substance? Mycologia 39:565–569
Waksman SA, Lechevalier HA, Harris DA (1949) Neomycin-production and antibiotic properties. J Clin Invest 28:934–939
Waksman SA, Lechevalier HA (1949) Neomycin, a new antibiotic active against streptomycin-resistant bacteria, including tuberculosis organisms. Science 25:305–307
Weber T, Welzel K, Pelzer S, Vente A, Wohlleben W (2003) Exploiting the genetic potential of polyketide producing streptomycetes. J Biotechnol 106:221–232
Weiss HJ, Rogers J, Brand H (1973) Defective ristocetin-induced platelet aggregation in von Willebrand’s disease and its correction by factor VIII. J Clin Invest 52:2697–2707
WHO (2009) Treatment of tuberculosis: guidelines for national programmes
Williams KJ (2009) The introduction of ‘chemotherapy’ using arsphenamine—The first magic bullet. J R Soc Med 102(8):343–348
Williams C (2013) Medicinal plants in Australia Vol 4: An antipodean apothecary. Rosenberg Publishing
Woodward RB, Doering WE (1945) The total synthesis of quinine. J. Amer Chem Soc 67:860–874
Würth I (2012) Geißler in Thüringen: Die Entstehung einer spätmittelalterlichen Häresie (Hallische Beiträge zur Geschichte des Mittelalters und der Frühen Neuzeit, Band 10
Zhanel GG, Walters M, Noreddin A, Vercaigne LM, Wierzbowski A, Embil JM, Gin AS, Douthwaite S, Hoban DJ (2002) The ketolides: a critical review. Drugs 62:1771–1804
Zimdahl RL (2015) Six chemicals that changed agriculture. 1st edn. Academic Press Elsevier
Acknowledgments
I would like to thank Prof. Dr. Christoph Friedrich, Bildarchiv des Instituts für Geschichte der Pharmazie der Universität Marburg (Germany), Prof. Dr. Ulrich Kück and Dr. Julia Böhm, Ruhr-University Bochum, Dr. Heinke Jäger, Restoration Ecologist Charles Darwin Foundation Puerta Ayora, Galápagos (Ecuador), and PD Dr. Joachim Wink for providing the nice pictures and Dr. Rolf Jansen for examination of the chemical structures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing AG
About this chapter
Cite this chapter
Mohr, K.I. (2016). History of Antibiotics Research. In: Stadler, M., Dersch, P. (eds) How to Overcome the Antibiotic Crisis . Current Topics in Microbiology and Immunology, vol 398. Springer, Cham. https://doi.org/10.1007/82_2016_499
Download citation
DOI: https://doi.org/10.1007/82_2016_499
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-49282-7
Online ISBN: 978-3-319-49284-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)